
Abstract
Background
Optimal treatment for patients with HCV genotype-3 infection and liver cirrhosis remains a medical priority. Daclatasvir+sofosbuvir and ribavirin is a recommended option for such patients, but clinical trial data are lacking for treatment >16 weeks.
Methods
This was a single-arm, Phase III study of daclatasvir+sofosbuvir+ribavirin for 24 weeks in patients with compensated cirrhosis and HCV genotype-3 infection. The primary end point was sustained virological response at post-treatment week 12 (SVR12); the primary objective was to demonstrate statistical superiority to historical SVR12 data for 12 weeks’ daclatasvir+sofosbuvir without ribavirin in genotype-3-infected patients with cirrhosis (95% CI lower bound >79.0%).
Results
A total of 78 patients were treated (54 treatment-naive, 24 treatment-experienced including 8 with prior sofosbuvir exposure). SVR12 was achieved by 87% (68/78; 95% CI 77.7, 93.7%) of patients in the primary analysis of central laboratory data. One additional patient achieved SVR12 by local testing resulting in an overall SVR12 rate of 88% (95% CI 79.2, 94.6%) and the lower bound of the 95% CI above the historical threshold. SVR12 rates were 93% (50/54) for treatment-naive and 79% (19/24) for treatment-experienced patients. Of the nine non-SVR12 patients, four were lost to follow-up, two relapsed (both sofosbuvir-experienced), two had end-of-treatment virological failure and one discontinued early. There were no unexpected safety signals; only one patient discontinued for an adverse event.
Conclusions
Daclatasvir+sofosbuvir+ribavirin for 24 weeks was well tolerated and efficacious in HCV genotype-3-infected patients with compensated cirrhosis, with SVR12 outcomes comparable to previously reported outcomes in patients treated with this regimen for 12–16 weeks.
Introduction
HCV genotype (GT)-3 is the second most common of the seven major HCV genotypes, and responsible for approximately 30% of global HCV infections [1], 30–40% of infections in parts of Europe and Australia [2–5], with an even higher prevalence (>60%) in parts of South East Asia [6]. GT-3 infection results in more rapid progression of HCV-associated liver disease and a higher risk of hepatocellular carcinoma, hospitalization and death than other genotypes [7–10], and has proven challenging to cure with oral direct-acting antiviral (DAA) regimens. Several DAA agents used successfully for the treatment of HCV GT-1 have poor antiviral activity in vitro against GT-3, including ledipasvir [11], simeprevir [12,13], dasabuvir [14] and asunaprevir [15]. Although the range of options for GT-3 infection is now expanding, this patient group remains in need of treatment options, particularly those with cirrhosis who have historically shown reduced response to a number of DAA-based regimens.
The pan-genotypic DAA combination of daclatasvir (DCV), an HCV non-structural protein (NS) 5A inhibitor, and sofosbuvir (SOF), a nucleoside analogue NS5B polymerase inhibitor, has been extensively studied in clinical trials of patients with HCV GT-3 infection [16,17]. Data from compassionate use programmes with a significant proportion of GT-3 patients have also been reported [18,19], as well as studies in a variety of at-need patient groups including those with advanced liver disease or prior liver transplant [18–20], and patients with HIV–HCV coinfection receiving a broad range of antiretroviral therapies [21–23]. The ALLY-3 study, which explored 12 weeks of DCV+SOF treatment in GT-3-infected patients with or without compensated cirrhosis, noted a lower rate of sustained virological response at post-treatment week 12 (SVR12) among cirrhotic patients (63% versus 96% without cirrhosis) [16]. The follow-up ALLY-3+ study demonstrated that the addition of ribavirin (RBV) to DCV+SOF significantly enhanced efficacy in GT-3-infected patients, with SVR12 rates of 83% and 89% among patients with compensated cirrhosis treated for 12 or 16 weeks, respectively [17]. However, there are no clinical trial data for the DCV+SOF+RBV combination administered for longer than 16 weeks, other than observational analyses from large, non-randomized, European early-access cohorts of patients with advanced liver disease. The largest of these analyses from the French ‘Autorisation Temporaire d'Utilisation’ (ATU) programme showed an 86% SVR12 rate in GT-3 patients with compensated or decompensated cirrhosis treated with DCV+SOF without RBV for a duration of 24 weeks, with no benefit seen with the addition of RBV (SVR12, 82%) [18]. However, RBV dosing was at physician's discretion and those who received RBV had more advanced baseline markers of disease than those who did not, which, along with the limitations of voluntary data submission for participating physicians, confounded any comparisons. Thus, there is a need for formal evaluation of 24-week treatment with DCV+SOF+RBV in advanced HCV GT-3 disease, including those with prior treatment experience who previously failed interferon- or SOF-based regimens.
Herein we report the results of a Phase III study of DCV+SOF+RBV for 24 weeks in HCV GT-3-infected patients with compensated cirrhosis.
Methods
Study design and patients
This was an open-label, single-arm, multicentre study (ALLY-3C; ClinicalTrials.gov ID NCT02673489) evaluating the safety and efficacy of 24 weeks’ oral treatment with DCV+SOF+RBV in treatment-naive or treatment-experienced patients with HCV GT-3 infection and compensated cirrhosis.
Eligible patients were HCV GT-3-infected adults ≥18 years of age with a screening HCV RNA ≥10,000 IU/ml and a body mass index between 18 and 40 kg/m2. All patients had compensated cirrhosis, defined by either a liver biopsy (Metavir >F3, Ishak >4 or equivalent) at any time prior to screening; a transient elastography (FibroScan) result ≥14.6 kPa within 1 year of baseline/day 1; or a screening serum FibroTest score ≥0.75 with a screening aspartate-aminotransferase-to-platelet ratio index (APRI) ≥2. Cirrhosis was defined via a hierarchical assessment in which biopsy data ranked above FibroScan results, which in turn ranked higher than FibroTest+APRI results.
Treatment-naive patients had no prior exposure to any type of HCV therapy; treatment-experienced patients were permitted to have received any form of HCV therapy except NS5A inhibitors, with all treatment concluded no less than 12 weeks before screening.
Coinfection with HCV and HIV-1 was permitted provided that patients had undetectable plasma HIV RNA (<40 copies/ml) on combination antiretroviral therapy. Permitted antiretrovirals were as previously described [21]; in addition, etravirine or cobicistat-boosted elvitegravir or protease inhibitors were also permitted in this study.
Major exclusion criteria were non-GT-3 or mixed genotype HCV infection; prior NS5A inhibitor exposure; HBsAg seropositivity; decompensated liver disease or hepatocellular carcinoma; uncontrolled diabetes (confirmed screening HbA1c ≥8.5) or hypertension, and prior discontinuation of SOF+RBV for intolerance or exacerbated anaemia. Amiodarone was not permitted during the study or within 60 days of study drug initiation. Women of child-bearing potential were required to use two methods of contraception during and for ≥6 months after the study (unless not heterosexually active), could not be breastfeeding and required a negative pregnancy test result within 24 hours of study drug initiation.
Patients received 24 weeks of treatment with DCV 60 mg once daily plus SOF 400 mg once daily, plus weight-based RBV 1,000 mg/day (<75 kg) or 1,200 mg/day (≥75 kg) in two divided doses, followed by 24 weeks of follow-up. DCV dosing was reduced to 30 mg/day for HIV–HCV-coinfected patients receiving boosted atazanavir, fosamprenavir or elvitegravir, and increased to 90 mg/day with efavirenz, nevirapine or etravirine.
The study was conducted at 19 clinical centres in the United States and Canada in accordance with the ethical principles originating in the Declaration of Helsinki. The study protocol was approved by the institutional review board or independent ethics committee at each study site, and all patients provided written informed consent prior to participation.
Study assessments
HCV genotype and subtype were assessed using the Versant HCV genotype 2.0 (LIPA) assay (Siemens Healthcare Diagnostics, Tarrytown, NY, USA). Plasma HCV RNA was determined at screening, baseline (day 1), weeks 1, 2, 4, 8, 12, 16, 20 and 24 or at end of treatment (EOT) if week 24 was not reached, and at post-treatment weeks 4, 12 and 24, using the Roche COBAS® AmpliPrep/COBAS® TaqMan® HCV Test v2.0 (Roche Molecular Systems, Pleasanton, CA, USA) with a lower limit of quantitation (LLOQ) of 15 IU/ml. On-treatment virological response was defined as HCV RNA <LLOQ without detectable target (<LLOQTND) and post-treatment virological response as HCV RNA <LLOQ with or without detectable target (<LLOQTD/TND). Virological failure was defined as virological breakthrough (confirmed ≥1 log10 IU/ml increase from nadir on treatment or confirmed HCV RNA ≥LLOQ if previously <LLOQTD/TND), relapse (confirmed HCV RNA ≥LLOQ following a <LLOQTND result at EOT) or HCV RNA ≥LLOQ at any time point not meeting the definition of virological breakthrough or relapse.
Resistance testing for NS5A and NS5B was performed by direct (population-based) sequencing with a sensitivity of ≥20% of the virus population, as previously described [24,25], and by next-generation sequencing (NGS) with a ≥1% sensitivity (DDL Diagnostics, Rijswijk, the Netherlands). Sequencing was performed on isolates collected at virological failure from patients with a post-failure HCV RNA ≥1,000 IU/ml. Baseline sequencing of the NS5A region was undertaken prospectively for all patients, whereas baseline sequencing of the NS5B region was performed retrospectively for patients with virological failure. Signature resistance-associated substitutions (RASs) in both target proteins were identified as previously described [25]. Phylogenetic analyses were performed on all available baseline and virological failure NS5A sequences (nucleotides 1–718) from this study and 13 published GT-3 reference sequences [26], as described previously [27].
Statistical analyses
The primary efficacy end point was the proportion of all treated patients achieving an SVR12 (HCV RNA <LLOQTD/TND at post-treatment week 12), analysed by a modified intention-to-treat assessment with missing data at post-treatment week 12 imputed from the next available HCV RNA measurement (next-value-carried-backward). The primary objective was to demonstrate that the SVR12 rate in ALLY-3C was greater than the historical rate derived from the 63% (95% CI 43.7, 78.9%) rate observed for GT-3-infected cirrhotic patients given 12 weeks of DCV+SOF without RBV in the ALLY-3 study. The study was deemed to have met its primary objective if the lower bound of the exact (Clopper–Pearson) [28] 95% CI around the observed SVR12 rate exceeded 79.0%, as derived from the upper CI bound from ALLY-3. For the planned sample size of 75 patients, a minimum observed SVR12 rate of 89.3% (67/75) would be required for the lower CI boundary to exceed 79%. A sensitivity analysis of the primary end point using observed values was also performed, where the denominator was based on treated patients with observed HCV RNA values at post-treatment week 12.
Key secondary end points included the proportion of patients achieving SVR12 in the presence or absence of baseline NS5A RASs, SVR at post-treatment week 24 and on-treatment safety.
Results
Patients
A total of 106 patients were enrolled across seven Canadian and 12 US clinical sites, of whom 78 were treated and analysed. Patient disposition is shown in Additional file 1. Of the 28 patients not treated, 26 did not meet one or more entry criteria, one missed the screening window and one missed the baseline visit. Baseline characteristics of the treated patients are shown in Table 1. Patients were predominantly White and male; 3 (4%) were HIV–HCV-coinfected; most patients (n=65, 83%) were categorized as cirrhotic on the basis of a FibroScan result ≥14.6 kPa, 5% (n=4) by biopsy and 12% (n=9) by a screening FibroTest+APRI result. A single treatment-experienced patient was infected with the GT-3b subtype; all others were infected with GT-3a, including five with an indeterminate GT-3 subtype at screening who were subsequently categorized by phylogenetic analysis. Among the 24 treatment-experienced patients, 15 had previously failed pegylated interferon (pegIFN)/RBV, eight had previously failed SOF-based regimens (SOF+RBV, n=7; SOF+pegIFN+RBV, n=1) and one patient had failed prior treatment with boceprevir+pegIFN/RBV.
Baseline characteristics
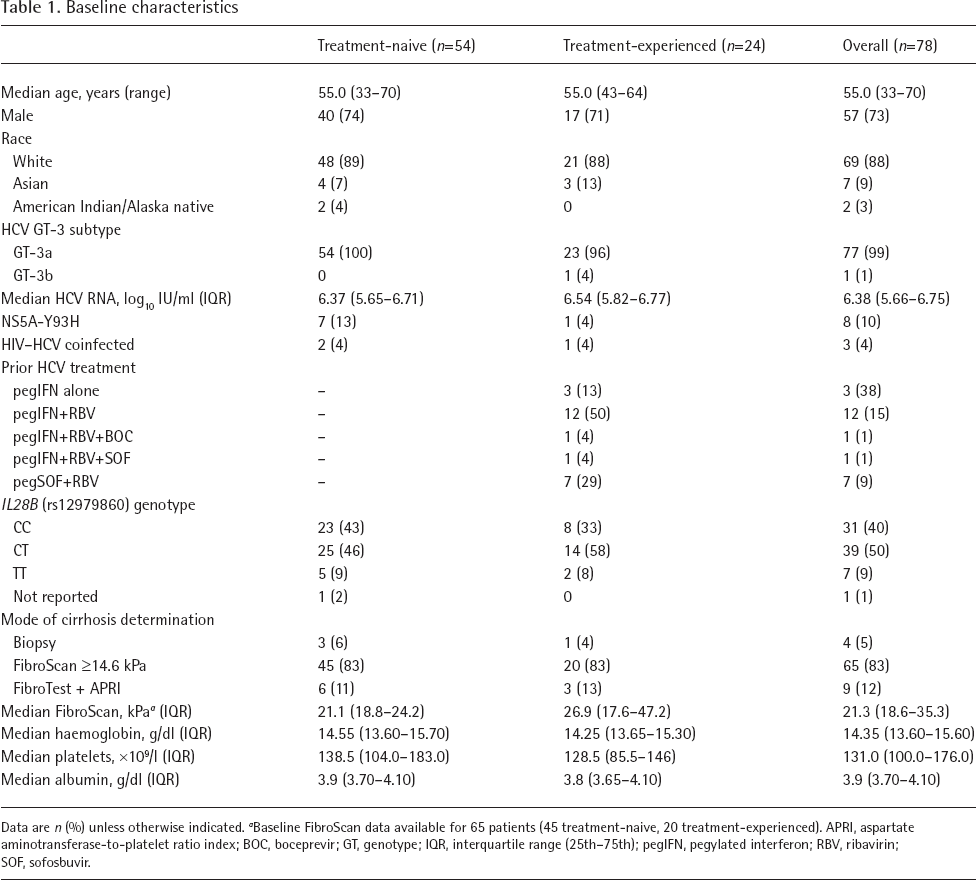
Data are n (%) unless otherwise indicated.
Baseline FibroScan data available for 65 patients (45 treatment-naive, 20 treatment-experienced). APRI, aspartate aminotransferase-to-platelet ratio index; BOC, boceprevir; GT, genotype; IQR, interquartile range (25th–75th); pegIFN, pegylated interferon; RBV, ribavirin; SOF, sofosbuvir.
Seventy (90%) patients completed the assigned 24-week treatment period. Of the eight who did not, one discontinued for an adverse event (AE), two discontinued at their own request, three were lost to follow-up, one moved to another location and one was incarcerated. No patient discontinued treatment for lack of efficacy. One treatment-naive patient completed 24 weeks of treatment but was subsequently lost to follow-up.
Virological response
Overall, SVR12 was achieved by 87% (95% CI 77.7, 93.7%) of patients per the primary analysis, which included only HCV RNA results from the central laboratory (Figure 1A). SVR12 rates were 93% in the treatment-naive group and 75% in the treatment-experienced group. One additional patient was shown to have achieved an SVR12 through local laboratory testing; with this measurement included in the analysis, the adjusted SVR12 rate was 88% (95% CI 79.2, 94.6%) and the lower bound of the 95% CI was above the historical threshold (Figure 1B). SVR12 rates using observed values were higher (94%; 67/71; 95% CI 86.2, 98.4%) due to the number of patients who were lost to follow-up at post-treatment week 12.

SVR12 rate overall and by prior treatment experience in the primary analysis
The characteristics of the 10 patients not achieving SVR12 by central laboratory testing are shown in Table 2. There were no virological failures in the treatment-naive patient group; four non-SVR12 patients were all <LLOQTND at their last available on-treatment visit but were subsequently lost to follow-up either during or immediately after treatment. Of the six non-SVR12 patients in the treatment-experienced group, two experienced relapse (both prior SOF-experienced), one was infected with HCV GT-3b and had detectable (<LLOQTD) HCV RNA at the week 24 (EOT) visit with subsequent HCV RNA samples >LLOQ at post-treatment visits, one had detectable HCV RNA at early discontinuation (day 24) per patient request, one had no on-treatment HCV RNA data due to an early (day 7) discontinuation and one was missing central laboratory data due to incarceration (as previously described).
Characteristics of patients who failed to achieve SVR12 by central laboratory testing

AE, adverse event; BL, baseline; EOT, end of treatment; F, female; GT, genotype; LLOQTD, lower limit of quantitation with detectable target; LLOQTND, lower limit of quantitation without detectable target; LTFU, lost to follow-up; M, male; RAS, resistance-associated substitution; SOF, sofosbuvir; SVR12, sustained virological response at post-treatment week 12; Tx, treatment.
Of the eight patients who had previously been treated with SOF-containing regimens, one patient who had failed SOF+pegIFN/RBV discontinued on day 7 due to a serious AE of depression. The remaining seven patients (including the incarcerated patient) had all failed prior SOF+RBV therapy; of these, five (71%) achieved SVR12 and two relapsed.
All three HIV–HCV-coinfected patients enrolled in the study achieved SVR12. Concordance between SVR12 and SVR at post-treatment week 24 was 100% in the 69 patients with data available at both time points.
Resistance-associated substitutions
Baseline NS5A resistance substitutions and impact on response
Among patients with GT-3a infection (n=77), baseline NS5A RASs at A30K or Y93H were present in 6 (8%) and 8 (10%) patients, respectively. No patient had both polymorphisms. The impact of these RASs on SVR12 was evaluated in a subset of 71 GT-3a patients (including the incarcerated patient) after excluding 6 with short treatment durations (≤4 weeks; n=2) or without observed or imputable SVR12 data following an EOT response (<LLOQTND; n=4). Baseline NS5A-A30K and -Y93H were each detected in 6/50 treatment-naive patients, and 100% of these patients with (12/12) or without (38/38) these RASs achieved SVR12. Among the treatment-experienced patients, only one had a baseline NS5A RAS (Y93H) and this patient did not achieve SVR12, whereas 95% (19/20) without this RAS achieved SVR12. A single treatment-experienced patient with HCV GT-3b infection had NS5A RASs A30K plus L31M detected at baseline. This combination has been associated with high-level resistance to DCV in vitro [24,29] and this patient did not achieve SVR12 with detectable (<LLOQTD) HCV RNA at the week 24 (EOT) visit as described above.
Emergent NS5A and NS5B substitutions
Five patients, all treatment-experienced, had virological failure and met the criteria for resistance testing. Both patients who discontinued treatment early at days 7 and 24 had no NS5A or NS5B RASs detected at baseline or virological rebound by NGS. One SOF-experienced patient with relapse had NS5A-Y93H at both baseline and failure, and also low-frequency NS5B RASs (L159F and V321A) at baseline that were not enriched at failure. The second SOF-experienced patient with relapse had emergent NS5A-Y93H, whereas low-frequency baseline NS5B RASs (L159F and F289L) detected by NGS were not observed at failure. The GT-3b-infected patient with baseline NS5A RASs A30K plus L31M had no emergent NS5A RASs, although emergent NS5B-C316R was observed in combination with baseline NS5B-T473S, as detected by direct sequencing. However, no NS5B RASs were detected by NGS at either baseline or post-failure.
SVR12 Subgroup Responses
Figure 2 shows SVR12 rates and 95% CIs for selected subgroups of gender, IL28B genotype, baseline HCV RNA, body mass index, presence of baseline NS5A-Y93H, RBV dose reductions or interruptions of >14 days and baseline platelet count. The overall SVR12 rate and 95% CIs are provided for comparison. No single factor had a clear effect on SVR12 rate. Trends towards lower SVR12 rates among patients with baseline NS5A-Y93H or baseline platelets <90x10 9 /l were observed, although the CIs were wide. Furthermore, the analysis includes one patient with NS5A-Y93 at baseline who achieved HCV RNA <LLOQTND at EOT but was subsequently lost to follow-up; if this patient was excluded, this trend may not have been observed. There was no apparent association between baseline FibroScan data and SVR12 in this patient group, although numbers were limited. The median FibroScan result for patients with SVR12 (n=56; 21.4 kPa [IQR 18.7-31.2]) versus those without SVR12 (n=9; 20.9 kPa [IQR 16.9-37.4]) were similar. FibroScan results for the two GT-3a-infected patients who experienced relapse were 36.3 and 75.0 kPa; the GT-3b-infected patient who had a detectable HCV RNA at EOT had a baseline FibroScan result of 16.0 kPa. All 17 patients who reduced or interrupted RBV achieved SVR12.
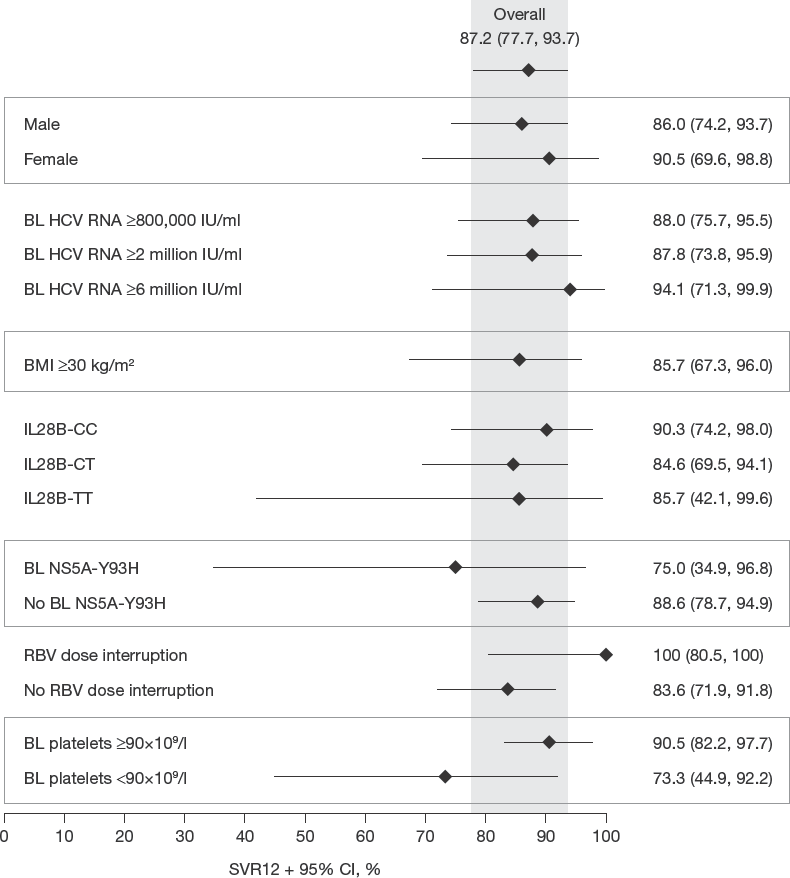
SVR12 by selected subgroups
Safety
On-treatment safety data are summarized in Table 3. Overall, treatment with DCV+SOF+RBV was well tolerated. A total of eight (10.3%) patients experienced one or more serious AEs, of which four were considered related to treatment (two RBV overdose; one SOF overdose; one worsening of depression that led to treatment discontinuation on day 7) and four were unrelated (bronchiolitis and ascites; streptococcal bacteraemia; chest discomfort; cholecystitis). The most common AEs occurring in >10% of patients were fatigue, headache, nausea, insomnia and anaemia. There were no deaths during the study. There were 19 grade 3 or 4 laboratory abnormalities on treatment, of which all were grade 3, except for three grade 4 toxicities, including two transient total lipase increases that were not associated with pancreatitis and one transient total lymphocyte decrease temporarily associated with cholecystitis. Overall, 17 patients reduced their RBV dose at least once over the course of treatment. All reductions, except one due to a dosing error, were for AEs, including one patient who both reduced RBV from 1,200 mg/day to 600 mg/day and underwent an RBV dosing interruption of 63 days following an episode of moderate anaemia.
Summary of on-treatment adverse events
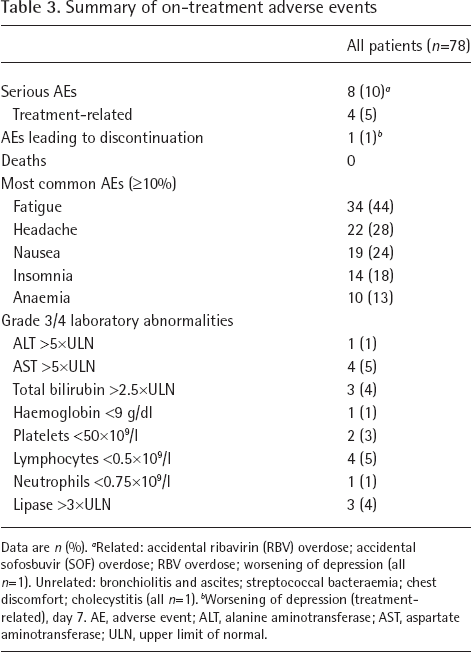
Data are n (%).
Related: accidental ribavirin (RBV) overdose; accidental sofosbuvir (SOF) overdose; RBV overdose; worsening of depression (all n=1). Unrelated: bronchiolitis and ascites; streptococcal bacteraemia; chest discomfort; cholecystitis (all n=1).
Worsening of depression (treatment-related), day 7. AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ULN, upper limit of normal.
Discussion
In this study of HCV GT-3-infected patients with cirrhosis, the combination of DCV+SOF+RBV achieved an SVR12 rate of 87% in the primary analysis. However, inclusion of an additional patient who achieved SVR12 through local laboratory testing resulted in an overall SVR12 rate of 88%, with the lower bound of the 95% CI above the historical threshold of 79% from ALLY-3 [16]. ALLY-3C complements and extends previous HCV GT-3 clinical data for DCV+SOF+RBV given for 12 or 16 weeks to 50 patients with advanced fibrosis or compensated cirrhosis (ALLY-3+) [17], and real-world ATU cohort data for 256 patients with compensated or decompensated cirrhosis given DCV+SOF±RBV for 24 weeks [18]. The SVR12 rate observed in ALLY-3C is consistent with the 83–89% overall cirrhotic SVR12 rates in ALLY-3+ (DCV+SOF+RBV for 12 or 16 weeks) and the 82–86% cirrhotic rates reported in the ATU (DCV+SOF with or without RBV for 24 weeks). All are numerically higher than the 63% rate in cirrhotic patients treated for 12 weeks without RBV in ALLY-3 [16], although only ALLY-3C was designed to compare with the historical threshold.
Subject to the limitations of cross-study comparisons, the overall data suggest that RBV plays an important role in optimizing the cirrhotic GT-3 response to DCV+SOF over shorter (12–16 weeks) treatment durations. The advantage of extending treatment with DCV+SOF+RBV beyond 12–16 weeks is unclear, as is the role of RBV when DCV+SOF is administered for 24 weeks rather than 12 weeks. The similar SVR12 rates to DCV+SOF+RBV when administered for 12 (83%), 16 (89%) or 24 weeks (87%) in ALLY-3+ and ALLY-3C make it difficult to establish an incremental advantage for longer treatment with RBV on the basis of the available data, particularly given that the numerical difference between the 12- and 16-week groups in ALLY-3+ was due to a single on-treatment death (unrelated to treatment) and not to treatment failure [17]. Likewise, the similarity of the SVR12 rate with RBV in ALLY-3C (87%) in which all patients had compensated cirrhosis, and the 86% rate seen after 24 weeks of DCV+SOF without RBV in cirrhotic GT-3-infected patients in the ATU cohort (18% decompensated) [18], make it difficult to establish a role for RBV in optimizing response at longer treatment durations.
A single GT-3b-infected patient with NS5A RASs A30K plus L31M, a combination associated with high-level resistance to DCV [24], was a treatment failure with detectable HCV RNA at EOT and subsequent HCV rebound off-treatment. This patient had low baseline HCV RNA (5.4 log10 IU/ml), normal platelet count (245x10 9 /l) and a baseline FibroScan result of 16.0 kPa. HCV RNA remained below the LLOQ from week 2 of treatment onwards, with an undetectable level at treatment week 20. Previous experience of treating GT-3b-infected patients with DCV+SOF is limited; however, a treatment-naive non-cirrhotic patient with GT-3b and baseline NS5A RASs A30K plus L31M was treated in study AI444-040 and achieved SVR12 [29]. There was no effect of baseline A30K as a single RAS in GT-3a-infected patients: 100% (n=6) of patients with only this polymorphism at baseline achieved SVR12 in ALLY-3C, which mirrored the 100% SVR12 rate for A30K alone (n=6) after shorter (12 weeks) treatment in ALLY-3+. Similarly, in the ENDURANCE-3 study, all GT-3-infected, non-cirrhotic patients with a baseline A30K RAS who received DCV+SOF (n=5) for 12 weeks achieved SVR12 [30]. In comparison, among those with this RAS at baseline receiving glecaprevir+pibrentasvir for 8 (n=16) or 12 (n=10) weeks, the SVR12 rates were 75% and 90%, respectively.
Data from the ALLY programme suggest a role for both RBV and extended treatment in optimizing SVR in GT-3-infected patients with cirrhosis who harbour baseline NS5A-Y93H; although with the caveat that any inference is based on small numbers. SVR12 was 25% (1/4) among Y93H cirrhotic patients in ALLY-3 following 12 weeks of DCV+SOF without RBV [16], whereas with RBV, SVR12 was 50% (1/2) after 12–16 weeks in ALLY-3+ [17] rising to 86% (6/7) after 24 weeks in ALLY-3C. Given the apparent effect of extended treatment on Y93H response, it is noteworthy that 24 weeks of treatment did not reduce the proportion of SOF-experienced patients in ALLY-3C who experienced virological failure compared with shorter treatment in ALLY-3+, although only a small number of SOF-experienced patients were enrolled in these studies.
Of four treatment-experienced GT-3a-infected patients who did not achieve SVR12, two discontinued treatment early with detectable HCV RNA at days 7 and 24, respectively. Thus, there were only two GT-3a patients who could be considered ‘true’ virological failures in having detectable HCV RNA following a therapeutically adequate duration of treatment (>4 weeks). Both of these virological failures were relapses in SOF-experienced patients, one of whom had baseline NS5A-Y93H and was the only virological failure with this substitution, and one of whom had no baseline NS5A RAS. The proportion of patients who achieved SVR12 in ALLY-3C (71%; 5/7) is similar to that for cirrhotic SOF-experienced patients in ALLY-3+ (67%; 4/6) [17], and in neither study did failure appear to be linked to low-frequency baseline HCV populations harbouring NS5A RASs. Thus, it would appear that in HCV GT-3-infected patients with cirrhosis, prior failure of a SOF-based regimen predisposes towards failure on DCV+SOF+RBV irrespective of treatment duration, for reasons other than the persistence of any detectable treatment-emergent SOF resistance. Indeed, similar results were observed with SOF+velpatasvir in the POLARIS-4 study, where an SVR12 rate of 85% was observed among GT-3-infected treatment-experienced patients, most of whom had received prior treatment with SOF-based regimens [31].
There were no unexpected or unusual safety observations in ALLY-3C compared with previous studies of DCV+SOF±RBV, and treatment-related AEs were uncommon.
The only other interferon-free combination studied for 24 weeks in cirrhotic patients with GT-3 infection has been SOF+RBV, which is established as a suboptimal combination for treatment-experienced patients with cirrhosis (SVR12 rates: 62–76%) [32,33]. Data for cirrhotic patients receiving glecaprevir+pibrentasvir in part 3 of the SURVEYOR-II study showed a 98% (39/40) SVR12 rate in treatment-naive patients receiving 12 weeks of treatment and a 96% (45/47) rate in treatment-experienced patients receiving 16 weeks of treatment [34]. These results have not been validated in larger real-world data sets, and this regimen is not indicated for patients beyond Child–Pugh class A cirrhosis due to the potential toxicity of the protease inhibitor glecaprevir. In the ASTRAL-3 study of SOF/velpatasvir for 12 weeks in GT-3 infection, an overall SVR12 rate of 93% (40/43) was observed in treatment-naive and 89% (33/37) in treatment-experienced patients with compensated cirrhosis; however, prior SOF exposure was a study exclusion criterion [35]. In the POLARIS-3 study, that included treatment-naive and treatment-experienced cirrhotic HCV GT-3 patients without prior exposure to DAAs, the SVR rate was 96% (105/109) in those treated with SOF+velpatasvir for 12 weeks [36]. There were only two virological failures, both of whom failed prior pegIFN/RBV and neither of whom had baseline NS5A RASs.
In conclusion, these data from the ALLY programme suggest that although 12–16 weeks of DCV+SOF+RBV will be sufficient for most GT-3-infected patients with compensated cirrhosis, those with baseline NS5A-Y93H single substitutions may benefit from 24 weeks of treatment, while there is a trend for lower SVR rates in those who have previously experienced failure of a SOF-containing regimen irrespective of treatment duration. Overall, 24 weeks of treatment with DCV+SOF+RBV in HCV GT-3-infected patients with compensated cirrhosis was efficacious and well tolerated, and resulted in SVR12 rates comparable to those previously observed among cirrhotic GT-3-infected patients given DCV+SOF for 12–16 weeks with RBV, or 24 weeks without RBV.
Footnotes
FP reports grant support, speaker fees and advisory board honorarium from Bristol-Myers Squibb. MLS reports grant support, speaker fees and advisory board honorarium from AbbVie, Bristol-Myers Squibb, Gilead, Intercept and Merck; grant support from Conatus, CymaBay, Exalenz, Galectin, Genfit, Immuron, NGM Biopharmaceuticals, Novartis and Shire; speaker fees from Bayer and Daiichi Sankyo; consulting fees from OptumRx. AW reports grant support, personal fees and non-financial support from Gilead; grant support and personal fees from AbbVie and Merck; personal fees from Bristol-Myers Squibb. GDH reports grant support from Bristol-Myers Squibb along with grant support, personal fees and consulting fees from Gilead during the conduct of the study; grant support, personal fees and consulting fees from Janssen, grant support and personal fees from ViiV, personal fees from Theratechnologies, grant support from Proteus outside of the submitted work. AR reports grant support and personal fees from AbbVie, Gilead and Merck; personal fees from Bristol-Myers Squibb, Celgene, Allergan, Janssen, Intercept, Lupin and Novartis. SDS reports grant support during the conduct of the study; grant support from AbbVie and Janssen, grant support and personal fees from Gilead and Merck, personal fees from Pfizer outside of the submitted work. FM is an employee of Bristol-Myers Squibb. RY is an employee and stockholder of Bristol-Myers Squibb. SN is an employee and stockholder of Bristol-Myers Squibb; stockholder of Merck/Schering-Plough and Johnson & Johnson. ML is an employee of Bristol-Myers Squibb. The remaining authors declare no competing interests.
Acknowledgements
This study was funded by Bristol-Myers Squibb. The authors would like to thank Eugene Scott Swenson, a BMS employee at the time that ALLY-3C was planned and undertaken, for his assistance with developing the study protocol and initiating the study. Editorial assistance with the manuscript was provided by Nick Fitch of Articulate Science (London, UK) and funded by Bristol-Myers Squibb.