
Abstract
Catalytic HIV type-1 (HIV-1) integrase (IN) and ribonuclease H (RNase H) domains belong to the polynucleotidyl transferase superfamily and are characterized by highly conserved motifs that coordinate two divalent Mg2+ cations and are attractive targets for new antiviral agents. Several structural features of both domains are now available. Drugs targeting the HIV-1 IN are currently approved for anti-HIV therapy, while no drug targeting the HIV-1 RNase H function is yet available. This review describes HIV-1 IN and the RNase H function and structures, compounds targeting their active sites and dual inhibition as a new approach for drug development.
Introduction
HIV type-1 (HIV-1) integrase (IN) and reverse transcriptase (RT)-associated ribonuclease H (RNase H) domains belong to the polynucleotidyl transferase superfamily and share common structural features. The catalytic sites of both enzymes contain a central core composed of three highly conserved amino acid residues forming the DDE motif or RNase H-like fold (Asp, Asp and Glu) [1]. These residues are essential for both enzymatic activities and their mutation leads to catalytic inactivation. Structurally, the core domain is formed by five-strand β sheets in both enzymes and four-to-six α-helices in HIV-1 IN and RNase H, respectively. The five strands of the HIV-1 IN β sheets super-impose closely with the corresponding HIV-1 RNase H sheets (Figure 1A and 1B) [2,3]. However, comparison of the helical regions shows several distinguishing features: IN α1 is displaced from the four β sheet by approximately 6 Å relative to the RNase H α1; IN α2 is a one-turn helix while RNase H α2 is a the three-turn helix; α3 is similarly positioned in both structures; α4 orientation is similar in the two structures, aligned parallel to β1; and in the HIV-1 IN structure, there are two additional helices that are not present in RNase H. IN α5 consists of residues 171–186 and is located adjacent and parallel to β3, whereas α6 extends in residues 196–208, is located on the same side as α1 and has an orientation of approximately 90° with respect to α5. Divalent metal ions and various metals, including Mn2+ and Mg2+, which are essential for the activity, have been observed in close proximity to active site residues [4].
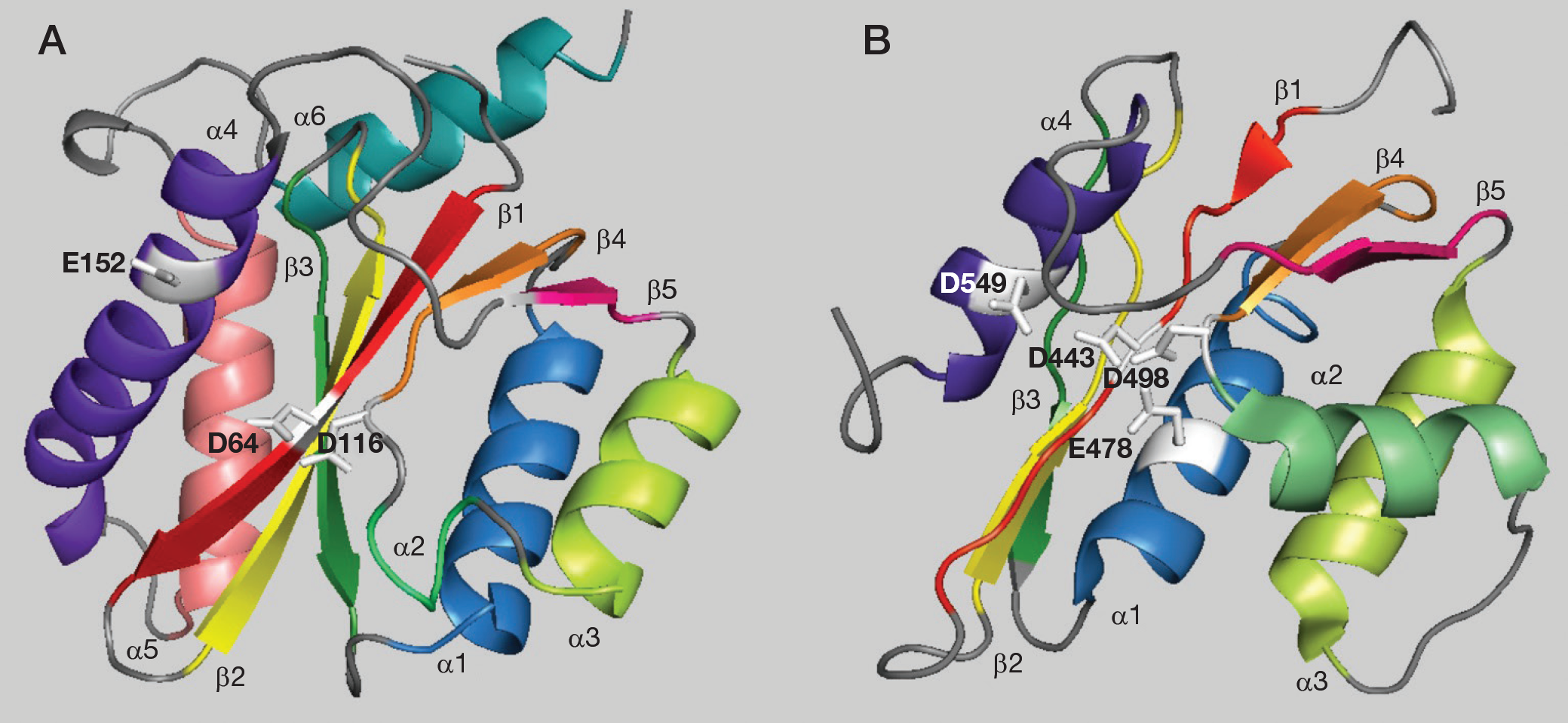
Crystallographic structures of the catalytic core domain of HIV-1 IN and RNase H
Currently, >25 different antiretrovirals have been licensed by the US Food and Drug Administration (FDA). The present therapeutic regimen, HAART, is composed of a combination of two nucleoside reverse transcriptase inhibitors (NRTIs) to which a third agent is added. As the third agent, a non-nucleoside reverse transcriptase inhibitor (NNRTI) or a protease inhibitor can be chosen. Effective inhibition of viral replication can be achieved in >90% of patients starting with such regimens. Despite such a large armamentarium, both acute and chronic toxicities limit the prolonged use of several antiretroviral agents, and this is even more a concern when patients become older and suffer from age-related comorbidities, as many HIV-negative subjects do. In addition, the selection of drug-resistant strains and the spreading of such strains in newly infected patients is also an increasing concern. Because HIV-1 IN inhibitors (INIs) are expected to retain efficacy against HIV-1 strains resistant to other classes of antiretroviral drugs, they represent a valid second-line therapy. In fact, many INIs with different chemical scaffolds have been developed. Compound (N-{2-[4-(4-fluorobenzylcarbamoyl)-5-hydroxy-1-methyl-6-oxo-1,6-dihydropyrimidin-2-yl] propan-2-yl}-5-methyl1,3,4-oxadiazole-2-carboxamide; raltegravir [RAL];
Overall, therefore, there is still a pressing need for the development of novel antiretroviral agents, particularly with new modes of action. Among the novel approaches aimed to identify new HIV-1 inhibitors, the development of dual-action inhibitors, single drugs that may act on two different enzymes, is mostly attractive for the possibility of reducing the number of administered drugs, reducing their chronic toxicity, maintaining the same efficacy and reducing the rapid selection of drug resistance virus strains having a higher resistance barrier. This review briefly describes the HIV-1 IN and the RNase H structures and functions as well as the compounds targeting the active sites of these enzymes. The key mechanism of action of these compounds is the ability to sequester the two divalent cations of the catalytic core, thereby inhibiting the enzymatic activity. In addition, because the two active sites share significant homologies, dual-acting inhibitors are described as new approach for HIV-1 drug development.
HIV-1 integrase: Structure and function
The HIV-1 double-stranded DNA (dsDNA) proviral integration into the chromosomal host cell is an essential step for viral replication that is carried out by the viral coded IN. HIV-1 IN is a 32 kDa protein of 288 amino acids composed of three functional domains: the N-terminal domain (amino acids 1–49), the catalytic core domain (amino acids 50–212) and the C-terminal domain (amino acids 213–288). The active form of the HIV-1 IN is a multimer [11]. Dimerization is required for the 3′-processing step, with tetrameric IN catalysing the strand-transfer reaction [12,13]. Dimeric models were built to reflect the specific contacts between IN and the viral long terminal repeat terminal CA/TG nucleotides identified in vitro [14,15]. During the viral infection, IN operates within large nucleoprotein assemblies, in a complex named the pre-integration complex, which is a macromolecular assemblage consisting of viral DNA, viral proteins and probably host cell proteins. After the pre-integration complex transits into the nucleus, chromatin gets involved through protein–protein as well as DNA–protein interactions, completing the integration process [16,17]. Among the implicated cellular factors is the human lens epithelium-derived growth factor (LEDGF)/p75, a nuclear protein that promotes IN chromatin linkage by allowing specific interaction between its IN-binding domain, represented by residues 347–429 at the C-terminal region of the p75 protein and the IN dimer [18].
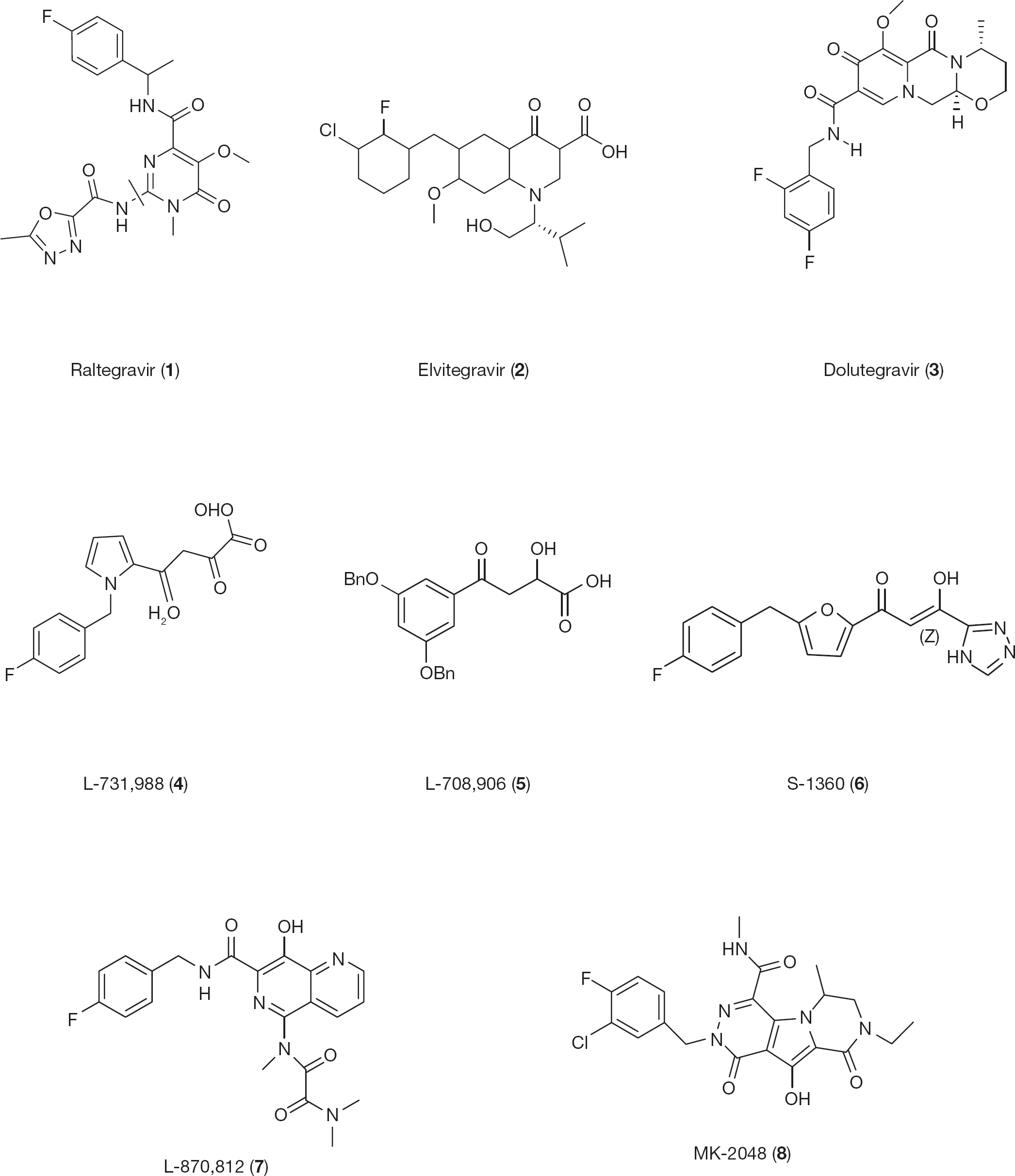
Chemical structures of first-generation and approved HIV type-1 integrase inhibitors
The HIV-1 IN N-terminal domain is characterized by a highly conserved zinc-binding H12H16C40C43 motif [19,20] involved in stabilizing the protein folding and allowing proper IN multimerization [21–23]. The catalytic core domain contains the catalytic D64D116E152 motif, highly conserved in all retroviral INs [19]. In addition to its indispensable role in the IN enzymatic activity, it contains functional elements and specific residues that are essential in mediating the integration complex nuclear import [24]. In fact, several residues, such as L102, A128, A129, W132, E170, T174, M178 and I365, as well as the K186R187K188 multimerization motif at the dimer:dimer interface [25–29], are involved in chemical bonding and in hydrophobic contacts with LEDGF/p75 [26,27] and show an indispensable role in IN nuclear distribution, essential for viral replication [23]. The IN C-terminal domain possesses a strong non-specific DNA-binding ability and is bound to both viral and cellular DNA with its minimal non-specific DNA binding region (amino acids 220–270) [30–33]. This domain is also involved in protein oligomerization, in some interactions with the HIV-1 RT [31] and LEDGF/p75 [30–33].
The multimer form of HIV-1 IN accomplishes the viral DNA integration process through a defined set of cutting (termed 3′-processing) and joining (termed strand-transfer) reactions. During 3′-processing, two conserved nucleotides are removed from each 3′-end of the long terminal repeat terminus of the viral DNA obtained at completion of the reverse transcription process. The 3′-processing reaction is carried out in the cytoplasm within the pre-integration complex [34]. After the pre-integration complex is transported through the nuclear pore into the nucleus, the second catalytic step can take place. The strand-transfer reaction catalyses the covalent ligation of the viral dsDNA into the host genome, leading to HIV-1 genome integration. The gap filling at the interfaces between viral and host DNA is then completed using the host DNA repair machinery via a mechanism that is not yet fully understood [35,36].
Although full-length HIV-1 IN has not been crystallized, individual IN domain or two domain fragments have been solved. The model deposited in the Protein Data Bank (PDB; 3OY9), represents two chains, A and B, including the catalytic core domain and the C-terminal domain IN fragments [28,37]. Structural snapshots of the integration process have been recently revealed by a series of crystal structures of the prototype foamy virus (PFV) IN, which is structurally closely related to HIV IN [37]. The first reported structures were those of a PFV IN intasome (a complex of the enzyme with two ends of the viral dsDNA after the 3′-end processing step). In this structure the protein is a tetramer (dimer of dimers) with inner subunits performing the catalysis, outer subunits with disordered N-terminal and C-terminal domains probably playing a structural role, and the DNA ends buried within the IN tetramer and their recessed 3′-ends located at the active sites. Figure 3A shows the PFV IN catalytic D128D185E221 motif within the IN catalytic core [37].
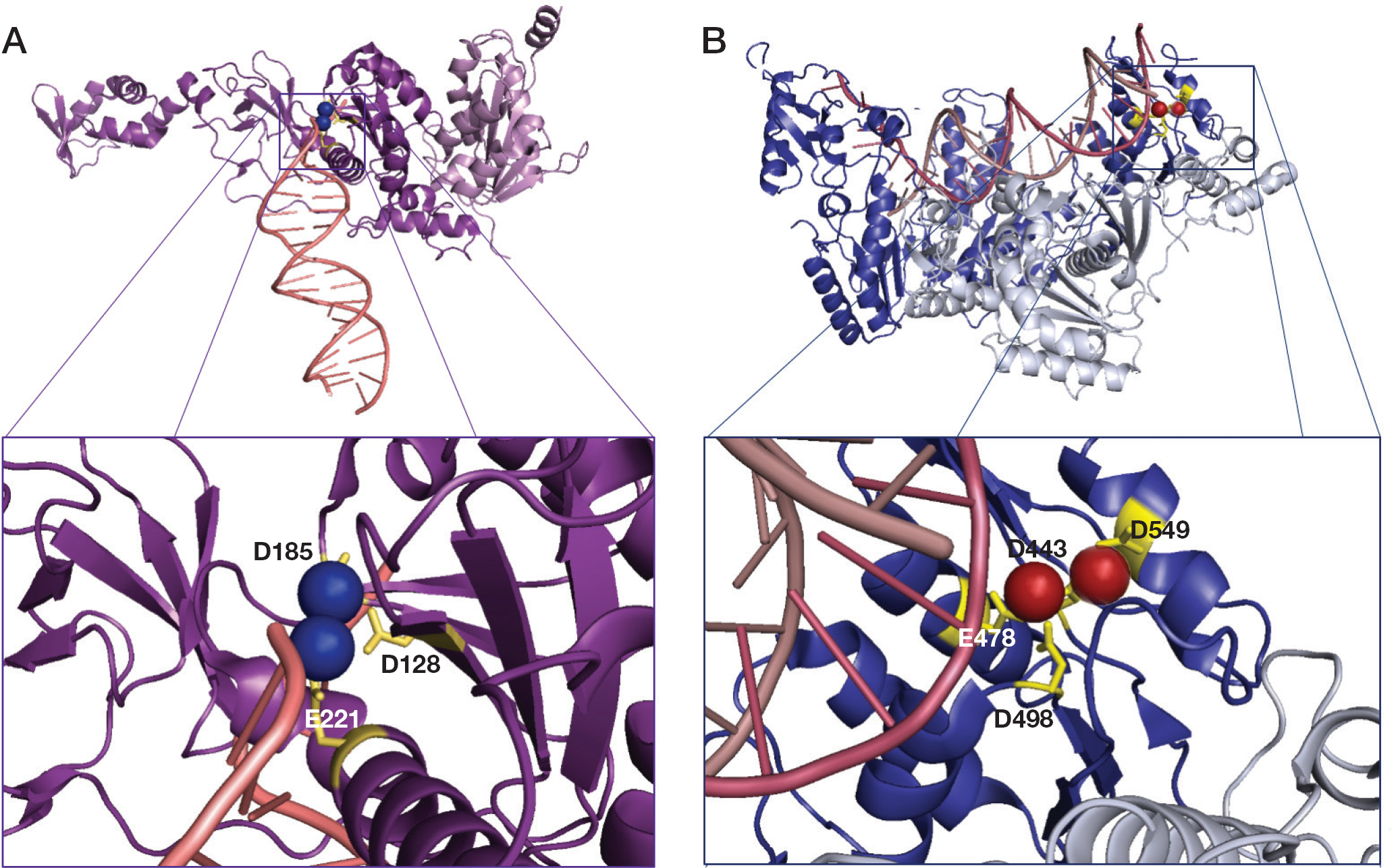
Schematic representation of prototype foamy virus integrase and HIV-1 reverse transcriptase active sites
HIV-1 RT-associated RNase H activity: Structure and function
The single-stranded RNA (ssRNA) HIV-1 genome ret-rotranscription into proviral dsDNA is an essential step for viral replication that is carried out by the viral coded RT, a multifunctional protein that catalyses different reactions among which are the RNA- and DNA-dependent DNA polymerase and RNase H activities, both essential for viral replication [38]. Structurally, HIV-1 RT is a heterodimer composed of two different subunits, the p66 (66 kDa) and the p51 (51 kDa), deriving from the Pol polyprotein processing by the viral protease [38]. The p66 subunit is arranged into two spatially distinct domains, polymerase and RNase H domains. The polymerase domain shows a characteristic highly conserved structure that resembles a right hand, consisting of fingers (amino acids 1–85 and 118–155), palm (amino acids 86–117 and 156–237) and thumb (amino acids 238–318) subdomains. This subunit also comprises the connection subdomain (residues 319–426) and the RNase H domain (residues 427–560) [39,40]. The p51 subunit lacks the RNase H domain, it does not present enzymatic activities but anchors the proper folding of the p66 subunit that performs all catalytic functions.
The HIV-1 RT-associated RNase H function is essential for virus replication (RNase H-deficient viruses are non-infectious) [9], and it is located at C-terminus of the p66 subunit, 60 Å far from the polymerase active site, equivalent to 17 nucleotides of a DNA:DNA duplex and/or 18 nucleotides of a RNA:DNA hybrid [41]. The RNase H domain catalyses the selective hydrolysis of the RNA strand of the RNA:DNA replication intermediate by a phosphoryl transfer through nucleophilic substitution reactions on phosphate ester. This action occurs through the deprotonation of a water molecule, with the production of a nucleophilic hydroxide group that attacks the scissile phosphate group on the RNA previously activated by coordination with the Mg2+ cofactor [42]. The RNase H cleavage specificity for the RNA portion of the RNA:DNA hybrid mainly relies on its particular minor groove width and its interaction with the ‘primer grip’ [38].
The RNase H active site contains a highly conserved, essential, DDE motif comprising the carboxylate residues D443, E478, D498 and D549 (Figure 3B) [3], which coordinate two divalent cations, consistent with the proposed phosphoryl transfer geometry [43].
Integrase inhibitors
In order to design INIs, both the unbound IN protein (3′-processing inhibitors) and the complex resulting from the IN association to viral DNA (strand-transfer inhibitors) have been considered viable strategies [44].
First-generation integrase inhibitors
Random chemical library screening resulted in the initial discovery of INIs. In 2000, Merck Pharmaceuticals identified a class of compounds that contain a diketo acid (DKAs) moiety and their derivatives were shown to inhibit HIV-1 IN by chelation of bound cations [44,45]. The chemical structures of DKAs and their derivatives comprise a common β-diketopropyl linker and different aromatic as well as acid portions. The DKA moiety is the key pharmacophore for enzyme inhibition and the aromatic group improves potency and selectivity of the compounds. Among analogues in which pyrrole groups were conjugated to the phenyl groups, the 4-[1-(4-fluorobenzyl)-1h-pyrrol-2-yl]-2,4-dioxobutanoic acid (L-731,988;
Integrase inhibitors that are approved and under clinical trial evaluation
The new phase in the INIs development came from the discovery of the naphthyridine derivatives by Merck Pharmaceuticals in which the carboxylate group was replaced with a suitable heterocycle which contained lone pair donor atoms such as an 8-hydroxy-1,6-naphthyridine linked to a benzoyl substituent [49]. The N′-{7-[(4-fluorophenyl)methylcarbamoyl]-8-hydroxy-1,6-naphthyridin-5-yl}-N,N,N′-trimethyloxamide (L-870,812;
The molecular basis of the action of a number of raltegravir derivatives was revealed through studies of their crystal structure in complex with the PFV intasome (PDB: 3L2V; Figure 4A) [51]. The pharmacophore comprises a rigid ring system with three oxygen or nitrogen atoms spaced to coordinate two metal ions at the active site. Indeed, as shown by crystal structures, these INIs act by blocking the active site and displacing the 3′-end of the viral DNA (Figure 4B). The second common element of raltegravir derivatives is a halogenated benzyl group. The benzyl ring and the ring system with ion-coordinating heteroatoms stack with the penultimate and last bases in the 3′-end of the viral DNA. Since raltegravir clinical approval in 2007, HIV-1 raltegravir-resistant strains emerged indicating three resistance pathways: Q148H/R/K combined with 138K or G140S; N155H along with L74M, E92Q and Q163R; and Y143R/C [52,53].
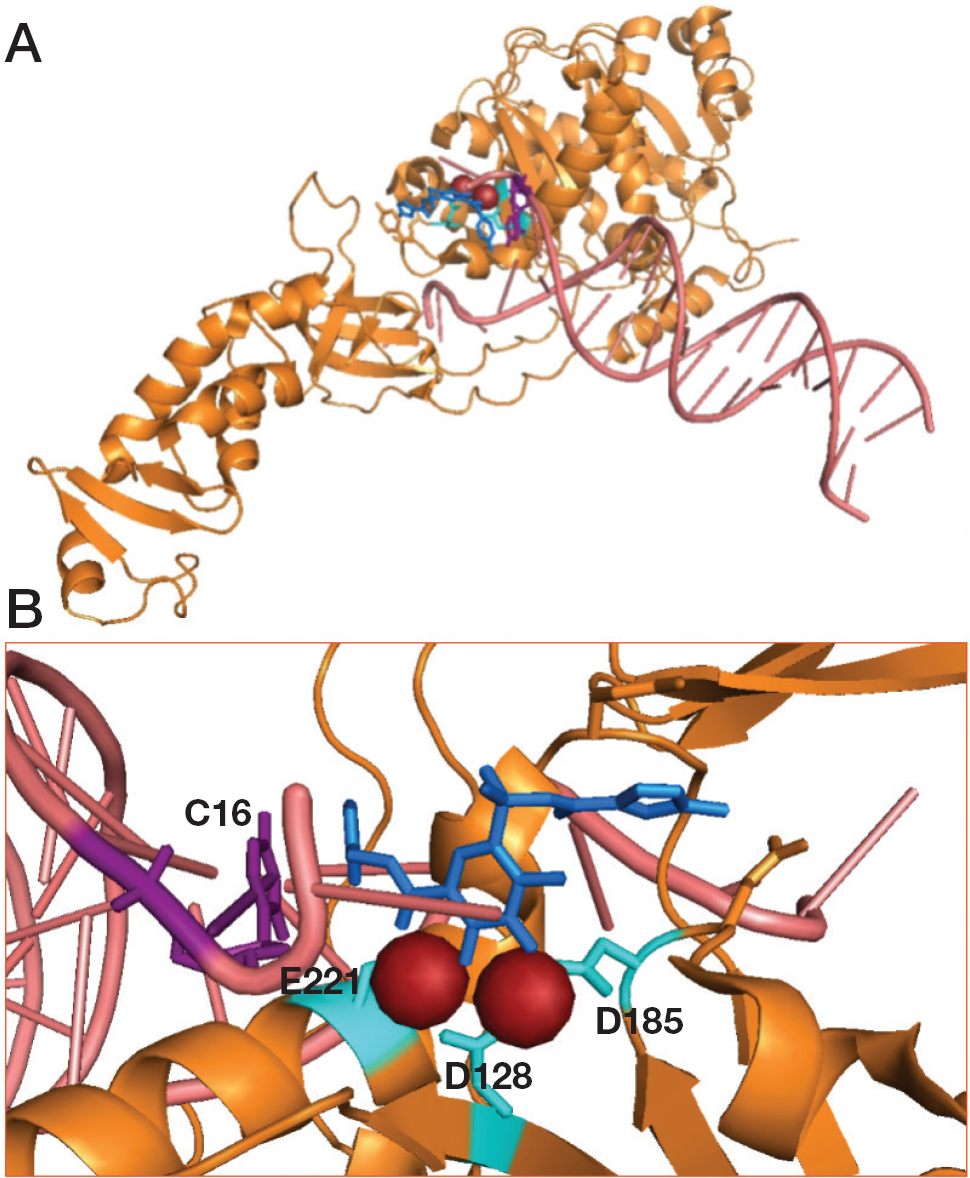
Representation of complex prototype foamy virus intasome and raltegravir
Researchers at Japan Tobacco Company provided an alternative DKA motif, the 4-quinolone-3-carboxylic acid motif with the coplanar monoketo acid motif in 4-quinolone-3-carboxylic acids and reported elvitegravir (
Shinogi in a collaboration with GlaxoSmithKline designed a different naphthyridinone scaffold as IN active site inhibitors that might be effective on raltegravir-resistant strains and can be considered as second-generation INIs [57]. Among them, dolultegravir (
Also in the search for second-generation INIs that might display novel patterns of resistance allowing their use in patients who have failed therapy with raltegravir or elvitegravir, Merck Pharmaceuticals optimized another series of INIs starting from the prototype compound tricyclic 10-hydroxy-7,8-dihydropyrazinopyrrolopyrazine-1,9-dione and leading to the identification of (6S)-2-[(3-chloro-4-fluorophenyl) methyl]-8-ethyl-10-hydroxy-N,6-dimethyl-1,9-dioxo-6,7-dihydropyrazino [5,6]pyrrolo [1,3 b]pyridazine-4-carboxamide (MK-2048;
Furthermore, the search for other second-generation INIs effective on first-generation drug resistance strains led to the identification of a number of new chemical scaffolds, among which the most interesting are the following.
Integrase inhibitors in preclinical development
Dihydroxypyrimidine-4-carboxamides known as HCV NS5B RNA polymerase inhibitors were identified as potent INIs [61]. Several modifications of this scaffold have been explored and led to the synthesis of N-benzyl-5,6-dihydroxy-2-(thiophen-2-yl)pyrimidine-4-carboxamide (
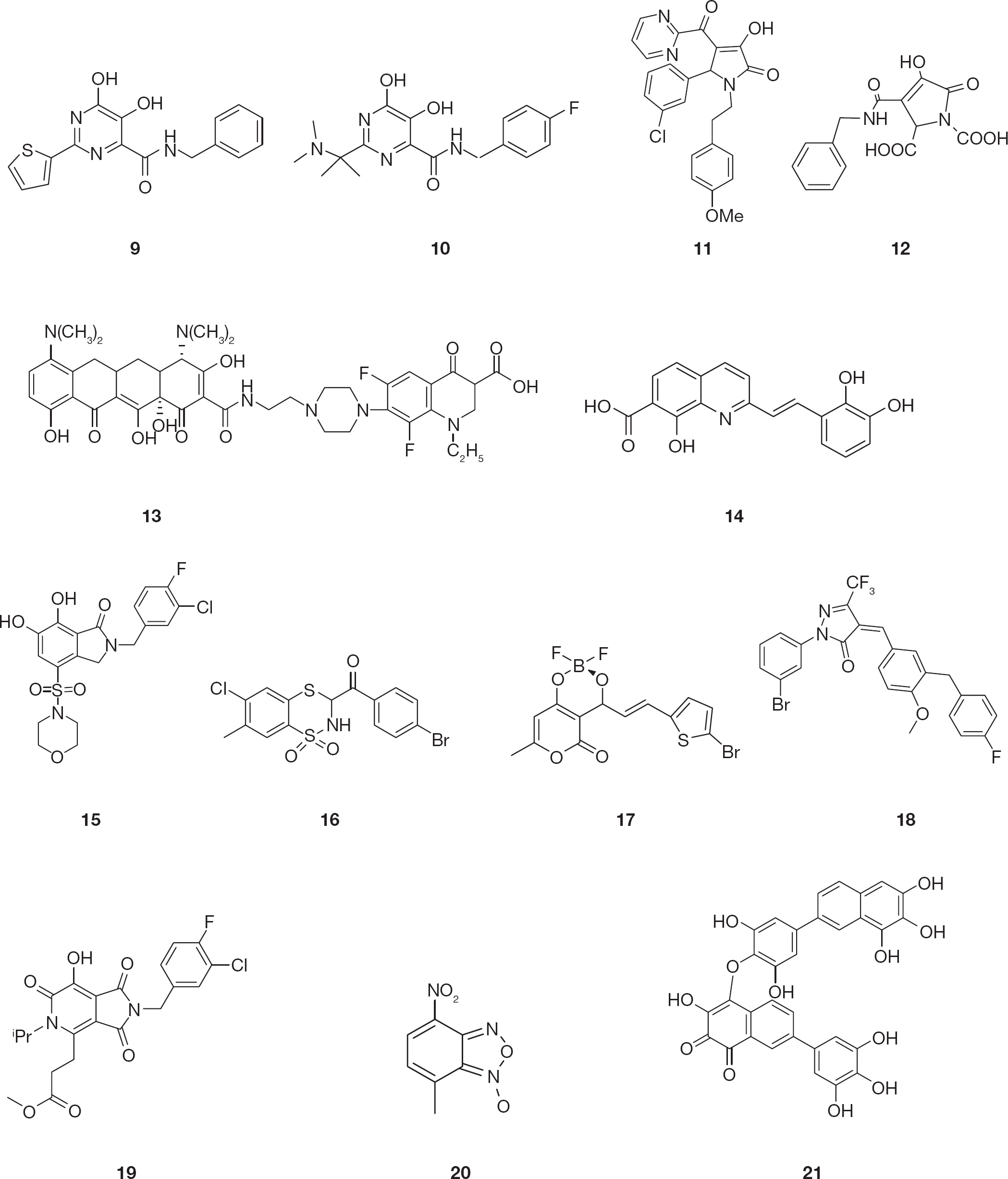
Chemical structure of HIV type-1 integrase inhibitors
The pyrrolinone moiety has been identified as a novel scaffold for suitable replacement of the DKA pharmacophore. Similar to DKAs, substituted pyrrolinones also incorporate three heteroatoms for chelating the metal ion in the IN active site. In addition, the pyrrolinone ring structure enforces coplanarity of the heteroatoms, which is believed to be important for metal chelation. Several pyrrolinones with various substitutions were identified through pharmacophore searching [64]. In particular, the compound 5-(3-chlorophenyl)-3-hydroxy-1-(4-methoxyphenethyl)-4-(pyrimidine-2-carbonyl)-1H-pyrrol-2(5H)-one (
Also tetracyclines have been shown to be a good scaffold for the synthesis of new INIs. In particular, the aryl piperazine derivatives displayed moderate potency against HIV-1 IN and antiviral activity, while being relatively non-cytotoxic with derivative 7-(4-{2-[(4S, 12aS)-4,7-bis(dimethylamino)-3,10,12,12a-tetrahydroxy-1,11-dioxo-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamido]ethyl}piperazin-1-yl)-1-ethyl-6,8-difluoro-4-oxo-1,2,3,4-tetrahydroquinolinone-3-carboxylic acid (
The pharmacophore properties of styrylquinoline derivatives are well known and were used to design INIs which are equally potent against both 3′-processing and strand transfer reactions [68–70]. Notably, differently from most INIs, which are non-selective against one of the two IN reactions, several styrylquinolines inhibited HIV-1 replication in cell-based assays at low micromolar concentrations. In particular, (E)-2-(2,3-dihydroxystyryl)-8-hydroxyquinoline-7-carboxylic acid (
Bicyclic 6,7-dihydroxyoxoisoindolin-1-one-based INIs revealed good potency and strand transfer selectivity in the presence of Mg2+ cofactor [72]. Importantly, the introduction of sulfonamide groups in the 4-position led to derivative 2-(3-chloro-4-fluorobenzyl)-6,7-dihydroxy-4-(morpholinosulfonyl)isoindolin-1-one (
Further drug design and structure activity relationship studies led to compounds of the 3-aroyl-2,3-dihydro-1,1-dioxo-1,4,2-benzodithiazine class in which the replacement of the benzodithiazine ring in position 7 with a methyl group determined different inhibitory outcomes, whereas partial hydrogenation led to a significant improvement in activity [74]. For example, the hydrogenated derivative (
Various substituted pyranones have been previously reported to exhibit anticancer, antimicrobial, anti-coagulant and anti-HIV-protease properties [75,76]. The class of substituted analogues of 3-acetyl-4-hydroxy-pyranone and their difluoridoborate complexes showed potent IN inhibitory effect. In particular, the difluoridoborate complex compound (
Computational modelling predicted that compounds possessing a pyrazolone scaffold could be an IN pharmacophoric model and these compounds were shown to inhibit the IN catalytic activities [78]. This scaffold had been modified in three sites. In particular, modification of the benzylidene ring of the pyrazolone increased the binding affinity to the metal ion into the IN active site leading to derivative (Z)-1-(3-bromophenyl)-4-[3-(4-fluorobenzyl)-4-methoxybenzylidene]-3-(trifluoromethyl)-1H-pyrazol-5(4H)-one (
Recently, tricyclic hydroxy-1H-pyrrolopyridine-tri-ones were reported as scaffolds for INIs [79]. Of note, the analogue methyl 3-[2-(3-chloro-4-fluorobenzyl)-7-hydroxy-5-isopropyl-1,3,6-trioxo-2,3,5,6-tetrahydro-1H-pyrrolo(3,4-c)pyridine-4-yl]propanoate (
A series of HIV-1 INIs containing a novel metal binding motif consisting of oxadiazole and triazole substituted naphthyridines were also reported. The design of the key structural components was based on a two-metal coordination pharmacophore [80]. In the 4-nitrobenzofuroxan derivative (
A series of new hydroxylated 2-arylnaphthalenes also showed good antiviral properties [82]. Notably, the 7-(3,4,5-trihydroxyphenyl) naphthalene-1,2,3-triol analogue was shown to be sensitive to oxidation thus leading to the dimer 4-[2,6-dihydroxy-4-(6,7,8-trihydroxynaphtalen-2yl)phenoxy]-3-hydroxy-7-(3,4,5-trihydroxyphenyl) naphthalene-1,2-dione derivative (
Dual integrase and RNase H inhibitors
The above described structural features shared by HIV-1 IN and RNase H led to the hypothesis that compounds that inhibited IN and/or other polynucleotidyl transferase enzymes could also act on RNase H and focused attention on the development of molecules that could be capable of dual IN/RNase H inhibition as a new approach that may reduce the number of drugs administered to HIV-positive patients, decrease the selection of drug-resistant strains and diminish drug toxicity.
The identification of compound 2-hydroxy-4H-isoquinoline-1,3-dione (
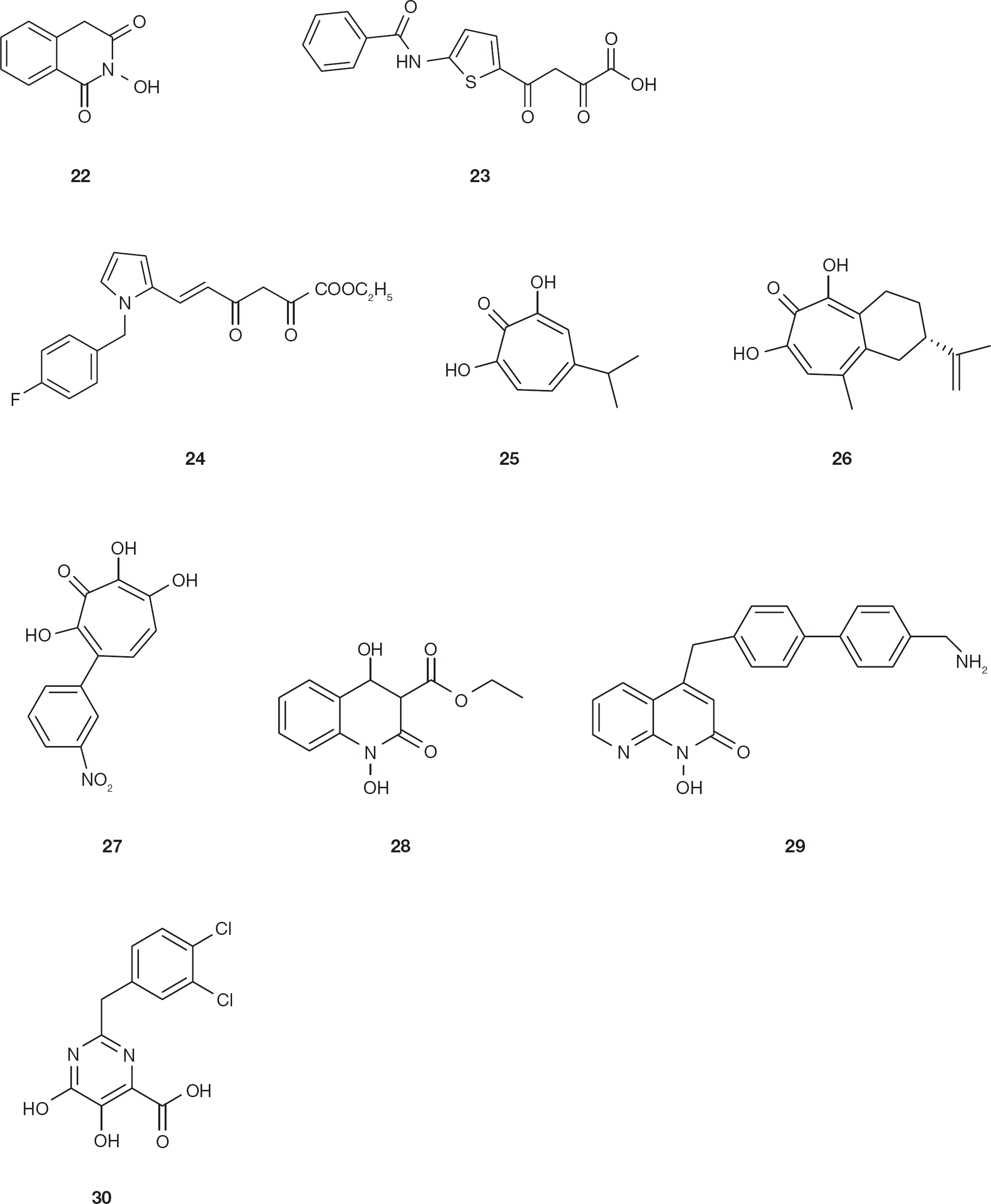
Chemical structures of dual HIV type-1 integrase and ribonuclease H inhibitors
As described above, compounds previously reported to inhibit both influenza virus endonuclease and HIV-1 IN were DKAs [44,88]. Consequently, DKA derivative 4-[5-(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid (BTDBA;
A high-throughput screening of a National Cancer Institute library of pure natural products led to the identification of compounds 2,7-dihydroxy-4–1(methylethyl)-2,4,6-cycloheptatrien-1-one (β-thujaplicinol,
The naphthyridine pharmacophore was initially reported to be important in INI development, also involving divalent ions coordination [100] and it was subsequently reported to inhibit also the HIV-1 RNase H activity [101]. The naphthyridine derivative ethyl 1,4-dihydroxy-2-oxo-1,2,3,4-tetrahydroquinoline-3-carboxylate (MK1;
Pyrimidinol carboxylic acids were reported to be stable HIV INIs that effectively mimic the α,γ-DKAs and α,γ-DK amides units [62] and their scaffold was used also for the synthesis of HIV-1 RNase H inhibitors [103]. In particular, 2-(3,4-dichlorobenzyl)-5,6-dihydroxypyrimidine-4-carboxylic acid (
RNase H inhibitors
Following the identification of the first DKAs as HIV-1 IN and RNase H inhibitors [89,94], a few other classes of compounds were identified as selective HIV-1 RNase H inhibitors.
5-nitro-furan-2-carboxylic acid carbamoylmethyl ester (NAMCE) derivatives were identified in a screening of small molecular weight compounds [105]. Compound 5-nitro-furan-2-carboxylic acid adamantan-1-carbamoylmethyl ester (
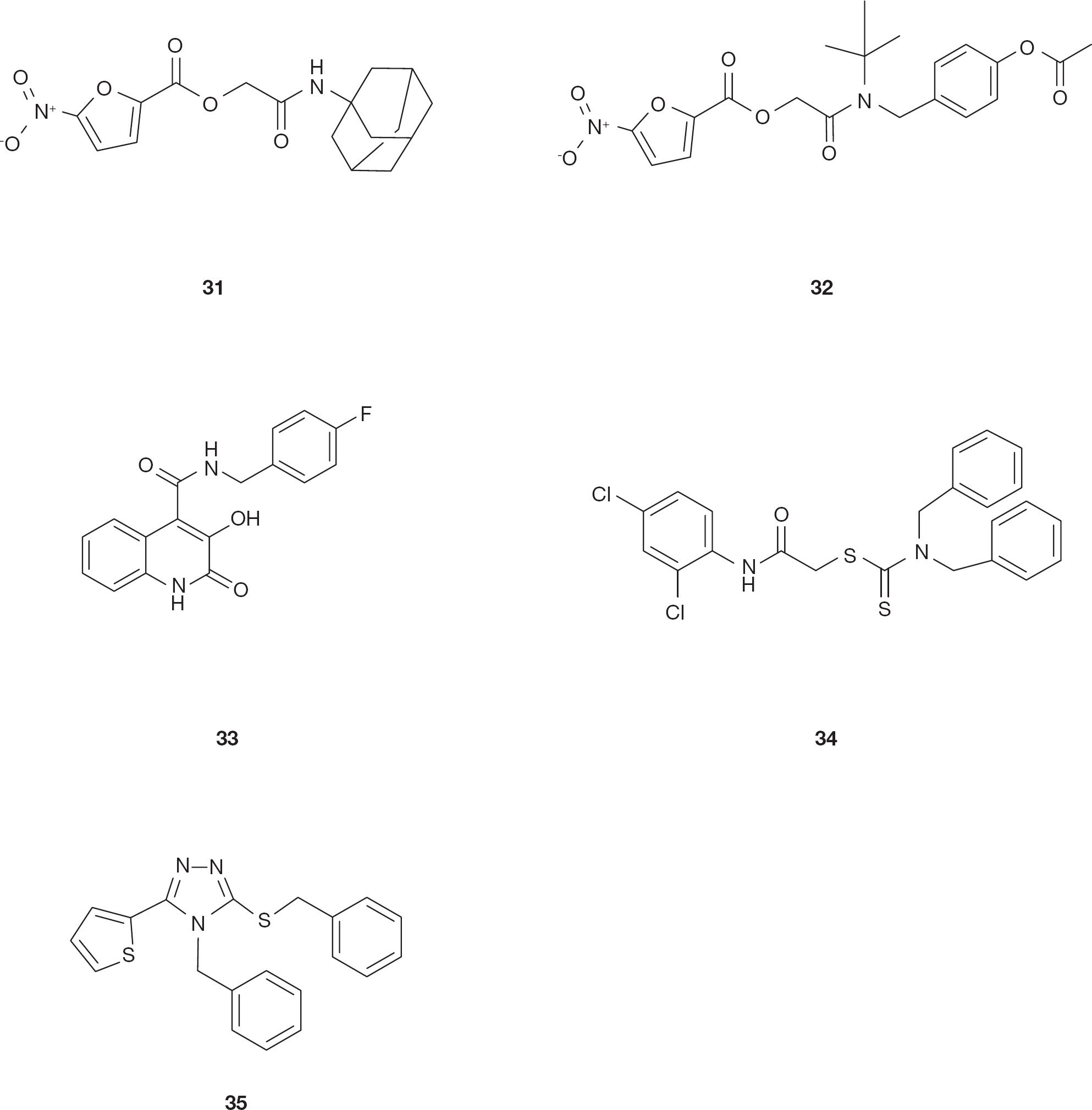
Chemical structure of HIV type-1 ribonuclease H inhibitors
By contrast with the previously described N-hydroxyimide derivatives [84,86,87], a recent series of substituted 3-hydroxyquinolin-2(1H)-ones was reported to selectively inhibit the HIV-1 RNase H in the low micromolar range while inactive on IN [107]. Compound N-(4-fluorobenzyl)-3-hydroxy-2-oxo-1,2-dihydroquinoline-4-carboxamide (
Thiocarbamates and triazoles were reported to inhibit the HIV-1 RNase H activity [108]. In fact, the thiocarbamate derivatives compound 2-[(2,4-dichlorophenyl) amino]-2-oxoethyl dibenzylcarbamodithioate (
Conclusions
In conclusion, after the approval of INIs for treatment of HIV-1 infections, several screenings were performed aimed at the development of second-generation INIs that may be effective on mutant HIV-1 strains resistant to the first-generation INIs. In addition, given the structural similarities between IN and RNase H active sites, a number of attempts have been made to identify compounds that may act on both enzymes. The identification and characterization of the molecular determinants that are essential for the interaction between the inhibitors and both IN and RNase H active site will be needed in order to be able to rationally develop compounds that may be truly dual inhibitors. This dual approach is certainly a valid rationale for drug development and a deeper understanding of differences and similarities between the two active sites will be of help to discover new potent HIV-1 inhibitors. In this regard it is of note that, recently, the crystal structure of the complex of HIV-1 RT, an NNRTI and an RNA/DNA hybrid was reported [109]. This is particularly important because, while >20 RT structures with nucleic acid substrate have been reported, only this structure contains an RNA/DNA hybrid. Hence, this information may be very useful for further studies aimed to determine how HIV-1 RNase H selects and binds its substrate and consequently it may open new avenues for identifying novel anti-HIV drug targets [109].
Footnotes
Acknowledgements
This work was supported by RAS grant LR 7/2007 CRP-24915 and MIUR PRIN 2010.
The authors declare no competing interests.