
Abstract
Background:
To improve in vitro antiviral activity and selectivity of stavudine (d4T), a range of its bi-functional prodrugs, 5′-O-myristoylated derivatives, have been synthesized.
Methods:
Stavudine 5′-O-myristoylated esters were synthesized using modified Parang's procedure. The cytotoxicity and anti-HIV activity was evaluated in the established MT-4 cell line. The level of p24 protein in culture medium was assayed, and EC50 and EC90 values were determined.
Results:
Excellent anti-HIV activity was obtained for stavudine derivatives 2′,3′-didehydro-2′,3′-dideoxy-5′-O-(11-thioethylundecanoyl) thymidine, 2′,3′-didehydro-2′,3′-dideoxy-5′-O-(12-thioethyldodecanoyl) thymidine and 5′-O-(12-azidododecanoyl)-2′,3′-didehydro-2′,3′-dideoxythymidine with C10 and C11 alkyl chains bearing thioethyl- and azido- substituents. These prodrugs were more potent than the parent stavudine, as is clear from their EC50 values: 2′,3′-didehydro-2′,3′-dideoxy-5′-O-(11-thioethylundecanoyl) thymidine (R=CO(CH2)10SC2H5, EC50 0.06 μM), 2′,3′-didehydro-2′,3′-dideoxy-5′-O-(12-thioethyldodecanoyl) thymidine (R=CO(CH2)11SC2H5, EC50 0.09 μM) and 5′-O-(12-azidododecanoyl)-2′,3′-didehydro-2′,3′-dideoxythymidine (R=CO(CH2)11N3, EC50 0.06 μM), while 50% cytotoxic concentration was >16.65 μM, >7.5 μM and >18.53 μM, respectively.
Conclusions:
Overall data demonstrate that compounds 2′,3′-didehydro-2′,3′-dideoxy-5′-O-(11-thioethylundecanoyl) thymidine, 2′,3′-didehydro-2′,3′-dideoxy-5′-O-(12-thioethyldodecanoyl) thymidine and 5′-O-(12-azidododecanoyl)-2′,3′-didehydro-2′,3′-dideoxythymidine are very potent and selective anti-HIV agents and could be useful in treatment of HIV infections of the central nervous system.
Introduction
Stavudine (2′,3′-didehydro-2′,3′-dideoxythymidine, d4T, Zerit) [1,2] is a thymidine nucleoside analogue approved for the treatment of HIV infection [3]. Its active metabolite, d4T-5′-triphosphate (d4TTP), is a competitive inhibitor of HIV reverse transcriptase, and acts as a chain terminator during DNA synthesis. Comparative studies demonstrated that, although d4T was less potent than zidovudine (AZT) against HIV, it had a better safety profile in a variety of in vitro studies [4] and was also found to be active against AZT-resistant strains of HIV [5].
Myristic acid analogues, probable anti-HIV agents, are found to be alternative substrates for myristoyl-CoA: protein N-myristoyltransferase (NMT) and it has been proposed that inhibition of post-translational myristoylation – an essential step in processing of viral precursor protein, and more specifically NMT – may be the probable mechanism of antiviral activity of myristic acid analogues. Therefore, NMT could be an attractive target for antiviral therapy to inhibit HIV replication in HIV-infected CD4 lymphocytes [6]. We therefore focused on studying prodrugs of stavudine with myristic acid and its analogues. Esterification of the 5′-hydroxyl of stavudine with myristic acid analogues is expected to produce compounds, which after penetration into the cells, will be cleaved by esterases to respective myristic acid analogues and stavudine, which, in turn, will be intracellularly metabolized to its active form, d4TTP. The prodrugs, bearing biodegradable ester linkages, produced will act as bi-functional HIV inhibitors with more potent antiviral activity and decreased cytotoxicity.
Materials and methods
Chemistry
High-resolution mass spectra were recorded on a Q-TOF MICROMASS spectrometer (Waters, Milford, MA, USA). High-resolution 1H NMR spectra were recorded in CDCl3 on a Varian 500 MHz (Varian Inc., Pablo Alto, CA, USA) with tetramethylsilane as an internal standard. Thin-layer chromatography was carried out using Merck silica gel F254 glass plates (Merck, Darmstadt, Germany) and the plates were developed with solvents (v/v): (A) dichloromethane-MeOH (95:5); (B) dichloromethane-ethyl acetate (60:40). The starting compound d4T (1), was prepared according to the procedure described by Mansuri et al. [7].
2′,3′-Didehydro-2′,3′-dideoxy-5′-O-(tetradecanoyl) thymidine (2)
To a suspension of d4T (100 mg, 0.45 mmol) and dimethyl aminopyridine (DMAP; 61 mg, 0.5 mmol) in dichloromethane (20 ml) under anhydrous conditions, myristoyl chloride (180 μl, 0.66 mmol) was added dropwise over 20 min at ambient temperature, and the reaction mixture stirred overnight. Dichloromethane (25 ml) was then added and the reaction mixture was washed with saturated sodium bicarbonate (2×15 ml) and water (3×10 ml). The organic layer was dried over anhydrous sodium sulfate and evaporated to dryness under vacuum. The dried mass was subjected to silica gel column chromatography (3×8 cm) and the product eluted using a gradient of methanol in dichloromethane (0–2%) to yield 180 mg (93%) of
General procedure for synthesis of 5′-O-esters of d4T
A solution of the desired fatty acid (0.44 mmol) and oxalyl chloride (63 μl, 0.66 mmol) in anhydrous toluene (8 ml) was stirred at ambient temperature for 1 h, and the solvent removed in vacuo. The resulting oil, dissolved in anhydrous toluene (5 ml), was added to a suspension of d4T (100 mg, 0.45 mmol) and DMAP (80 mg, 0.66 mmol) in anhydrous toluene (10 ml). The reaction mixture was stirred at room temperature for 1 h and then heated under reflux for 3 h. The mixture was then cooled to about 24°C and an additional amount of toluene (10 ml) was added. The organic phase was washed with saturated sodium bicarbonate (2×15 ml) and water (2×10 ml), dried over anhydrous sodium sulfate, and evaporated to dryness under vacuum. The dried mass was subjected to silica gel column chromatography (3×15 cm) and the product eluted using a gradient of methanol in dichloromethane (0–2%).
5′-O-(12-Bromododecanoyl)-2′,3′-didehydro-2′,3′-dideoxythymidine (3)
Yield 60%; Rf=0.73 (A), 0.44 (B); 1H NMR (500 MHz, CDCl3) 8.31 (br, 1H, NH), 7.23 (d, 1H, H6), 6.99 (d, 1H, H1′, J1′,2′=1.94 Hz), 6.27 (dt, 1H, H3′, J1′,3′=1.44 Hz), 5.90 (dt 1H, H2′, J2′,3′=6.02 Hz, J2′,4′=1.60 Hz), 5.04 (m, 1H, H4′), 4.43 and 4.22 (two dd, 1H each, H5′, H5″, J5′,4′=4.10 Hz, J5″,4′=3.08 Hz, J5′,5″=12.41 Hz), 3.53 (t, 2H, CH2Br), 2.32 (t, 2H, CH2CO), 1.92 (d, 3H, 5-CH3), 1.76 (quintet, 2H, CH2CH2Br), 1.65–1.59 (m, 2H; CH2CH2CO), 1.32–1.25 (br, m; 14H, (CH2)7); HR-MS m/z for C22H34BrN2O5, calcd 485.16456; found 485.16433 [M + H]+, Anal calcd for C22H34 BrN2O5: C 54.46, H 7.04, N 5.77. Found C 54.30, H 7.00, N 5.70.
2′,3′-Didehydro-2′,3′-dideoxy-5′-O-(11-thioethylundecanoyl) thymidine (4)
Yield 68%; Rf=0.76 (A), 0.44 (B); 1H NMR (500 MHz, CDCl3 8.37 (br, 1H, NH), 7.23 (d, 1H, H6), 6.99 (d, 1H, H1′, J1′,2′=1.88 Hz), 6.27 (dt, 1H, H3′, J1′,3′=1.36 Hz), 5.90 (dt, 1H, H2′, J2′,3′=5.99 Hz, J2′,4′=1.58 Hz), 5.04 (m, 1H, H4′), 4.43 and 4.22 (two dd, 1H each, H5′, H5″, J5′,4′=4.08 Hz, J5″,4′=3.06 Hz, J5′,5″=12.40 Hz), 2.52 (q, 2H, CH3CH2S), 2.51 (t, 2H, CH2CH2S,), 2.32 (t, 2H, CH2CO), 1.92 (d, 3H, 5-CH3), 1.65–1.54 (m, 4H; CH2CH2S and CH2CH2CO), 1.32–1.26 (br, m, 12H, (CH2)6), 1.25 (t, 3H, CH3); HR-MS m/z for C23H37N2O5S calcd 453.24177; found, 453.24126 [M + H]+, Anal calcd for C23H37N2O5S: C 60.95, H 8.23, N 6.18. Found C 60.90, H 8.19, N 6.00.
2′,3′-Didehydro-2′,3′-dideoxy-5′-O-(12-thioethyldodecanoyl) thymidine (5)
Yield 64%; Rf=0.78 (A), 0.47 (B); 1H NMR (500 MHz, CDCl3) 8.48 (s, 1H, NH), 7.23 (d, 1H, H6), 6.99 (d, 1H, H1′, J1′,2′=1.85 Hz), 6.27 (dt, 1H, H3, J1′,3′=1.35 Hz), 5.90 (dt, 1H, H2′, J2′,3′=6.0 Hz, J2′,4′=1.55 Hz), 5.04 (m, 1H, H4′), 4.43 and 4.22 (two dd, 1H each, H5′, H5″, J5′,4′=4.07 Hz, J5″,4′=3.06 Hz, J5′,5″=12.40 Hz), 2.53 (q, 2H, CH3CH2S), 2.51 (t, 2H, CH2CH2S), 2.32 (t, 2H, CH2CO), 1.92 (d, 3H, 5-CH3), 1.63–1.54 (m, 4H, CH2CH2S, CH2CH2CO), 1.32–1.26 (br, m, 14H, (CH2)7) 1.25 (t, 3H, CH3); HR-MS m/z for C24H39N2O5S calcd 467.25742; found, 467.25701 [M + H]+, Anal calcd for C24H39N2O5S: C 61.69, H 8.41, N 6.00. Found C 61.60, H 8.35, N 6.14.
2′,3′-Didehydro-2′,3′-dideoxy-5′-O-(12-methoxydodecanoyl) thymidine (7)
Yield 69%; Rf=0.62 (A), 0.31 (B); 1H NMR (500 MHz, CDCl3) 8.23 (br, 1H, NH), 7.23 (d, 1H, H6), 6.99 (d, 1H, H1′, J1′,2′=1.95 Hz), 6.27 (dt, 1H, H3′, J1′,3′=1.43 Hz), 5.90 (dt, 1H, H2′ J2′,3′=6.02 Hz, J2′,4′=1.55 Hz), 5.04 (m, 1H, H4′), 4.43 and 4.22 (two dd, 1H each, H5′, H5″, J5′,4′=4.08 Hz, J5″,4′=3.09 Hz, J5′,5″=12.41 Hz), 3.36 (t, 2H, CH2OMe), 3.33 (s, 3H, OCH3), 2.32 (t, 2H, CH2CO), 1.92 (d, 3H, 5-CH3), 1.66–1.53 (m, 4H; CH2CH2OMe and CH2CH2CO), 1.35–1.25 (br, m; 14H, (CH2)7); HR-MS m/z for C23H37N2O6 calcd 437.26461; found, 437.26410 [M + H]+, Anal calcd for C23H37N2O6: C 63.18, H 8.53, N 6.41. Found C 63.09, H 8.50, N 6.35.
5′-O-(12-Azidododecanoyl)-2′,3′-didehydro-2′,3′-dideoxythymidine (6)
To a solution of 5′-O-(12-bromododecanoyl)-2′,3′-didehydro-2′,3′-dideoxythymidine (
Virology
In vitro cytotoxicity of the investigated compounds was determined using MT4 and CEM T4 cell lines. The materials and related reagents were obtained through the NIH Reagent Program, Germantown, MD, USA, Division of AIDS, NIAID, NIH [8].
To determine the anti-HIV-1 inhibitory effects of compounds, MT-4 cells (1×106 cells/ml) were incubated for 24 h in RPMI medium supplemented with 10% fetal calf serum (FCS) and a known concentration of the tested compound. The cell cultures were inoculated with syncytia-inducing HIV-1 laboratory isolates. For each concentration of the tested compounds, cell cultures were prepared in triplicate. The positive control contained an identical concentration of HIV-1 inoculated cells maintained in RPMI with 10% FCS. The HIV inhibitory effect was estimated by measurement of p24 protein in the media after eight days of culture.
Efficacy of inhibition of HIV-1 replication by tested compounds was compared to controls cultured in medium without tested compounds, as well as to cells cultured in media enriched with a known amount of d4T (
Results
d4T (
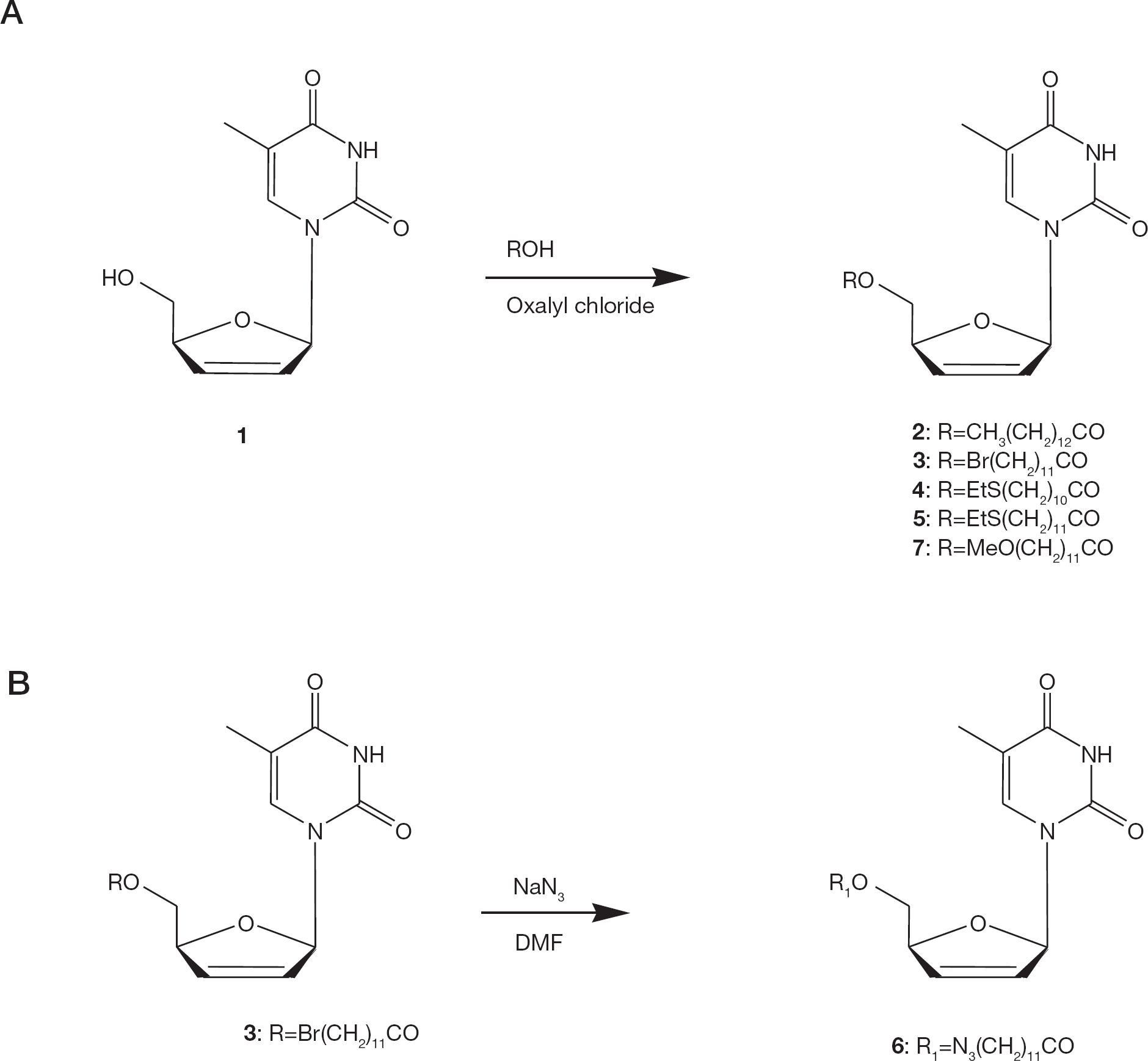
Synthesis of 5′-O-esters of stavudine (
Compounds
Data presented in Table 1 clearly indicates that nearly all 5′-O-myristoylated compounds, especially 2′,3′-didehydro-2′,3′-dideoxy-5′-O-(12-azidododecanoyl) thymidine (
Inhibitory effects of 5′-O-myristoylated esters of stavudine on HIV-1 replication in MT-4 cells
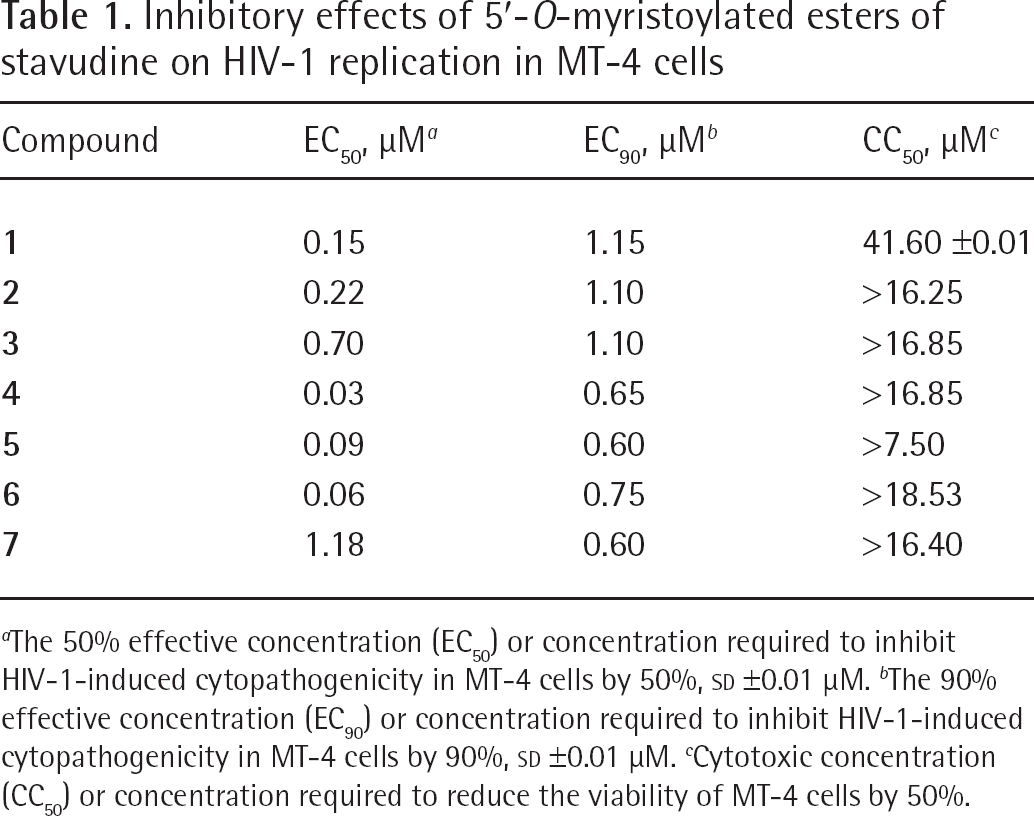
The 50% effective concentration (EC50) or concentration required to inhibit HIV-1-induced cytopathogenicity in MT-4 cells by 50%, SD ±0.01 μM.
The 90% effective concentration (EC90) or concentration required to inhibit HIV-1-induced cytopathogenicity in MT-4 cells by 90%, SD ±0.01 μM.
Cytotoxic concentration (CC50) or concentration required to reduce the viability of MT-4 cells by 50%.
The highest in vitro antiviral activity (in nanomolar range) and low cytotoxicity (in micromolar range) was obtained for 5′-O-myristoyl derivatives of stavudine
Discussion
The highly lipophilic bi-functional stavudine derivatives are expected to show better cellular uptake than stavudine and readily penetrate into macrophages as well, which are reservoirs of HIV particles in HIV-infected patients [10]. Moreover, enhanced lipophilicity of myristoylated derivatives of stavudine would increase their ability to cross the blood–brain barrier through diffusion. Improved brain permeability is also observed when increased lipophilicity is exhibited by compounds with molecular weight not exceeding 400 amu [11]. Our stavudine derivatives possess molecular weights comparable to this value.
The better anti-HIV activity shown by the bi-functional stavudine derivatives can be explained on the basis that they are effective inhibitors of HIV replication at two different stages: reverse transcription and post transcriptional processing of different proteins.
Antiviral activity (EC50) of our compounds, estimated in MT-4 cells, is approximately 112–1,305x higher than those reported recently by Agarwal et al. [12]. This difference is probably due to the different HIV strains used by Agarwal (lymphocytotropic strain IIIB and monocytotropic strain BaL) and a single round infection assay. To clarify this surprising discrepancy, we also estimated antiviral activity in CEM T4 cells (data not shown). In this culture, EC50 values were nearly identical to those determined in MT4 cells.
In conclusion, our findings indicate that lipophilic bi-functional stavudine derivatives
Footnotes
Acknowledgements
This study was supported in part by INSA Exchange Fellowship, India.
The authors declare no competing interests.