
Abstract
Background:
Human HBV and HIV integrate their retro-transcribed DNA proviruses into the human host genome. Existing antiretroviral drug regimens fail to directly target these intrachromosomal xenogenomes, leading to persistence of viral genetic information. Retinazone (RTZ) constitutes a novel vitamin A-derived (retinoid) thiosemicarbazone derivative with broad-spectrum antiviral activity versus HIV, HCV, varicella-zoster virus and cytomegalovirus.
Methods:
The in vitro inhibitory action of RTZ on HIV-1 strain LAI, human HBV strain ayw, HCV-1b strain Con1, enhanced green fluorescent protein-expressing Ebola virus Zaire 1976 strain Mayinga, wild-type Ebola virus Zaire 1976 strain Mayinga, human herpesvirus 6B and Kaposi's sarcoma-associated herpesvirus replication was investigated. The binding of RTZ to human glucocorticoid receptor was determined.
Results:
RTZ inhibits blood-borne human HBV multiplication in vitro by covalent inactivation of intragenic and intraexonic viral glucocorticoid response elements, and, in close analogy, RTZ suppresses HIV-1 multiplication in vitro. RTZ disrupts the multiplication of blood-borne human HCV and Ebola Zaire virus at nanomolar concentrations in vitro. RTZ has the capacity to bind to human glucocorticoid receptor, to selectively and covalently bind to intraexonic viral glucocorticoid response elements, and thereby to inactivate human genome-integrated proviral DNA of human HBV and HIV.
Conclusions:
RTZ represents the first reported antiviral agent capable of eradicating HIV and HBV proviruses from their human host. Furthermore, RTZ represents a potent and efficacious small-molecule in vitro inhibitor of Ebola virus Zaire 1976 strain Mayinga replication.
Introduction
Tremendous efforts were initiated to counteract the devastating effect that the blood-borne human viruses, notably HIV, human HBV and human HCV, have on a significant part of the world's population. Approximately 46 million people have died from AIDS-related illnesses since 1981, and 33.3 million individuals (in 2009) were estimated to be currently infected with diverse HIVs [1]. For hepatitis viruses, approximately 370 million (around 6.5% of the world's population) individuals are infected chronically with HBV [2], and 170 million (around 3% of the world's population) individuals are infected chronically with HCV [2]. Infection with the hepatotropic (liver-targeting) Hepacivirus (Flaviviridae) HCV can result in chronic disease involving liver cirrhosis and/or primary human hepatocellular carcinoma (HCC) [3]. The latter characteristic shares resemblance to the Orthohepadnavirus (Hepadnaviridae) human HBV, a completely different pathogen which also can manifest itself chronically with progression to HCC [4]. The burden of mortality from viral hepatitis is still increasing, as exemplified in the United States [5]. A clinical challenge is the increasingly high incidence of HIV-1/HBV/HCV triple coinfections [6]. Clearly, a pharmacological agent endowed with simultaneous activity versus HIV-1, HBV and HCV would be highly desirable.
The human herpesvirus 6B (HHV-6B) [7] and the Kaposi's sarcoma-associated herpesvirus (KSHV, human herpesvirus 8 [HHV-8]) [8] are amongst typical blood-borne viral pathogens affecting humans. HHV-8 was recognized as the aetiological agent of Kaposi's sarcoma, a strongly growth factor-dependent neoplastic condition of the skin with high meta-static potential [8]. AIDS-related Kaposi's sarcoma often develops in HIV-1-infected individuals as an opportunistic cancer [8]. HHV-6B, like many other human herpesviruses, causes life-long latent persistence (latency) in its human host, and HHV-6B coinfection is often detected in HIV-1-infected individuals [7]. By analogy, a pharmacological agent endowed with simultaneous activity versus HIV-1, HBV, HCV, HHV-6B and HHV-8 would be most desirable.
Ebola virus (EBOV) was first recognized during the 1976 (northwestern) Zaire (Yambuku, Mongala District, now Democratic Republic of the Congo) epidemic [9]. EBOV 1976 Zaire causes a severe haemorrhagic fever with extremely high mortality (around 90%), and is rated amongst the most hazardous human pathogens [9]. The EBOV 1976 Zaire strain Mayinga is named after Mayinga N'Seka, a nurse who died during the Yambuku outbreak on 20 October 1976. Because fruit bats (Hypsignathus monstrosus, Epomops franqueti and Myonycteris torquata) serve as natural reservoirs of EBOV in equatorial Africa [10], future outbreaks of Ebola haemorrhagic fever are certainly to be expected. A clinically useful, safe and efficacious antiviral agent targeting EBOV would be of major public interest, especially due to the threat of the utilization of EBOV-like viruses as potential bioterrorist attack weapons.
To address these challenges, a vitamin A-derived (retinoid) thiosemicarbazone derivative, retinazone (RTZ; formerly called RTSC-Na 5.5 H2O; Figure 1A) was prepared from all-trans retinyl acetate (vitamin A–acetate) by chemical synthesis [11] and proved to exhibit broad-spectrum antiviral activity [11] including the viruses HIV type-1 strain LAI (HIV-1LAI) [12], HCV subtype 1b strain Con1 (HCV-1b Con1) [13], human varicella-zoster virus (VZV, human herpesvirus 3 [HHV-3] strain Ellen), and human cytomegalovirus (HCMV, human herpesvirus 5 [HHV-5] strain AD169). New data indicated suppressing activity of RTZ on enhanced green fluorescent protein (eGFP)-expressing Ebola virus Zaire (eGFP–EBOV Zaire 1976 strain Mayinga [14]) multiplication, on wild-type Ebola virus Zaire (EBOV Zaire 1976 strain Mayinga) virus yield, on human herpesvirus 6B (HHV-6B strain Z29) replication and on Kaposi's sarcoma-associated herpesvirus (HHV-8 strain BCBL-1) production from latency.
Materials and methods
Materials
The synthesis and the analytical data of RTZ were reported previously [11]. Chemicals were of analytical or best available quality. 1,2-Phenylenediamine was purchased from Merck–Schuchardt OHG (Hohenbrunn, Germany). Vitamin A–acetate (purity 95%) oily concentrate (approximately 0.327 g all-trans retinyl acetate/g oil) and glacial acetic acid (HOAc) 100% pro analysi were purchased from AppliChem GmbH (Darmstadt, Germany). Hydrocortisone micronized Ph. Eur. 6.5 was purchased from Caelo (Caesar & Loretz GmbH, Hilden, Germany). The lambda (λ) bacteriophage λcIts857 ind1 Sam7 [15] double-stranded DNA (dsDNA; 5 A260 units, 0.25 μg/μl in 10 mM Tris–HCl [pH 8.0] +1 mM EDTA [TE buffer]; 1,000 μl) was from Roche Diagnostics Deutschland GmbH (Roche Applied Science, formerly Boehringer Mannheim GmbH, Mannheim, Germany). The 48,502 bp (32,300 kDa) λ dsDNA with cos sticky ends carries the following mutations in comparison to wild-type λ virus: 37,589 C→T (ind1); 37,742 C→T (cI857); 43,082 G→A (ts); 45,352 G→A (Sam7). The temperature (70°C) for the experiment in Covalent modification of λ DNA by RTZ in Additional file 1 was selected 10 K below the reported melting temperature (80°C) of λcIts857 ind1 Sam7 dsDNA [16]. The analysis of the cos (cohesive sticky ends) oligomer composition of λ dsDNA was published [17].
Virology
HIV-1 replication reverse transcriptase assay
HIV-1LAI (=HIV-1BRU = LAV-1) was assayed in primary human peripheral blood lymphocyte (PBL) cells in the presence of a drug being evaluated. The parameter for antiviral activity was reduction of reverse transcriptase (RT) activity in the cell supernatant after Triton X–100-mediated lysis of released virions, as measured by [5α-3H]dTTP (5α-tritiated thymidine 5′-triphosphate) incorporation into poly(rA) • poly(dT) directed by the primed RNA template poly(rA) • oligo(dT). It should be noted that the assay did not detect RT inhibition by potential RT inhibitors per se, but indirectly quantified the amount of released HIV-1 in the supernatant. The detailed assay methodology was reported by Schinazi et al. [18], as based on an older assay system of Spira et al. [19]. The experiments were conducted in triplicate and treated statistically by regression curve analysis (r2 coefficient of determination). The RT inhibitor zidovudine (AZT, 3′-azido-3′-deoxythymidine; RETROVIRTM) served as a positive control. Cytotoxicity on PBL cells exerted by the test compounds was determined as described by Stuyver et al. [20], by application of the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega Corp., Madison, WI, USA). Briefly, the phenazine ethosulfate (PES)-coupled reduction of the tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) to a purple, water-soluble formazan by living, undamaged cells was measured.
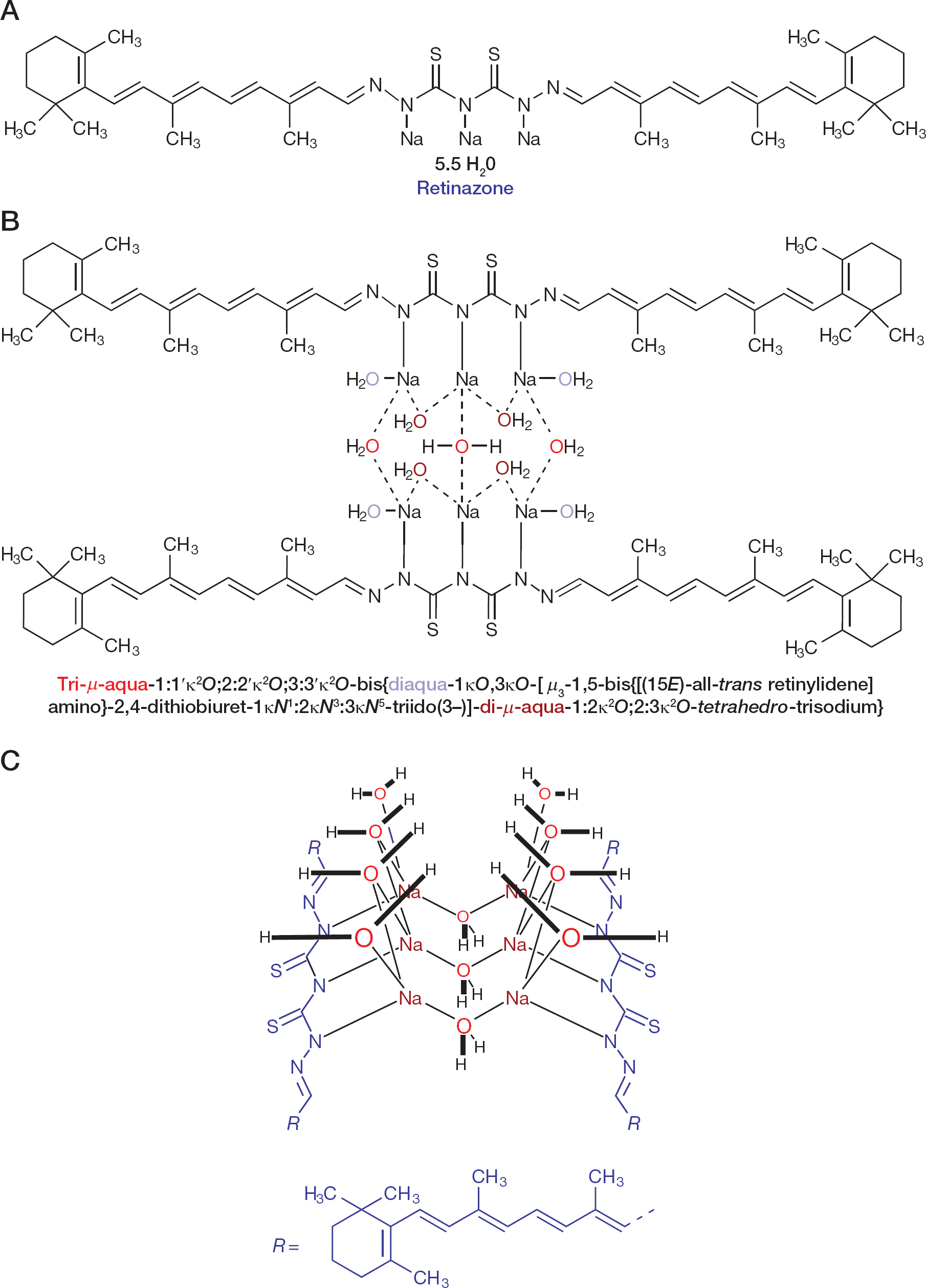
Chemical structure of RTZ
Extracellular (virion) HBV ayw DNA dot blot hybridization assay
The general assay methodology was described previously [21,22]. The assays were conducted in HepG2 2.2.15 HBV ayw producer HCC cell line, harbouring a persistently human chromosome-integrated HBV ayw genome, which was cultured as described previously [21,22]. Briefly, after isolation of HBV DNA [21], extracellular (virion) HBV ayw episomal covalently closed circular (cccDNA) levels were assessed by quantitative dot blot hybridization. The probe used was a gel-purified, restriction endonuclease EcoRI-digested (linearized) HBV ayw DNA 3.2 kb (3,182 bp) fragment [23], which was phosphorus-32 (32P)-labelled by DNA polymerase I-mediated nick translation with [α-32P]dCTP ([α-32P]-2′-deoxycytidine 5′-triphosphate). The relative quantification of the 32P signal hybridized to the sample was compared to the signal hybridizing to known quantities of HBV ayw DNA standards, as included with each nitrocellulose filter (dot blot). Standard curves, generated by multiple positive control analyses, were used to correlate relative counts-per-minute (cpm) measurements with standard HBV ayw DNA cpm. Host cellular (HepG2 2.2.15) toxicity was determined by neutral red lysosomal uptake assay as described previously [21,22].
Extracellular (virion) HBV ayw DNA TaqMan® RT-qPCR assay
The general TaqMan® RT-qPCR assay methodology was described previously as liver HBV DNA assay [24]. The anti-HBV assay developed at Southern Research Institute employs real-time quantitative PCR (RT-qPCR, TaqMan®) to directly measure extracellular HBV DNA copy number. Briefly, HepG2 2.2.15 cells were plated in 96-well microtitre plates. On the following day, the HepG2 2.2.15 cells were washed and the medium was replaced with complete medium containing various concentrations of a test compound in triplicate. The antiretroviral drug (−)-lamivudine (3′-thia-2′,3′-dideoxy-L-cytidine, 3TC; EPIVIR™, ZEFFIX™) was used as the positive control, while medium alone was added to cells as a negative control (virus control). Three days later, the culture medium was replaced with fresh medium containing the appropriately diluted drug. Six days following the initial administration of the test compound, the cell culture supernatant was collected, treated with pronase, and then used in a TaqMan® RT-qPCR assay to determine HBV DNA copy numbers. The fluorescent dye (6-FAM)/quencher dye (TAMRA)-labelled TaqMan® RT-qPCR probe utilized spanned from nt 2,333 to nt 2,363 of HBV ayw DNA and was composed of and labelled with: 5′-(6-FAM)–CGGAAACTACTGTTGTTAGACGACGAGGCAG–(TAMRA–[6-FAM])-3′ ([6-FAM] = 6-carboxyfluorescein; TAMRA = 6-carboxytetramethyl-rhodamine). The forward primer spanned from nt 2,305 to nt 2,325 of HBV ayw DNA: 5′-CCAAATGCCCCTATCCTATCA-3′. The reverse primer spanned from nt 2,392 to nt 2,370 of HBV ayw DNA: 5′-GAGGCGAGGGAGTTCTTCTTCTA-3′. FullVelocity™ quantitative PCR (qPCR) Master Mix (Stratagene, Inc., La Jolla, CA, USA) was used. The assay was run with a series of dilutions of DNA isolated from HepG2 2.2.15 cells to obtain a standard curve, with the x-axis as the threshold cycle (CT) value, and the y-axis as the log10 dilution of the standard. Obtained r2 values were used to measure the quality of the curve and, deduced from it, the efficiency of the TaqMan® RT-qPCR. The r2 regression curve correlation values were always above 0.98 (the average r2 value of the TaqMan® RT-qPCR procedure was 0.99). Mean CT values were obtained for duplicates of each sample. The mean CT values of each sample were used to obtain the relative log10 DNA value by using a formula of the fit line of the standard curve. Antiviral activity was calculated from the reduction in HBV DNA levels relative to the controls. The cells were used to assess cytotoxicity 50% with CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) kit (Promega) according to manufacturer's protocol.
HCV Huh7 ET (luc-ubi-neo/ET) replicon
The anti-HCV activity of RTZ was assessed using a subgenomic HCV replicon. The Huh7 ET cell line was kindly provided by Ralf Bartenschlager (Department of Molecular Virology, University of Heidelberg, Heidelberg, Germany) [25,26]. The ET cell line is a human HCC cell line (derived from Huh7) that contains a Con1 (genotype 1b) bicistronic subgenomic replicon. The subgenomic replicon contains a stable luciferase reporter gene, three cell culture-adaptive mutations (E1202G, T1280I, K1846T; numbered according to GenBank accession number AJ238799.1 [HCV-1b Con1 complete genome, 9,605 bp]) [26], the genuine HCV-1b internal ribosomal entry site (IRES), and the first amino acid codons of the HCV core protein which drive the production of luciferase, ubiquitin and neomycin phosphotransferase fusion protein. The murine encephalomyocarditis virus (EMCV, Picornaviridae, Cardiovirus) IRES element directs the translation of the second cistronic unit which encodes the non-structural proteins NS3, NS4A, NS4B, NS5A and NS5B. Cells were grown in Dulbecco's modified essential medium (DMEM) with 10% fetal bovine serum (FBS), 1% penicillin–streptomycin, 1% L-glutamine, 1% non-essential amino acids and 250 μg/ml G418 (geneticin) in a 5% CO2 incubator at 37°C.
HCV Huh7 ET (luc-ubi-neo/ET) replicon luciferase reporter assay
The general assay methodology was described previously [27,28]. Assessments of antiviral activity and cytotoxicity were carried out in parallel in clear-bottomed 96-well plates according to Southern Research Institute's standard format. The reporter replicon cells were seeded into two identical sets of 96-well plates at a density of 5×103 cells per well in 0.1 ml of DMEM without selection antibiotic (G418) in a humidified atmosphere supplemented with 5% CO2 at 37°C. RTZ was initially solubilized in DMSO. Half-log10 serial dilutions of RTZ, typically spanning the range of 63 nM to 20 μM, were then prepared in DMEM + 5% FBS and then applied to the corresponding wells. Human recombinant interferon-α2b (PBL Biomedical Laboratories, New Brunswick, NJ, USA) was included as a positive control at half-log10 dilutions in DMEM typically spanning the range of 0.006 to 2.0 IU/ml. Following 72 h incubation, one set of the cells was processed to assess the replicon-derived luciferase activity with the Steady-Glo® Luciferase Assay System (Promega) according to manufacturer's instructions. The activity of the luciferase reporter is proportional to HCV-1b Con1 RNA levels. The percent inhibition was plotted against the nominal concentration of compound to derive values for effective inhibitory concentration 50% (EC50). Cytotoxicity in the subgenomic replicon assay was assessed by using the CytoTox-ONE™ Homogeneous Membrane Integrity Assay Kit (Promega). The CytoTox-ONE™ Assay is a rapid, fluorescent measurement of the release of lactate dehydrogenase (LDH) from cells with a damaged membrane. LDH released into the culture medium is measured with a 10 min coupled enzymatic assay that results in the conversion of resazurin into resorufin in the presence of diaphorase. The CytoTox-ONE™ Reagent mix does not damage healthy cells; therefore the reactions to measure released LDH can be performed directly in a homogeneous format in assay wells containing a mixed population of viable and damaged cells.
HCV Huh7 ET (luc-ubi-neo/ET) replicon TaqMan® RT-qPCR assay
The general assay methodology was described previously [27,28]. The cell culture conditions were identical to the luciferase reporter assay. Cells were processed after a 72-h incubation at 37°C. The diluted compounds were applied to appropriate wells in the 96-well plates. Human recombinant interferon-α2b (PBL Biomedical Laboratories) was included as a positive control. The intracellular RNA from each well was extracted using the RNeasy® 96 Kit (Qiagen, Inc., Valencia, CA, USA). The level of HCV RNA was determined by a real-time reverse transcriptase PCR assay using TaqMan® One-Step RT-qPCR Master Mix Reagents (Applied Biosystems, Foster City, CA, USA) and an ABI PRISM® 7900 sequence detection system (Applied Biosystems). The cytotoxic effects were measured with TaqMan® Ribosomal RNA Control Reagents (Applied Biosystems) as an indication of cell numbers. The amounts of HCV RNA and ribosomal RNA (rRNA) were then plotted into Southern Research Institute's antiviral evaluation format to derive the EC50 and cytotoxic concentration 50% (CC50).
eGFP–EBOV Zaire 1976 Mayinga antiviral assay
The general assay methodology was described previously [29]. EBOV Zaire 1976 Mayinga expressing the eGFP was derived by reverse genetics to generate a full-length cDNA clone inserted with a foreign reporter gene eGFP (eGFP–EBOV Zaire 1976 Mayinga) [14]. Vero E6 cells (VERO C1008; Vero 76, clone E6) were seeded at 4×104 cells/well (100 μl) in black 96-well plates and incubated at 37°C and 5% CO2 with Eagle's minimum essential medium (EMEM; Invitrogen™/Life Technologies™, Carlsbad, CA, USA) complete with 10% FBS + 5% L-glutamine and HEPES buffer (EMEM–C). After 24-h incubation of seeded cells, and proper dilution of the compounds dissolved in DMSO with EMEM, a suitable amount of test compound (4× concentrated, 50 μl) from the dilution plate was transferred to two cell plates; one cell plate was used for the eGFP–EBOV Zaire 1976 Mayinga infection and the second for the cytotoxicity assay. The plates used for the eGFP–EBOV Zaire 1976 Mayinga infection were transferred into Bio-Safety Level (BSL)-4 containment, whereas those used for the cytotoxicity assay remained at BSL-2 containment until the 48 h time point. At BSL-4, 50 μl of eGFP–EBOV Zaire 1976 Mayinga virus diluted in EMEM was added to each infection plate at a multiplicity-of-infection (MOI) of 1. Infected eGFP–EBOV Zaire 1976 Mayinga assay plates incubated for 48 h at 37°C and 5% CO2; after incubation, plates were read for eGFP fluorescence (excitation 485 nm, emission 515 nm, cutoff 495 nm; 6 reads/well) on a Gemini™ EM spectrofluorometer (Molecular Devices, Sunnyvale, CA, USA). At the same time, plates remaining in BSL-2 were used for the cytotoxicity assay. The RTZ experiment was run in triplicate and treated statistically by Kruskal–Wallis one-way analysis of variance (ANOVA) on ranks with Bonferroni correction post-test (as compared to the associated virus control). Internal controls were +cells/+virus/-RTZ and +cells/-virus/-RTZ. The effect of RTZ on eGFP–EBOV Zaire 1976 Mayinga multiplication was expressed as a percentage of these controls and plotted against the RTZ concentration.
eGFP–EBOV Zaire 1976 Mayinga cytotoxicity assay
The CellTiter–Glo® Luminescent Cell Viability Assay (Promega) is a method of determining the number of viable cells based on the quantitation of adenosine 5′-triphosphate (ATP) with ATP-dependent Ultra–Glo™ Recombinant Luciferase (from the firefly Photuris pennsylvanica), which is an indirect quantification of the state of metabolically active cells (Promega). CellTiter–Glo® Buffer and lyophilized CellTiter–Glo® Substrate were removed from −20°C storage into −4°C for thawing 24 h before use. After proper thawing and equilibration to room temperature, the CellTiter–Glo® buffer (100 ml) was added to the CellTiter–Glo® Substrate and mixed by inverting the closed amber bottle to reconstitute the lyophilized enzyme/substrate mixture, making the CellTiter–Glo® Reagent mixture. All media and compound concentrations added to the assay plate (200 μl), as described before, were discarded by flicking into a waste container after 48 h incubation with compounds. CellTiter–Glo® reagent mixture (100 μl) and DMEM (Invitrogen™/Life Technologies™; 100 μl) were added. After addition of the reagent and media, the samples were mixed using an orbital shaker for 2 min; after mixing, plates were incubated at room temperature for 10 min to stabilize the luminescence signal. Plates were then read for fluorescence (excitation 485 nm, emission 515 nm, cutoff 495 nm; 6 reads/well) on a Gemini™ EM spectrofluorometer (Molecular Devices).
EBOV Zaire 1976 Mayinga (wild-type) antiviral assay
The assay was performed in Vero cells with wild-type EBOV Zaire 1976 Mayinga virus as described previously [30]. The utilized positive control drug was the guanine nucleobase antimetabolite 3-deazaguanine (6-amino-1,5-dihydro-4H-imidazo[4,5-c]pyridin-4-one, 3-DG) [31]. The applied assay method was the determination of virus yield reduction (VYR) utilizing crystal violet staining. The VYR assay determines actual virus yield in the presence and absence of the test compound; this is the confirmatory assay for antiviral activity. The effective concentration 90% (EC90) was determined by VYR for both 3-deazaguanine and RTZ. The cytotoxicity 50% exerted on Vero cells by both 3-deazaguanine and RTZ was determined by neutral red lysosomal uptake cell viability assay [32].
HHV-6B Z29 DNA hybridization assay
The general assay methodology was described previously [33,34]. In detail, uninfected MOLT-3 cells were added to 96-well plates at a concentration of 1×104 cells/well. Infection was initiated by adding HHV-6B-infected MOLT-3 cells at a ratio of approximately 1 infected cell for every 10 uninfected MOLT-3 cells. Drugs were serially diluted and added to triplicate wells. The anti-DNA-viral agent cidofovir (1-[(2S)-3-hydroxy-2-(phosphonomethoxy)-1-propyl]-1H-cytosine, CDV; VISTIDE™) was included as positive control. Drug-free medium was added to the wells that contained the cell and virus controls. The plates were incubated for seven days at 37°C. After incubation, 100 μl of 3× denaturation buffer (4.5 M NaCl, 1.2 M NaOH) was added to each well, and 50 μl was added to the wells of a Bio-Dot® apparatus (Bio-Rad Laboratories, Inc., Hercules, CA, USA) containing an Immobilon™ nylon membrane (Millipore, Bedford, MA, USA). A 50-μl aliquot of denatured DNA from the infected cells was aspirated through the membrane, and then 50 μl of denaturation buffer and 50 μl of phosphate-buffered saline (PBS) were added. The membrane was allowed to air dry, equilibrated in 2×SSC (1×SSC is 0.15 M NaCl plus 15 μM sodium citrate)–0.1% (m/v) sodium dodecyl sulfate (SDS), and prehybridized in DIG Easy Hyb (Roche Applied Science, Indianapolis, IN, USA) for 30 min at 37°C. A specific HHV-6A digoxigenin (DIG)-labelled probe was prepared by PCR (Roche Applied Science) from HHV-6A strain U1102 DNA using suitable primers. The forward primer spanned from nt 37,820 to nt 37,839 of HHV-6A strain U1102 DNA (GenBank accession number X83413.1): 5′-TGGGATTGGGATTAGAGCTG-3′. This HHV-6A forward primer corresponds to HHV-6B strain Z29 DNA from nt 38,936 to nt 38,955: 5′-GTCTCTTAGGATTGGAGCTG-3′. The reverse primer spanned from nt 38,418 to nt 38,399 of HHV-6A strain U1102 DNA: 5′-CCTTGATCATTCGACCGTTT-3′. This HHV-6A reverse primer is identical to HHV-6B strain Z29 DNA from nt 39,534 to nt 39,515. Therefore, a segment from nt 38,936 to nt 39,534 of HHV-6B strain Z29 DNA was detected by the HHV-6A strain U1102 probe. This fragment is located in the U27 gene coding for polymerase processivity factor (NCBI Reference Sequence accession number NC_000898.1). The denatured probe was allowed to hybridize to the viral DNA on the filter overnight at 37°C. The membrane was washed once at 37°C with 2xSSC–0.1% SDS, three times with 0.2xSSC–0.1% SDS, and once with 0.1xSSC–0.1% SDS. Detection of specifically bound DIG probe was performed with anti-DIG antibody using the manufacturer's protocol (Roche). An image of the film was captured digitally and quantified with Quantity One® software (Bio-Rad), and EC50 values were calculated as previously described [33]. CC50 was evaluated by application of the CellTiter–Glo® Luminescent Cell Viability Assay (Promega) as previously described [35,36].
HHV-8 BCBL-1 DNA TaqMan® RT-qPCR assay
Antiviral activity against HHV-8 was evaluated by methods reported previously in concise form [37]. In detail, BCBL-1 cells (NIH, NIAID, Division of AIDS, AIDS Research and Reference Reagent Program, Germantown, MD, USA) were maintained in growth medium consisting of RPMI 1640 supplemented with 10% FBS, penicillin, gentamicin, and L-glutamine, and 2×104 cells were added to each well in a 96-well plate. Compounds were diluted in triplicate wells of a 96-well plate, with the highest dilution yielding a concentration of 60 μM. The anti-DNA-viral agent cidofovir was included as positive control. The BCBL-1 cells were induced to undergo a lytic infection by the addition of phorbol 12-myristate 13-acetate (Promega) at a final concentration of 100 ng/ml. Plates were incubated for 7 days at 37°C in a humidified CO2 incubator. Total DNA was prepared with a Wizard® SV 96 Genomic DNA Purification System 96-well purification kit (Promega), and viral DNA was quantified by TaqMan® RT-qPCR. The fluorescent dye (6-FAM)/quencher dye (TAMRA)-labelled TaqMan® RT-qPCR probe utilized spanned from nt 14,144 to nt 14,160 of HHV-8 GK18 DNA and was composed of and labelled with: 5′-(6-FAM)–CCTACGTGTTCGTCGAC–(TAMRA)-3′. The forward primer spanned from nt 14,120 to nt 14,142 of HHV-8 GK18 DNA: 5′-TTCCCCAGATACACGACAGAATC-3′. The reverse primer spanned from nt 14,182 to nt 14,166 of HHV-8 GK18 DNA: 5′-CGGAGCGCAGGCTACCT-3′. Plasmid pMP218 containing a DNA sequence corresponding to nt 14,120 to nt 14,182 (HHV-8 GK18, GenBank accession number AF148805.2) was used to provide absolute quantification of viral DNA. Compound concentrations sufficient to reduce viral DNA accumulation by 50% (EC50) were calculated by standard methods. CC50 was evaluated in a duplicate plate containing BCBL-1 cells with the same exposure as to the test compounds by application of the CellTiter–Glo® Luminescent Cell Viability Assay (Promega) as previously described [35,36].
Cell biology
LanthaScreen® TR–FRET glucocorticoid receptor coactivator assay
The LanthaScreen® time-resolved Förster (fluorescence) resonance energy transfer (TR–FRET) glucocorticoid receptor coactivator assay (Invitrogen™/Life Technologies™) is based on quantifying the binding of the fluorescein-labelled coactivator peptide SRC1–4 (19-mer C-terminal part of steroid receptor coactivator-1 [SRC-1] [38], aa 1,423–1,441 [GPQTPQAQQKSL
LanthaScreen® TR–FRET glucocorticoid receptor competitive binding assay
The LanthaScreen® TR–FRET glucocorticoid receptor competitive binding assay (Invitrogen™/Life Technologies™) is based on monitoring the competitive displacement (by an agonist or antagonist) of the fluorescein-labelled glucocorticoid agonist Fluormone™ GS1 Green (exact structure not disclosed by Invitrogen™/Life Technologies™) from a GST fusion protein with hGRα LBD by TR–FRET utilizing a sensitized Tb3+-chelate-labelled anti-GST antibody. All general conditions were identical to the coactivator assay. A serial dilution of compounds was prepared in 100×DMSO, starting from the maximum desired concentration which was 200 μM for RTZ. Each 100× solution was diluted to 2× concentration with TR–FRET nuclear receptor buffer F, yielding a final concentration of 1% (v/v) DMSO in each well. A volume of 10 μl of 2× solution was added to a black low volume 384-well assay plate (Corning), followed by addition of 5 μl 4×hGRα LBD/Tb3+-chelate-labelled anti-GST antibody and 5 μl of 4× (80 nM) Fluormone™ GS1 Green (25× stock solution is 500 nM in 75% [v/v] aqueous methanol, to be diluted to 4× with TR–FRET nuclear receptor buffer F). Dexamethasone was included as a positive control at the high concentration of 20 μM, securing 100% Fluormone™ GS1 Green displacement from hGRα LBD. A negative control with 2×DMSO was present to account for any solvent vehicle effects. The hGRα LBD and the sensitized Tb3+-chelate-labelled anti-GST antibody were premixed in light protecting vials prior to use. The final concentrations of 5 mM DTT and of 1×hGRα-stabilizing peptide (10×hGRα-stabilizing peptide is 1 mM in 10 mM potassium phosphate pH 7.4, exact identity not disclosed by Invitrogen™/Life Technologies™) were used in the assay buffer in order to prevent protein degradation. Plates were incubated at room temperature protected from light for 2 hours prior to TR–FRET measurement. The vehicle control was set as 0% Fluormone™ GS1 Green agonist displacement from hGRα LBD, and the 20 μM dexamethasone experiment was treated as 100% Fluormone™ GS1 Green agonist displacement from hGRα LBD. The percentage values of agonist displacement from the hGRα LBD by RTZ were calculated in relation to these control values from the TR–FRET signal ratios 520 nm/495 nm, and were plotted against the logarithmic (log10) RTZ concentrations. All experiments were run in triplicate and treated statistically by two-way analysis of variance (ANOVA) with Bonferroni correction post-test (as compared to the associated vehicle control).
Results
The chemical structure of RTZ and its purity
The chemical structure of RTZ was elucidated as retinoid dimer-of-a-dimer sodium hydrate coordination complex [11] (Figure 1B and 1C), as based on prior experiences with structurally related sodium hydrate clusters [40]. RTZ was synthesized in ≥98% (n/n) purity according to proton nuclear magnetic resonance (1H NMR) spectroscopy (700.430 MHz proton nuclear magnetic resonance [1H NMR] spectrum of RTZ dissolved in DMSO-d6 in Additional file 1). RTZ is a remarkably stable compound, despite containing a retinoid polyene, due to intramolecular cessation of oxygen-initiated radical chain reactions by the intrinsic sulfur content. An impurity of RTZ, the content of which was diminished during purification in concentration, the so-called OBH acetate, was synthesized independently from vitamin A–acetate in the form of OBH chloride hydrate. The structure of OBH chloride hydrate was elucidated by NMR spectroscopy (700.430 MHz proton nuclear magnetic resonance [1H NMR] spectra of OBH chloridex1/3 H2O in Additional file 1) as stable cyclic oxonium salt retaining the vitamin A diterpenoid (C20) carbon scaffold. Its structure was verified unequivocally by independent synthesis as stable cyclic oxonium salt OBH chloridex1/3 H2O, similar in structure to the recently reported cyclic oxonium salts oxatriquinane [41] and 1,4,7-trimethyloxatriquinane [42]. OBH chloride hydrate was non-toxic to cultured cells (CEM and Vero cells, respectively: CC50>100 μM) and did not exhibit antiviral activity against HIV-1LAI in primary human PBL cells (EC50>100 μM; CC50>100 μM).
The antiviral spectrum of RTZ
The antiviral effects exerted by RTZ on HIV-1LAI and HCV-1b Con1 were confirmed and extended by data including HBV subtype ayw (HBV ayw; Figure 2A), HHV-6B strain Z29, HHV-8 strain BCBL-1 and wild-type EBOV Zaire 1976 Mayinga (Table 1). Additionally, the antiviral spectrum of RTZ was assessed versus 26 virus species from differently selected RNA and DNA virus families towards which RTZ was found to be inactive (Assessing the antiviral spectrum of RTZ in Additional file 1). In detail, RTZ was inactive versus Picornaviridae, Flaviviridae, Togaviridae, Coronaviridae, Rhabdoviridae, Paramyxoviridae, Arenaviridae, Bunyaviridae and Orthomyxoviridae, all of which represent RNA viruses. RTZ was also inactive versus Adenoviridae, Polyomaviridae and Poxviridae, all of which represent DNA viruses. Inactivity of RTZ was additionally detected towards HHV-1 (herpes simplex virus type 1), HHV-2 (herpes simplex virus type 2) and HHV-4 (Epstein–Barr virus).
Antiviral in vitro activities of RTZ
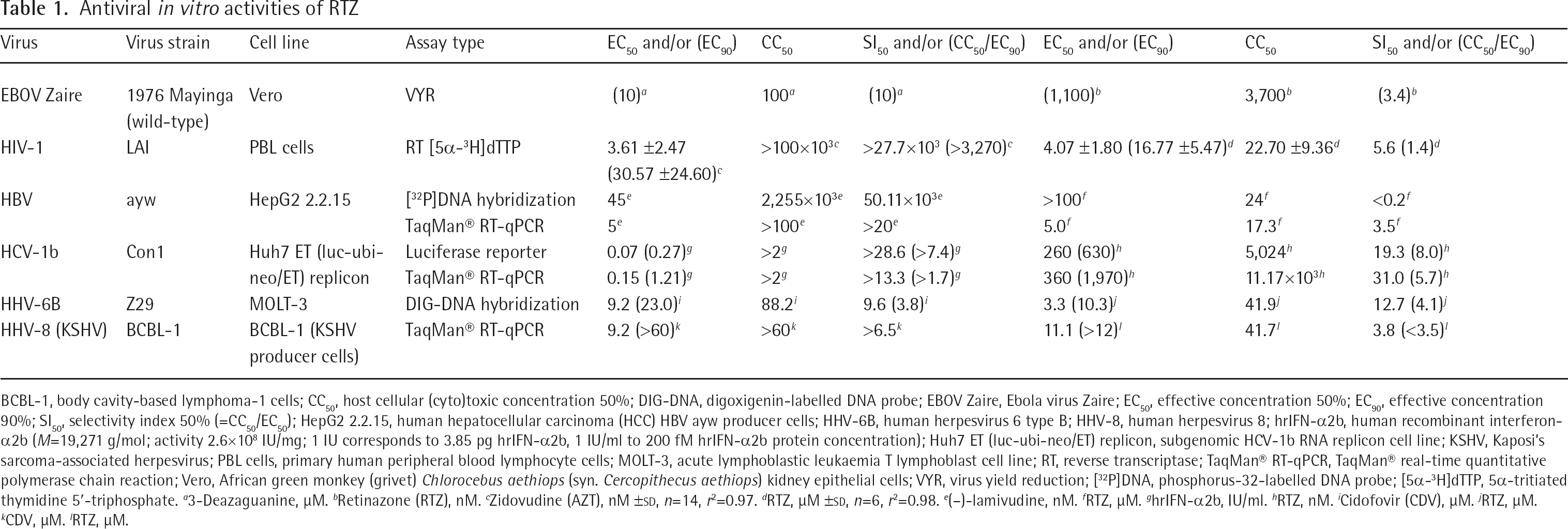
BCBL-1, body cavity-based lymphoma-1 cells; CC50, host cellular (cyto)toxic concentration 50%; DIG-DNA, digoxigenin-labelled DNA probe; EBOV Zaire, Ebola virus Zaire; EC50, effective concentration 50%; EC90, effective concentration 90%; SI50, selectivity index 50% (=CC50/EC50); HepG2 2.2.15, human hepatocellular carcinoma (HCC) HBV ayw producer cells; HHV-6B, human herpesvirus 6 type B; HHV-8, human herpesvirus 8; hrIFN-α2b, human recombinant interferon-α2b (M=19,271 g/mol; activity 2.6×108 IU/mg; 1 IU corresponds to 3.85 pg hrIFN-α2b, 1 IU/ml to 200 fM hrIFN-α2b protein concentration); Huh7 ET (luc-ubi-neo/ET) replicon, subgenomic HCV-1b RNA replicon cell line; KSHV, Kaposi's sarcoma-associated herpesvirus; PBL cells, primary human peripheral blood lymphocyte cells; MOLT-3, acute lymphoblastic leukaemia T lymphoblast cell line; RT, reverse transcriptase; TaqMan® RT-qPCR, TaqMan® real-time quantitative polymerase chain reaction; Vero, African green monkey (grivet) Chlorocebus aethiops (syn. Cercopithecus aethiops) kidney epithelial cells; VYR, virus yield reduction; [32P]DNA, phosphorus-32-labelled DNA probe; [5α-3H]dTTP, 5α-tritiated thymidine 5′-triphosphate.
3-Deazaguanine, μM.
Retinazone (RTZ), nM.
Zidovudine (AZT), nM ±sd, n=14, r2=0.97.
RTZ, μM ±sd, n=6, r2=0.98.
(–)-lamivudine, nM.
RTZ, μM.
hrIFN-α2b, IU/ml.
RTZ, nM.
Cidofovir (CDV), μM.
RTZ, μM.
CDV, μM.
RTZ, μM.
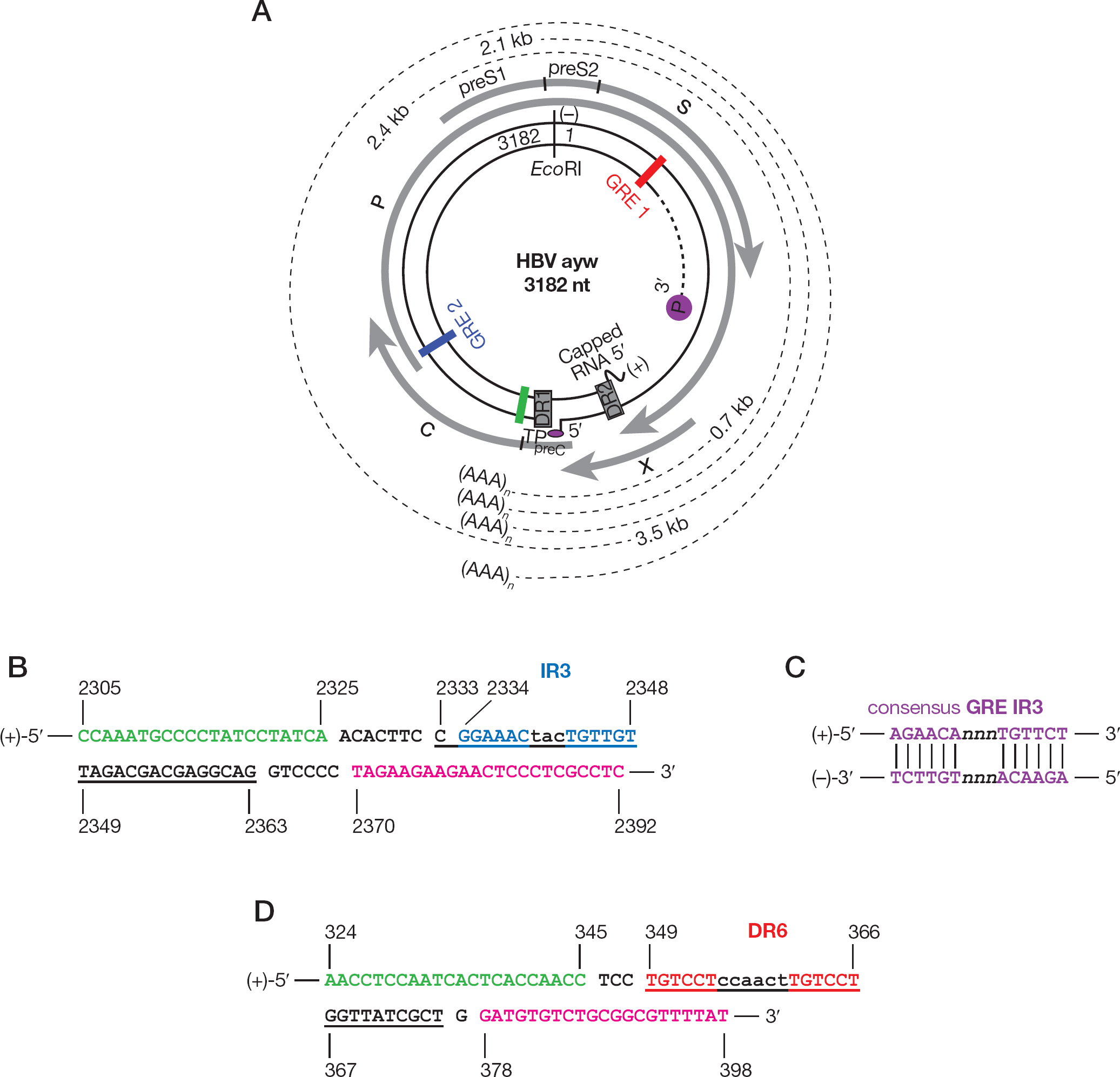
The established GRE 1 and the putative GRE 2 in the 3,182 nucleotide partially double-stranded DNA genome of HBV
The differential effects of RTZ on HBV ayw
Interestingly, we found antiviral activity of RTZ (EC50=5.0 μM) on HBV ayw only in the quantitative polymerase chain reaction (qPCR) assay, whereas in the Southern blot DNA hybridization assay we could not detect antiviral activity of RTZ. The difference between the two assays is that in qPCR a TaqMan® real-time qPCR (TaqMan® RT-qPCR) probe was used with suitable forward and reverse primers, whereas in the DNA hybridization assay a 3.2 kb EcoRI-digested full-genomic (Figure 2A) 32P-labelled HBV DNA probe [23] was applied. Therefore, we conclude that HBV DNA was fully produced in the presence of RTZ, but was not functional (that is, not functional as readable template). This means that RTZ must have covalently modified the HBV DNA in the sequence region complementary to the TaqMan® RT-qPCR probe. The qPCR probe utilized spanned from nucleotide (nt) 2,333 to nt 2,363 of HBV ayw DNA (Figure 2B). In this region we identified a good putative glucocorticoid nuclear receptor (GR) response DNA cis-element (glucocorticoid response element [GRE]) inverted repeat 3 (IR3),
The covalent modification of λ bacteriophage DNA by RTZ
To verify the reaction of RTZ with DNA, the covalent modification of λ bacteriophage double-stranded DNA (dsDNA) by RTZ under catalysis of aqueous acetic acid (HOAc) at room temperature (RT) was examined. Firstly, without HOAc no reaction was observed at RT (Covalent modification of λ DNA by RTZ in Additional file 1) or 70°C (Covalent modification of λ DNA by RTZ in Additional file 1), indicating that RTZ is non-mutagenic under these conditions. Secondly, under HOAc catalysis production of hydrazinothio-carbonyl-DNA (HTC-DNA) was observed as detected by thin-layer chromatography (TLC; Covalent modification of λ DNA by RTZ in Additional file 1). The acid-sensitive by-product all-trans retinal (vitamin A–aldehyde) was degraded by HOAc to OBH acetate. Oligomeric λ dsDNA was de-polymerized and reacted with (5RS)-5-(14-apo-β-caroten-14-yl)-1,3,4-oxadiazolidine-2-thione (ACO) by trans-thiocarbamoylation of its lact(hi)one ring. ACO is formed by splitting of RTZ under HOAc catalysis into the components (2E)-2-(all-trans retinylidene)hydrazinecarbothioic O-acid (which spontaneously cyclized to ACO) and (15E)-all-trans retinal thiosemicarbazone (ATR-TSC; Splitting of RTZ by acid catalysis in Additional file 1). The ATR-TSC was isolated from HOAc-treated RTZ and identified by NMR spectroscopy (see Additional file 1). These elucidations suggest that RTZ can modify viral target DNA (Covalent modification of HBV subtype ayw DNA by RTZ in Additional file 1) under proton catalysis. A direct (covalent) binding of RTZ to the glucocorticoid hydrocortisone could be excluded by investigating the chemical reaction of RTZ with hydrocortisone in aqueous ethanol at elevated temperature under presence of HOAc. Only ATR-TSC in admixture with OBH acetate could be isolated (see Additional file 1). This pointed to chemical inertness of hydrocortisone towards RTZ.
The mechanism of the anti-HBV ayw activity exerted by RTZ
The anti-HBV effect of RTZ should be mediated by GR, since the HBV genome contains at least two GR trans-activation targets (GREs). We propose that RTZ binds to human GR (hGR), is transported through the nuclear pore complex [46] to the intra-nuclear HBV genome by the RTZ-liganded hGR, the RTZ–hGR complex binds to GREs, and, finally, the co-transported RTZ switches to the HBV DNA and covalently modifies amine group-containing nucleobases (cytosine, guanine, adenine) by trans-thiocarbamoylation (Splitting of RTZ by acid catalysis and Covalent modification of HBV subtype ayw DNA by RTZ in Additional file 1). This could also happen for host cellular GREs, but we think there is selectivity for intraexonic GREs, which are not typical for the human genome [47], because viral intraexonic GREs are subject to different epigenetic regulation involving chromatin remodelling events [48] in comparison to host cellular GREs [47,48]. Human GREs are located in the promoter regions upstream of transcription initiation sites [47], with only few exceptions where the GRE is found within a human gene intron [49]. Additionally, it should be mentioned that human GREs are in most instances imperfect (GRE sequence degeneracy), that is, their sequences do not match the perfect GRE consensus sequence given (Figure 2C) [50]. In spite of these imperfect GRE sites, their glucocorticoid responsiveness is retained [47,50]. The linearized, full-genomic 3.2 kb HBV DNA probe could have hybridized to the RTZ-modified HBV DNA because two mutant nucleotides (in GRE 2) out of 3,182 are too minimal in extent (0.6285‰) to prevent hybridization.
The anti-HIV-1LAI activity of RTZ
For the case of HIV-1LAI a striking analogy to HBV ayw is predicted. HIV-1LAI proviral DNA (The proviral DNA genome of HIV-1
LAI
in Additional file 1) contains four recognized GREs (GRE 1, GRE 2, GRE 3 and GRE 4) [51]. GRE 1 (−264 to −259), GRE 2 (−6 to −1) and GRE 3 (+15 to +20) are located within the 5′-long terminal repeat (5′-LTR) [52] (The proviral DNA genome of HIV-1LAI in Additional file 1). The GRE 4,
Correlation of the antiviral spectrum of RTZ with the presence of viral GREs
Evidence for an involvement of human GR and its corresponding GREs in the antiviral activity spectrum of RTZ provided the comparison of complete genome data of RTZ-susceptible and RTZ-insensitive viruses with DNA stage (Table 2). In a systematic computational complete genome search all available RTZ-susceptible and RTZ-insensitive virus genomes were examined for intraexonic GREs of the consensus GRE type (Figure 2C), the HIV-1LAI vif GRE-related type, and the HBV ayw GRE 2-related type (Table 2). The GRE hits clearly indicated that in all RTZ-susceptible virus genomes at least one GRE was found within a gene essential for in vitro virus replication, whereas in all RTZ-insensitive virus genomes no GRE was found in genes essential for in vitro virus replication (Table 2).
Correlation of the antiviral spectrum of RTZ with the presence of intragenic/intraexonic GRE IR3 elements in RTZ-susceptible viruses
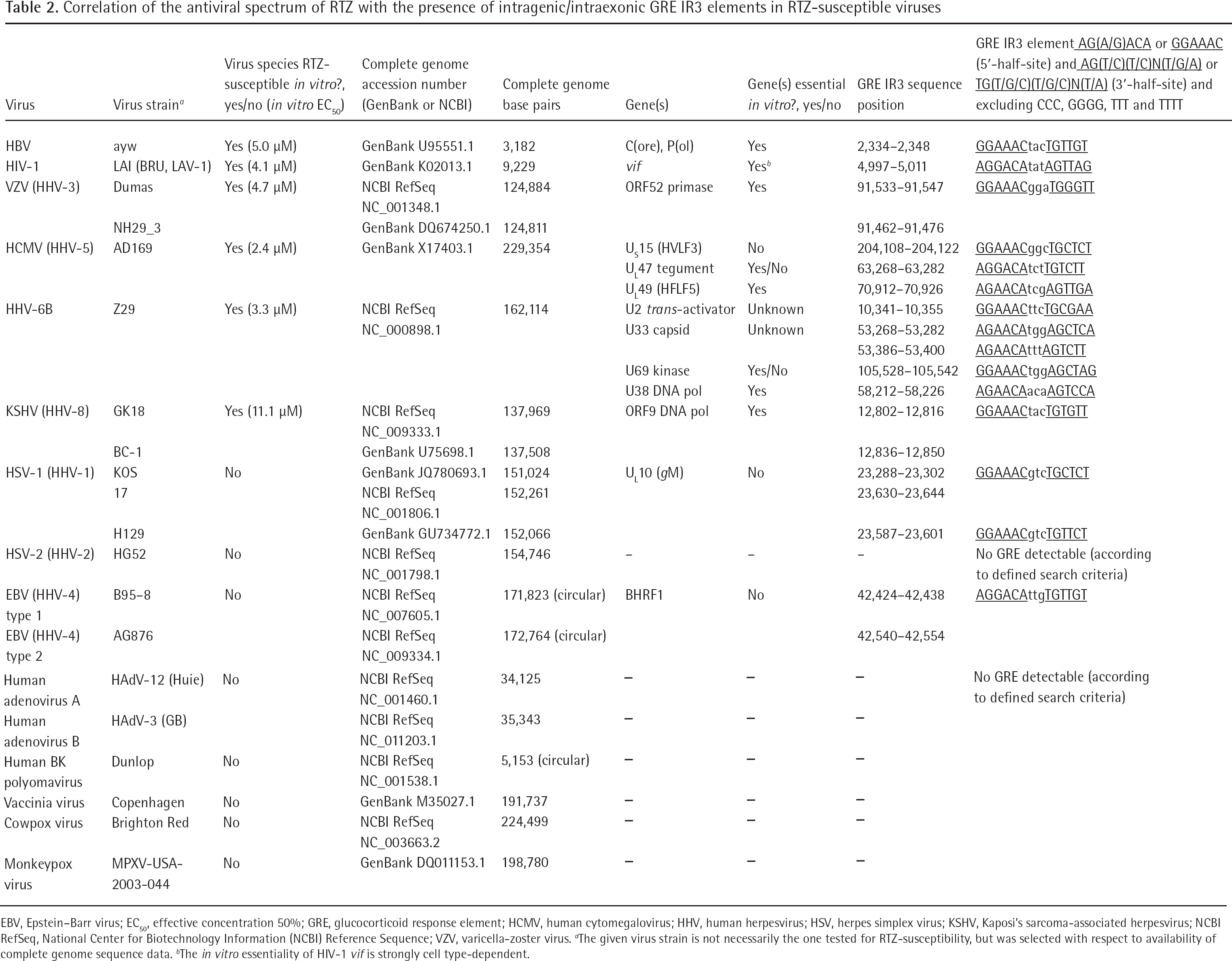
EBV, Epstein–Barr virus; EC50, effective concentration 50%; GRE, glucocorticoid response element; HCMV, human cytomegalovirus; HHV, human herpesvirus; HSV, herpes simplex virus; KSHV, Kaposi's sarcoma-associated herpesvirus; NCBI RefSeq, National Center for Biotechnology Information (NCBI) Reference Sequence; VZV, varicella-zoster virus.
The given virus strain is not necessarily the one tested for RTZ-susceptibility, but was selected with respect to availability of complete genome sequence data.
The in vitro essentiality of HIV-1 vif is strongly cell type-dependent.
In detail, the essentiality (indispensable function) or non-essentiality (dispensable function) of the gene(s) which contain the intragenic/intraexonic GRE elements for in vitro replication of the virus is indicated in Table 2. Included in the search algorithm were the consensus GRE
RTZ is a weak glucocorticoid antagonist and binds to human GRα
To obtain verification for the previous postulations, the binding of RTZ to hGR isoform α (hGRα) ligand (steroid)-binding domain (hGRα LBD) was examined (Figure 3). Indeed, RTZ did bind to hGRα LBD as determined by LanthaScreen® time-resolved TR–FRET glucocorticoid receptor coactivator assay. This steroid receptor coactivator-1 (SRC-1) [38] assay decides between agonist or antagonist activity of a test compound at hGRα LBD. RTZ acted as a weak antagonist at hGRα LBD, as compared to the potent hGRα LBD antagonist mifepristone (RU486; Figure 3A). The result was statistically significant (P<0.01). Subsequently, the concentration-dependent displacement of the fluorescein-labelled glucocorticoid agonist Fluormone™ GS1 Green from hGRα LBD by RTZ was interpolated to show an EC50 of 96 μM, as determined by the LanthaScreen® TR–FRET glucocorticoid receptor competitive binding assay (Figure 3B). The result was statistically significant (P<0.001). Therefore, it is reasonable to assume that RTZ binds to hGRα, acting as a weak, but detectable glucocorticoid nuclear receptor antagonist.
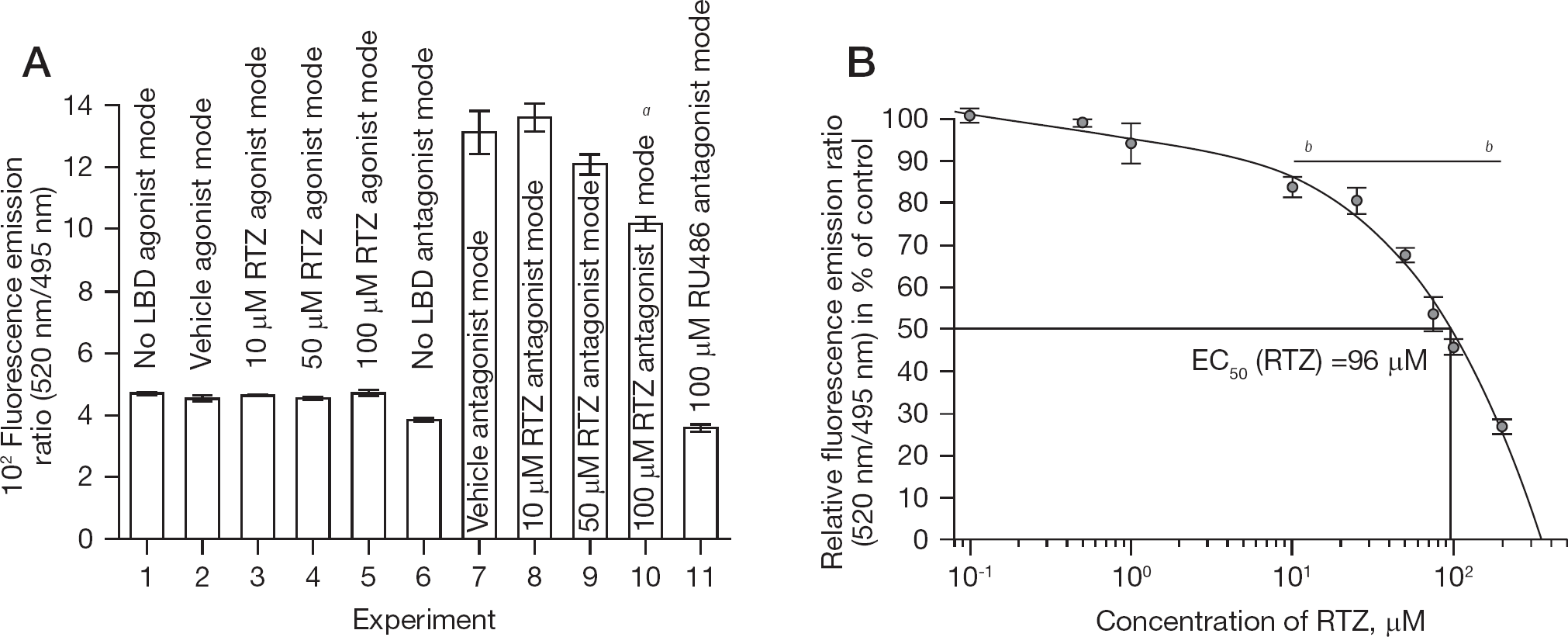
RTZ represents a weak glucocorticoid antagonist
Effects of RTZ on EBOV Zaire replication
The inhibition of eGFP–EBOV Zaire 1976 Mayinga multiplication by RTZ was examined in more detail (Figure 4). The EC50 of RTZ was interpolated to 830 nM (Figure 4). In comparison, the bis-benzamidine small molecule FGI-103 [64] was reported to suppress eGFP–EBOV Zaire 1976 Mayinga recombinant virus multiplication with an EC50 of 3.8 μM [64], and to suppress wild-type EBOV Zaire 1976 Mayinga virus yield with an EC50 of 100 nM [64]. Because EBOV inhibitors are rather scarce (Reported Filovirus inhibitors in Additional file 1), the activity of RTZ is interpreted to represent a useful result. The host CC50 exerted on Vero E6 cells by RTZ was interpolated to 5.67 μM, yielding a selectivity index 50% (SI50=CC50/EC50) of 6.8. Interestingly, non-tumorigenic HepG2 human HCC cells were completely insensitive to RTZ-mediated toxicity at 13.2 μM concentration, whereas Vero E6 (Vero 76, clone E6) cells were rather sensitive towards RTZ-mediated cellular toxicity, as will be discussed. To verify the results with eGFP–EBOV Zaire 1976 Mayinga recombinant virus, the suppression of wild-type EBOV Zaire 1976 Mayinga virus yield by RTZ was examined in Vero cells. The virus yield was suppressed by RTZ with an effective concentration 90% (EC90) of 1,100 nM (Table 1). The CC50 exerted on Vero cells by RTZ was determined to be 3,700 nM by the neutral red method, confirming the high sensitivity of Vero-type cells towards RTZ. The corresponding EC50 of RTZ regarding suppression of wild-type EBOV Zaire 1976 Mayinga virus yield could be interpolated to 48.8 nM yielding a SI50 of 75.8. Therefore, RTZ is comparable in antifiloviral potency to FGI-104 and FGI-103 when comparing the suppression of eGFP–EBOV Zaire 1976 Mayinga recombinant virus in vitro replication by FGI-104 (EC50 = 10 μM; CC50>50 μM; SI50>5) [29] and by FGI-103 (EC50=3.8 μM; CC50≥50 μM; SI50≥13.2) [64] with the corresponding data of RTZ (EC50=0.83 μM; CC50=5.67 μM; SI50=6.8). The data of RTZ regarding the in vitro suppression of wild-type EBOV Zaire 1976 Mayinga virus yield (EC50=0.0488 μM; CC50=3.7 μM; SI50=75.8) are of compelling evidence for the antifiloviral in vitro efficacy of RTZ.
Inhibition of eGFP–EBOV Zaire 1976 Mayinga multiplication by RTZ
The targeting of the RNA viruses HCV-1b and EBOV Zaire by RTZ
Regarding the antiviral mechanism-of-action of RTZ versus HCV-1b Con1 and EBOV Zaire 1976 Mayinga (HCV and Ebola virus genomes in Additional file 1), it is inherently clear that it cannot be being mediated by GRs, since both HCV and EBOV incorporate no DNA stages in their life cycles. It was recently reported that thiosemicarbazones (5,6-dimethoxyindan-1-one thiosemicarbazone) target bovine viral diarrhoea virus type 1 (BVDV-1; strain NADL, Flaviviridae, Pestivirus) NS5B protein RNA-dependent RNA polymerase [65]. BVDV-1 is commonly regarded as a suitable surrogate for HCV, because both Flaviviridae polyprotein sequences are closely related (maximal sequence identity 39%). Because the anti-HCV-1b activity of RTZ was determined with the HCV RNA replicon cell line Huh7 ET (luc-ubi-neo/ET) which only codes for the non-structural HCV proteins NS3, NS4A, NS4B, NS5A and NS5B, the inhibiting action of RTZ must be confined to these five genes and/or gene products.
The toxicity of RTZ on cultured cells
Regarding the toxicity of RTZ on cultured cell lines, the following observations should be added. RTZ (NSC-751959) showed antineoplastic activity within the 60-cancer cell panel included in the Developmental Therapeutics Program of the National Cancer Institute. The RTZ-induced average growth inhibition 50% (GI50 ±standard deviation [sd]) was found to be 1.73 ±0.48 μM (n=57), the RTZ-induced average growth inhibition 100% (TGI, GI100 ±sd) 5.57 ±0.49 μM (n=57), and the RTZ-induced average lethal concentration 50% (LC50 ±sd, the concentration at which 50% of cells die by apoptosis and/or necrosis) was 19.00 ±0.45 μM (n=53) on 57 tested cancer cell lines (Dose-response graphs of the antineoplastic action of RTZ and The experimental analysis of the antineoplastic action of RTZ in Additional file 1). Therefore, the observed in vitro toxicity exerted by RTZ on cultured cells reflects an intrinsic antineoplastic potential which is of considerable interest regarding the oncogenic nature of HIV-1, HBV, HCV and HHV-8. Cell lines especially sensitive to RTZ are, for example, CCRF-CEM (CEM, acute lymphoblastic leukaemia T lymphoblast cells) with a mean CC50 (RTZ) ±sd of 1.67 ±0.42 μM (n=6), and all Vero cell derivatives (Vero, Vero 76, Vero E6) with a mean CC50 (RTZ) ±sd of 5.83 ±4.58 μM (n=6) for Vero cells. The primary human PBL cells (containing primary human T lymphocytes) are relatively insensitive towards RTZ with a mean CC50 (RTZ) ±sd of 22.70 ±9.36 μM (n=6; Table 1).
Discussion
In the case of an infection with HIV-1, current HAART regimens are capable of achieving prolonged suppression of HIV-1 plasma viraemia in HIV-1-infected individuals [66,67]. However, it has not been possible to eradicate HIV-1 through HAART because HIV-1 seems to persist in latent retroviral reservoirs like follicular dendritic CD4+ T-cells located deep within the lymphatic system [66,67]. The mechanism of HIV-1 retroviral latency in persistent reservoirs unamenable to HAART is poorly defined and understood [66,67]. From a theoretical point of view the HIV-1 proviruses integrated in the human host chromatin of latent retroviral reservoirs should be responsible for the severe relapse of HIV-1 viraemia and replication following cessation of HAART in HIV-1-infected patients. By the application of reverse transcription-preceded TaqMan® RT-qPCR assays, the detection of single copies of HIV-1 genomic (+)-ssRNA in small volumes of blood plasma is possible [67]. By this technique it could be shown that low plasma viraemia (n<50 HIV-1 genomic RNA copies/ml blood plasma) persists in most HIV-1-infected individuals receiving HAART. It was demonstrated that residual plasma viraemia correlates with the size of the CD4+ T-lymphocyte cell viral reservoir [67], but not with markers of immune activation, suggesting that reactivation of this latent viral reservoir may not be the only source of residual plasma viraemia. This could explain why HIV-1 infection persists even after years of HAART. Although HAART can suppress viral replication greatly, and reduces viraemia to less than 50 HIV-1 genomic RNA copies per ml of blood plasma, proviral latency established within the host genome remains largely unaffected by HAART, and can replenish severe systemic infection following interruption of therapy. Pharmacological strategies which not only target viral replication per se, but also deplete proviral host genome infection, are certainly required for successful clearance of HIV-1 infection [66]. Novel therapeutic strategies aimed at targeting the source of residual viraemia are additionally necessary to achieve viral eradication [67].
We have shown that the vitamin A-derived thiosemicarbazone derivative RTZ has the ability to specifically inactivate human DNA-integrated proviruses of HBV and HIV-1 by exploiting the presence of human glucocorticoid nuclear receptor-binding DNA sequences, the glucocorticoid (hormone receptor) response elements, in the host chromatin-integrated HBV and HIV-1 proviral DNA genomes. These genomes are specifically inactivated by covalent attachment of N-HTC moieties to amine group-containing nucleobases (cytosine, guanine, adenine). These HTC-DNA lesions are lethal for the corresponding proviral genome, because they interrupt both DNA replication and RNA transcription from these proviruses. In addition, these lesions probably withstand repair by eukaryotic DNA repair systems like base excision repair [68], and could induce apoptosis in the infected host cell resulting in ultimate clearance of the viral xenogenomes. This opens the profound possibility to cure the blood-borne diseases chronic hepatitis B and AIDS. For the first time a pharmacological intervention could be found, in the form of the vitamin A-derived agent RTZ, which is able to covalently inactivate host chromatin-integrated HBV and HIV-1 proviral DNA genomes. This is a consequence of the affinity of RTZ-liganded human glucocorticoid nuclear receptor to proviral glucocorticoid response elements.
Moreover, HCV and EBOV Zaire, both also causing severe blood-borne human illnesses, are susceptible to life cycle disruption by RTZ. The HCV target of RTZ presumably represents the RNA-dependent RNA polymerase NS5B enzyme protein of HCV, because it carries a common thiosemicarbazone binding site [65]. The EBOV Zaire target of RTZ could be, for example, the filoviral matrix protein VP40. Binding of RTZ to EBOV Zaire proteins is expected to disrupt virus morphogenesis and budding, virion structure and transcriptional regulation of EBOV Zaire. The nanomolar activity of RTZ in inhibiting wild-type EBOV Zaire multiplication points to an efficacious interruption of the life cycle of EBOV Zaire by RTZ.
Regarding the observed in vitro toxicity of RTZ, there is evidence that this RTZ-mediated toxicity could be largely reduced in vivo, since RTZ is assumed to bind to and be transported with human plasma retinol-binding protein (hRBP) in human blood circulation. The in vitro toxicity of RTZ is assumed to be largely a consequence of RTZ interaction with host cellular lipid membranes leading to altered membrane fluidity status [69]. It was reported that N-retinylidene amine Schiff bases like RTZ bind to the chromophore binding site of hRBP in human blood plasma [70]. It is expected that the RTZ–hRBP in vivo complex distributed in human circulation mediates largely reduced toxicity when compared to the direct in vitro impact of RTZ on host cells utilized in our present study. Further work is clearly required to clarify this issue.
Footnotes
Acknowledgements
The authors thank T Westfeld, E-M May, D Wiegel and W Wuebbolt (Currenta GmbH & Co. OHG, Leverkusen, Germany) for analytical services. We are deeply indebted to BE Korba (Georgetown University Medical Center, Washington, DC, USA) for conducting the HBV DNA hybridization assay, to GG Olinger and C Scully (US Army Medical Research Institute of Infectious Diseases, Fort Detrick, MD, USA) for performing the eGFP–EBOV experiments, and to CKH Tseng (National Institutes of Health [NIH], National Institute of Allergy and Infectious Diseases [NIAID], Division of Microbiology and Infectious Diseases [DMID], Virology Branch) for supervising the wild-type EBOV experiments. We cordially thank R Koshy (NIH, NIAID, DMID, Enteric and Hepatic Diseases Branch) for helpful discussions. This work was supported in part by NIH CFAR grant 2P30–AI–50409 (to RFS) and by the Department of Veterans Affairs (to RFS). This project has been funded in whole or in part with Federal Funds from the DMID, NIAID, NIH, Department of Health and Human Services, under contract HHSN272201100012I. The Trinity Biomedical Sciences Institute is supported by a capital infrastructure investment from Cycle 5 of the Irish Higher Education Authority's Programme for Research in Third Level Institutions (PRTLI). DKN would like to thank the Irish Research Council and Dell Ireland for funding. LC expressed her thanks to the Health Research Board (HRB/2007/2) for funding. We are obliged to K Hecker (HEKAtech GmbH, Wegberg, Germany) for expert elemental analyses. LC and DKN contributed equally to this work.
The authors declare no competing interests.