
Abstract
This review article focuses on the anti-herpesvirus agents effective against herpes simplex virus, varicella-zoster virus and cytomegalovirus, which have either been licensed for clinical use (idoxuridine, trifluridine, brivudin, acyclovir, valaciclovir, valganciclovir, famciclovir and foscarnet) or are under clinical development (CMX001 [the hexadecyloxypropyl prodrug of cidofovir], the helicase-primase inhibitor BAY 57–1293 [now referred to as AIC316], FV-100 [the valine ester of Cf 1743] and the terminase inhibitor letermovir [AIC246]).
Idoxuridine and trifluridine
The antiviral drug era started with the discovery of idoxuridine (IDU) and trifluridine (TFT; Figure 1), which were both launched, and are still used today, for the topical treatment of herpetic eye infections (that is, herpetic keratitis) due to herpes simplex virus (HSV). IDU was first described by Prusoff [1] in 1959 as a potential anti-tumour agent, whereas both IDU and TFT were subsequently described as anti-HSV agents for the treatment of herpetic keratitis by Kaufman et al. [2–5].
Brivudin and BVaraU
IDU and TFT could be considered as the starting point for the synthesis of various new 5-substituted 2′-deoxyuridine derivatives, the most prominent of this series being brivudin (BVDU; Figure 2), which proved to be a highly specific inhibitor of HSV-1 as well as varicella-zoster virus (VZV) [6,7]. The arabinosyl counterpart of BVDU (BVaraU; Figure 2) was subsequently found to be equally potent as, if not more so than, BVDU against VZV [8].
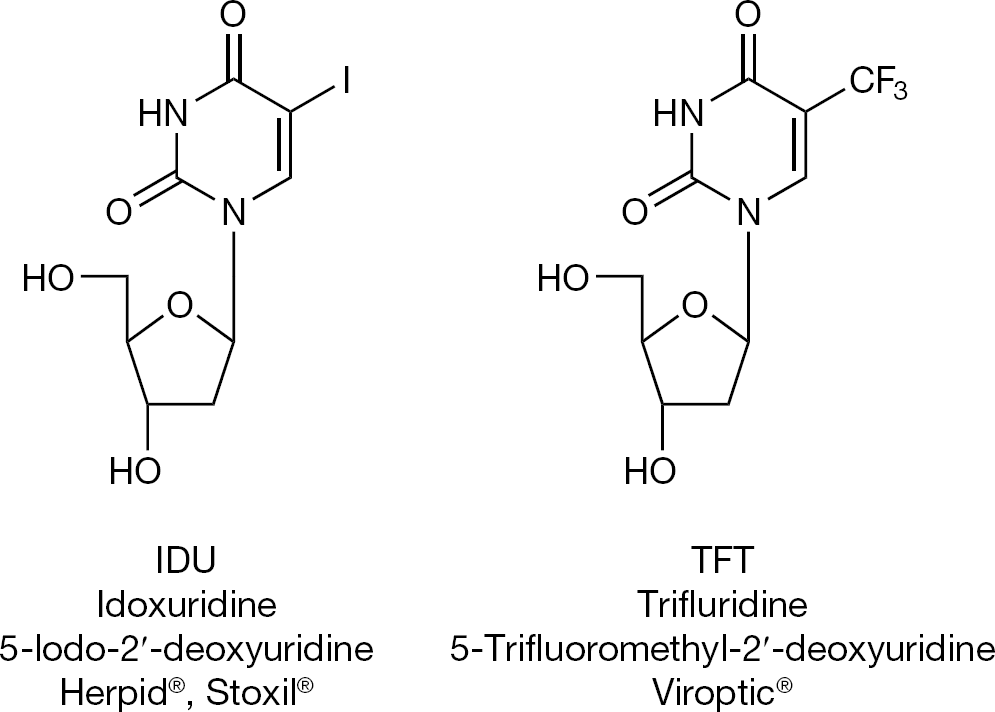
The first antivirals: 5-substituted 2′-deoxyuridines
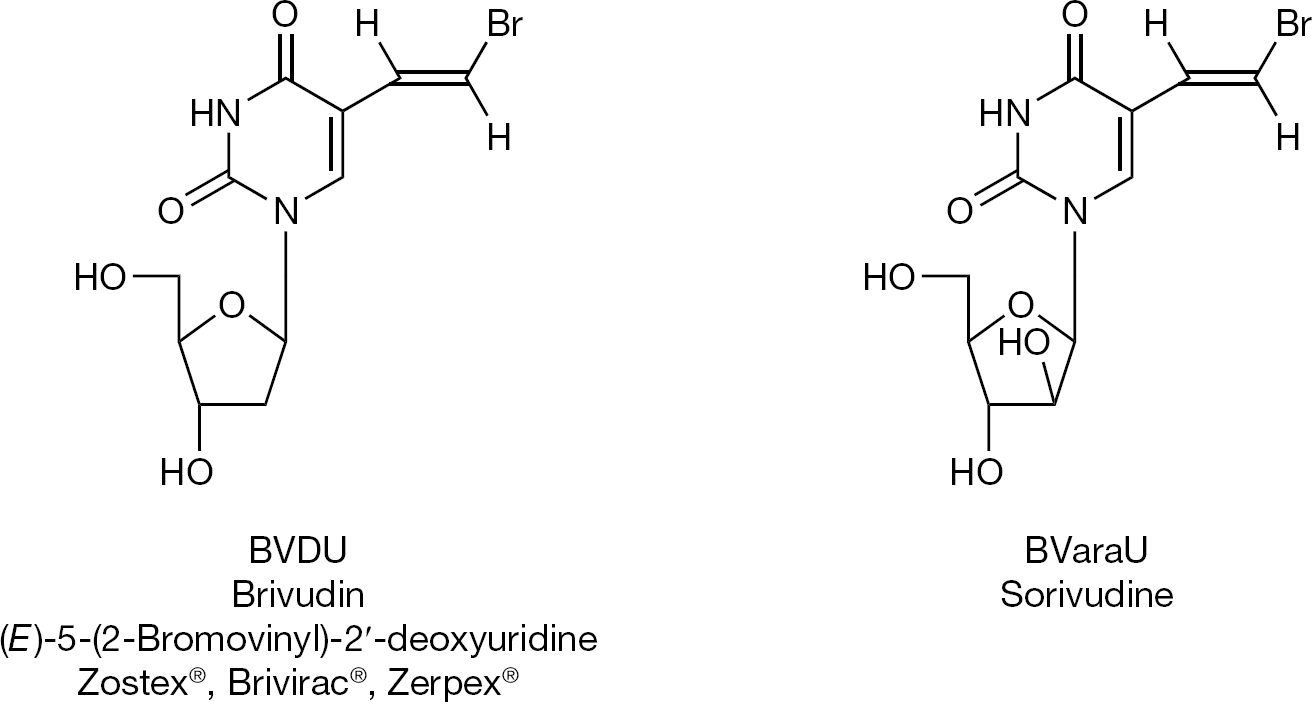
The (E)-5-(2-bromovinyl) component marking highly potent activity against varicella-zoster virus
However, BVDU and BVaraU can be cleaved by hydrolases to release the free aglycon, 5-bromovinyluracil (BVU), which acts as a powerful inhibitor of dihydrothymine dehydrogenase, the enzyme that catalyzes the hydrogenation of uracil and its 5-substituted derivatives (such as 5-fluorouracil), the first step in the catabolism of the uracil derivatives (Figure 3). The conversion of BVDU to BVU primarily occurs in the liver (and is reversible); that of BVaraU to BVU takes place in the gastrointestinal lumen (by the gastrointestinal bacteria) [9], is essentially irreversible, and has been shown to lead to some casualties, due to the enhanced toxicity of 5-fluorouracil [10,11]. Consequently, BVaraU was not further developed for the treatment of VZV infections, whereas BVDU was marketed for this indication in several European countries (including Germany, Italy and Belgium), with the warning, however, that, as a precaution, it should not be combined with 5-fluorouracil (or any of its prodrugs).
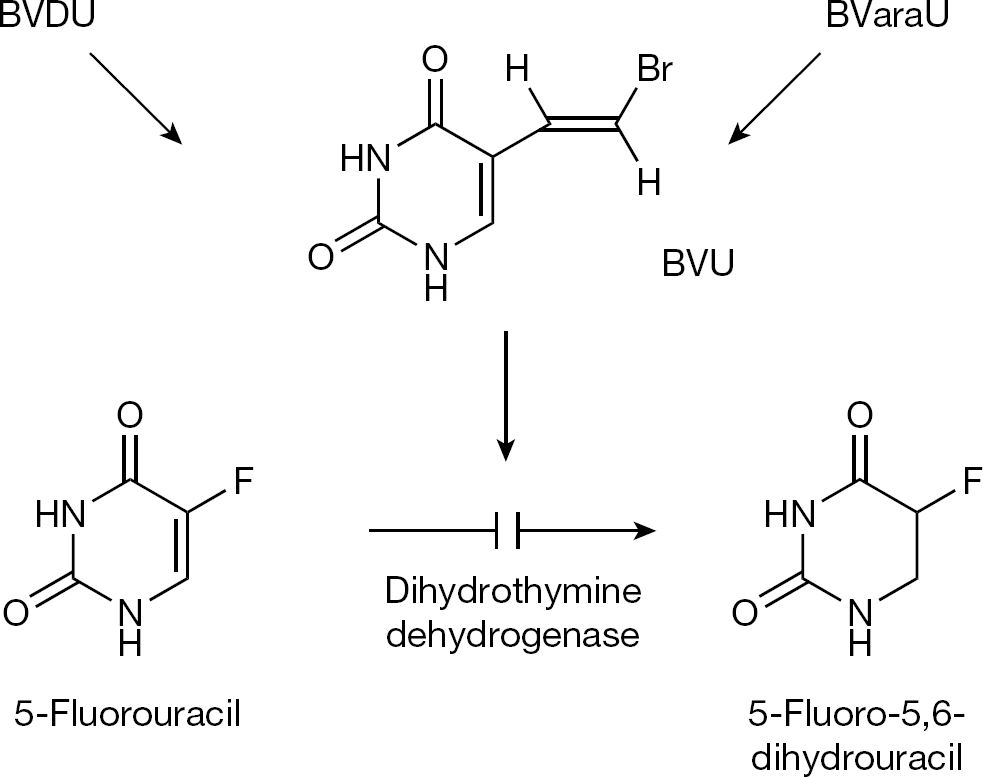
(E)-5-(2-bromovinyl)uracil inhibits degradation of 5-fluorouracil
Vidarabine
The first antiviral drug used for the systemic treatment of herpesvirus (HSV and VZV) infections was vidarabine (Figure 4). Its clinical use was prompted thanks to the pioneering efforts of Whitley et al. [12–14]. Vidarabine was later abandoned for a number of reasons, including relative insolubility in aqueous medium, and rapid deamination, by the ubiquitous adenosine deaminase, to its inactive inosine counterpart, the principal reason being that it was surpassed in potency by acyclovir, which, incidentally, was discovered in the original attempts to inhibit the rapid deamination of vidarabine.
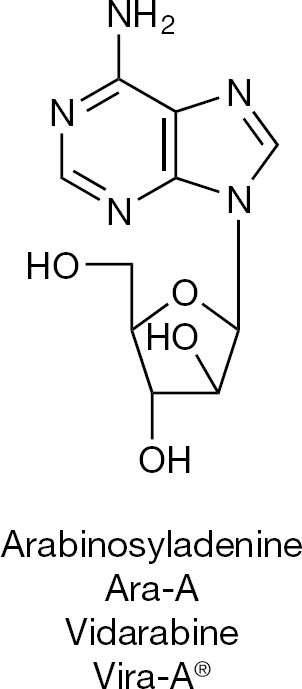
Vidarabine, the first antiviral drug for systemic use
Acyclovir
The antiviral drug era was greatly boosted by the discovery of acyclovir (Figure 5) as a selective antiherpetic agent [15]. In two papers that appeared in December 1977 [16] and April 1978 [17], acyclovir was described as a selective antiherpetic agent, whose antiviral activity depended on a specific phosphorylation by the virus-induced thymidine kinase (apparently, the guanosine analogue acyclovir shared sufficient structural requirements with thymidine to be recognized as a substrate by the HSV-induced thymidine kinase, which also acts as a deoxycytidine kinase) [18,19]. Both BVDU and acyclovir were then shown to prevent the establishment of latent HSV infection in mice [20], and the molecular basis for the resistance of HSV against acyclovir was further characterized by Field and Darby et al. [21–23].
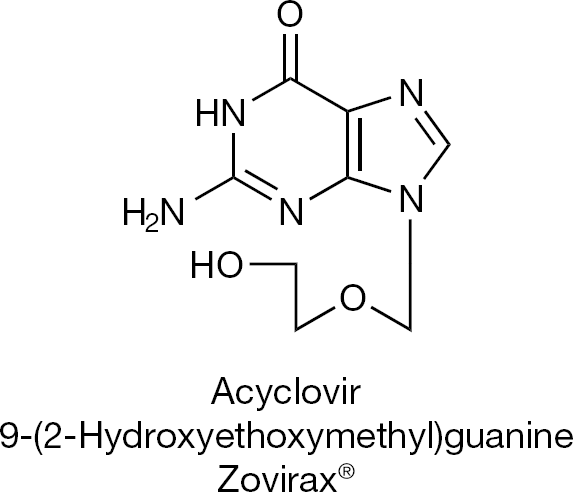
Acyclovir, the ‘gold’-standard for treatment of herpes simplex virus infections
Valaciclovir
In attempts to increase the aqueous solubility of acyclovir (so as to make it applicable as eye drops [instead of ointment], or amenable to intramuscular [instead of intravenous] injection), amino acid esters of acyclovir were synthesized (that is, glycyl- and alanyl-acyclovir; Figure 6) [24]. As an additional bonus, the valine ester (valaciclovir) also showed increased oral bioavailability and this finding [25] prompted the design of prodrugs of other antivirals as well.
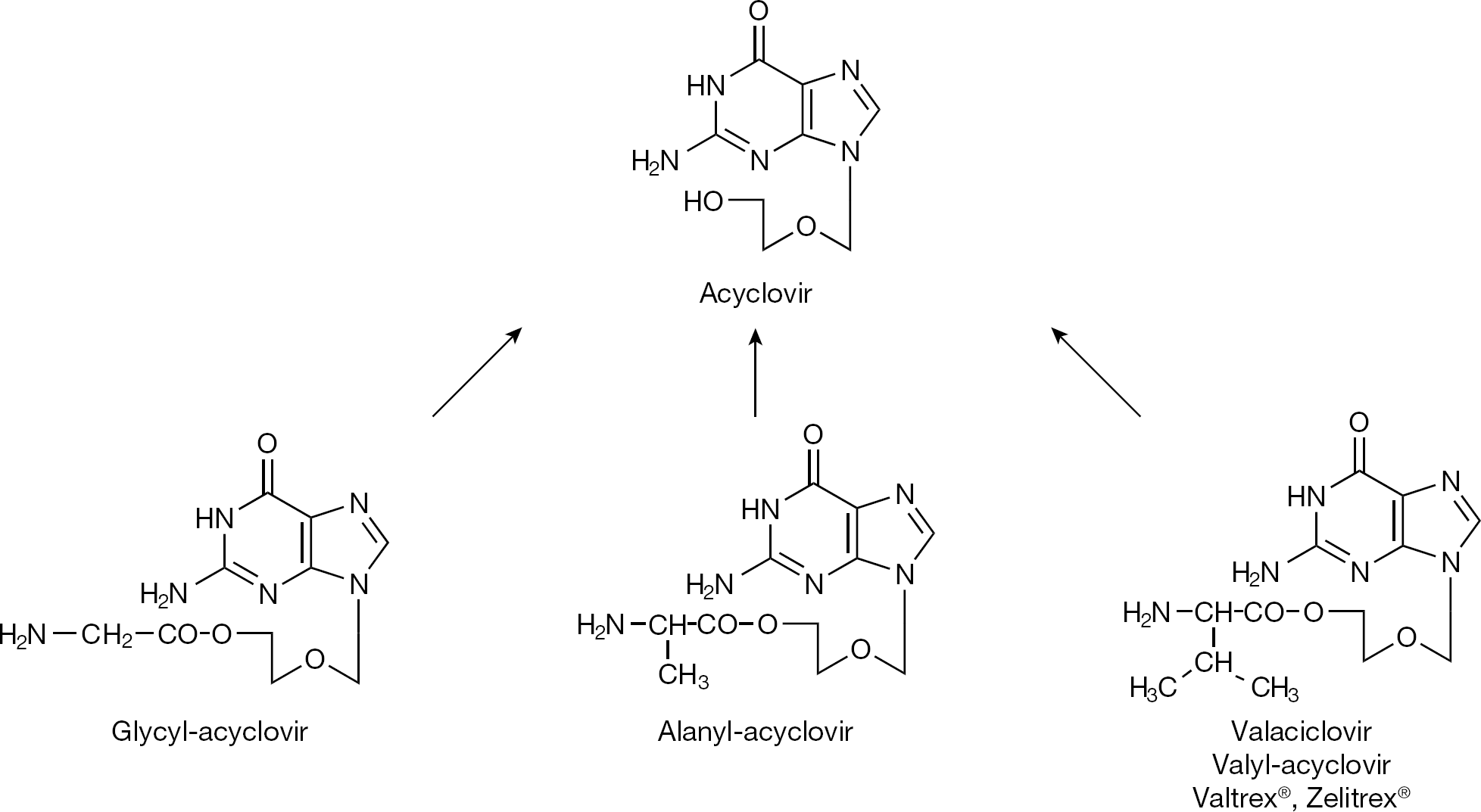
Amino acyl prodrugs of acyclovir
Famciclovir, valganciclovir, CMX001 and MIV-606
Following the finding of increased oral bioavailability of valaciclovir, synthesized prodrugs included: famciclovir, the double diacetyl precursor of 6-deoxy penciclovir (Figure 7); valganciclovir, the valine ester prodrug of ganciclovir (Figure 8); CMX001, the hexadecyloxypropyl prodrug of cidofovir (Figure 9), and MIV-606, the valomaciclovir stearate prodrug of H2G (Figure 10). All these antiviral prodrugs fulfil an essential requirement for antiviral efficacy, that is they possess an increased oral bioavailability [26]. Single-day famciclovir (1,000 mg twice daily) has been recommended for recurrent genital prophylaxis [27], and valganciclovir and valaciclovir have been recommended for preemptive prophylaxis of cytomegalovirus (CMV) disease (after renal transplantation) [28].
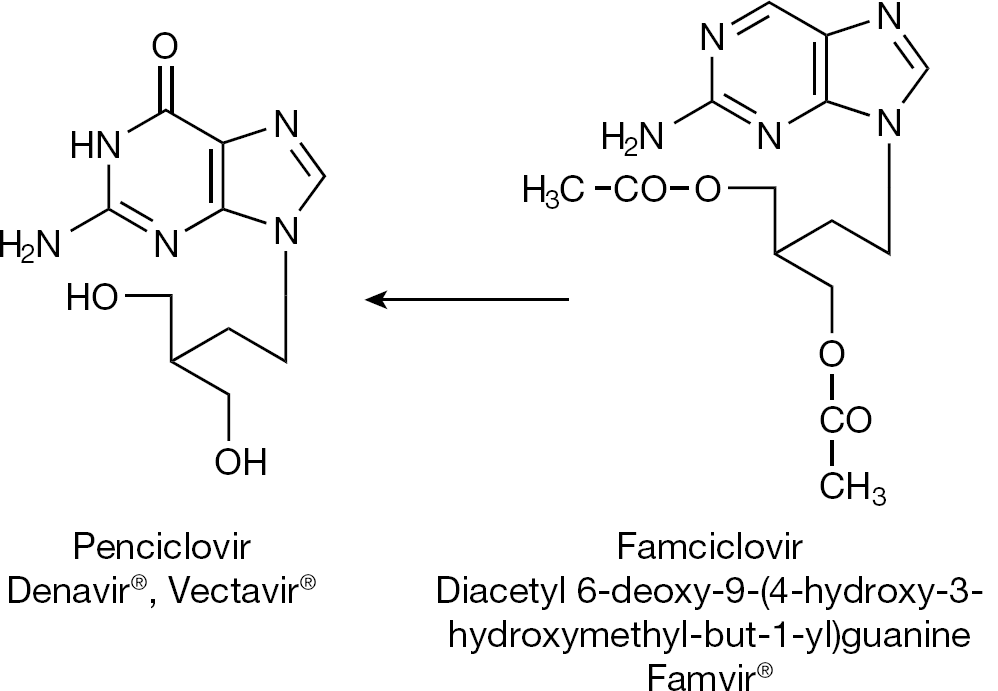
Famciclovir, the orally bioavailable prodrug of penciclovir
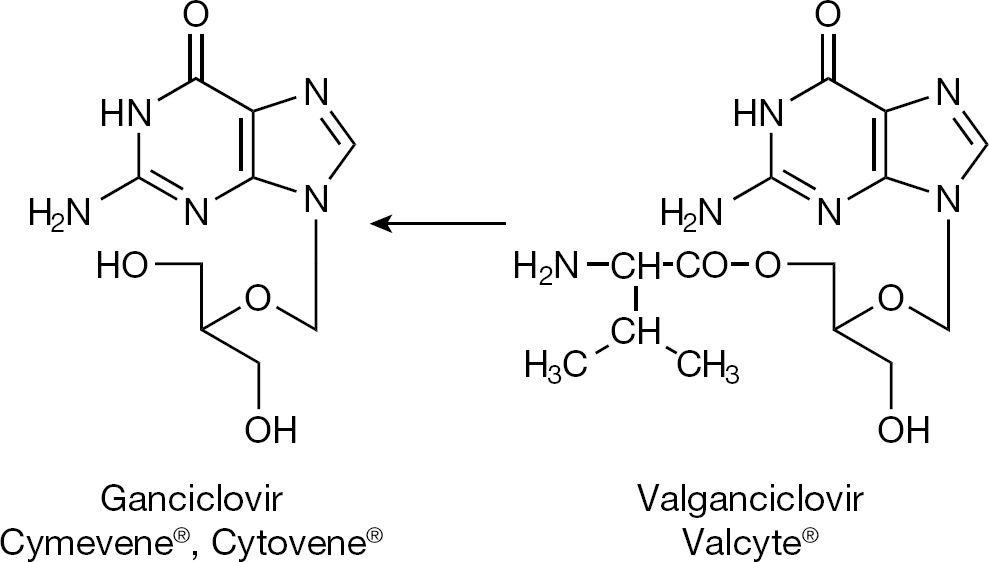
Valganciclovir, the orally bioavailable prodrug of ganciclovir
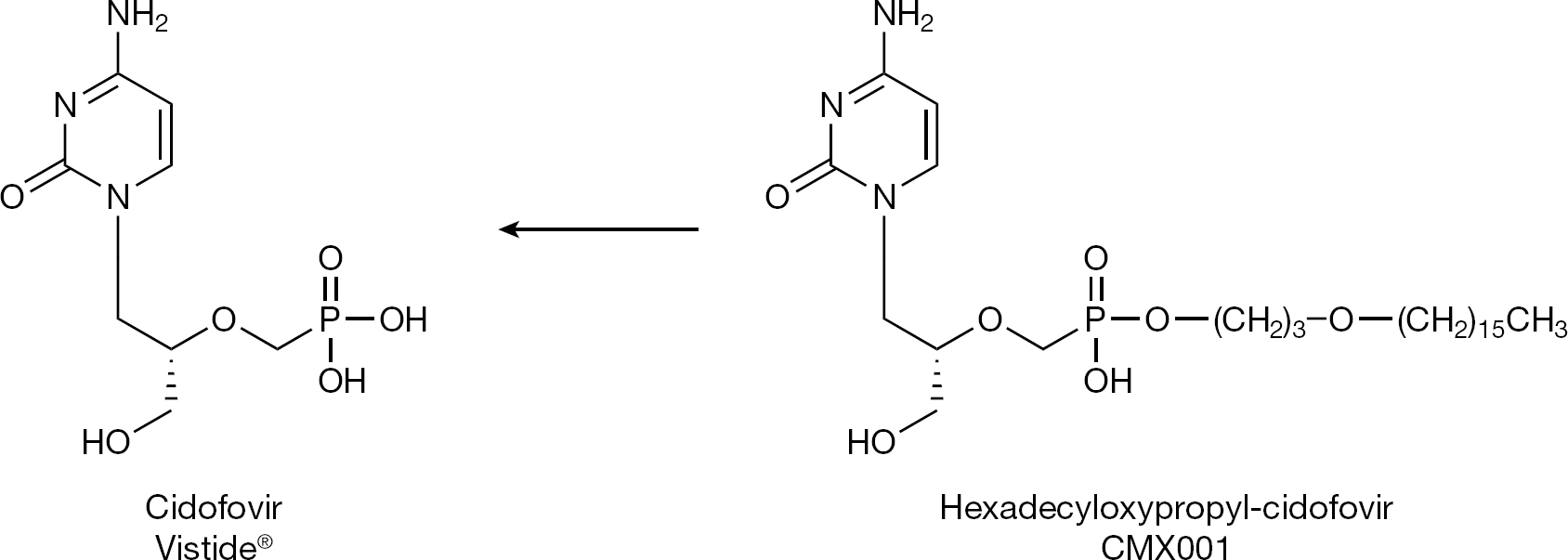
CMX001, the orally bioavailable prodrug of cidofovir
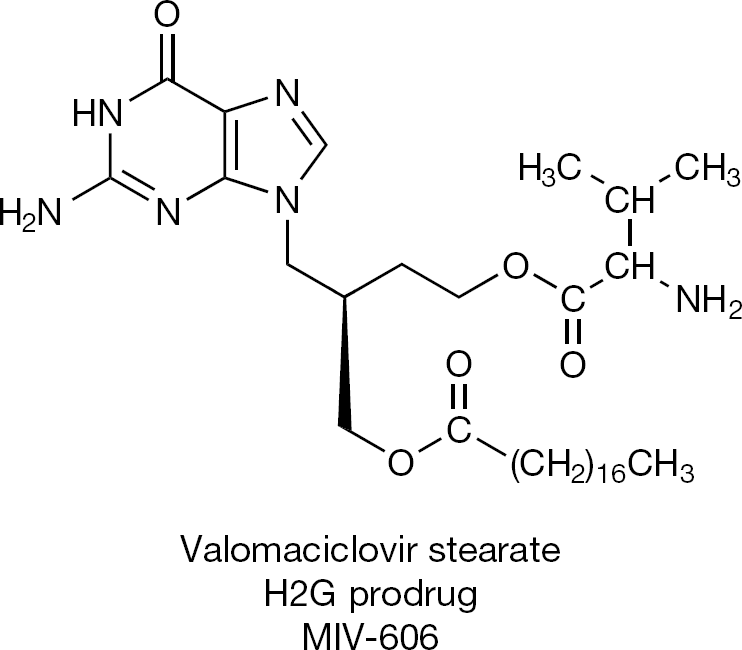
The orally bioavailable H2G prodrug
Foscarnet
The smallest of all antivirals, which, for parenteral use, must be injected intravenously, is foscarnet (phosphonoformic acid [PFA]). PFA has been approved for the treatment of CMV infections, but can also be used, off label, for the treatment of HSV and VZV infections that are resistant to acyclovir. Characteristic for PFA is the presence of a phosphonate (P–C) bridge (Figure 11), a hallmark that guarantees stability against (spontaneous and enzymatic) degradation and which is also found in the acyclic nucleoside phosphonates (that is, cidofovir, adefovir and tenofovir).
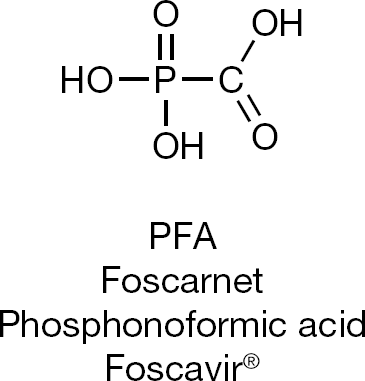
Foscarnet, the smallest of all antiviral drugs
Current state of the art: Approved antiherpesvirus drugs
The antiviral agents have been the subject of a fact file first published by De Clercq et al. [29] in 2006, followed by a second edition in 2008 [30–32]. For the treatment of herpesvirus infections (HSV, VZV, CMV and Epstein-Barr virus), the systemic antiviral agents, as currently approved by the US Food and Drug Administration, are listed in Table 1. This list includes acyclovir, valaciclovir and famciclovir for the treatment of HSV and VZV infections, and ganciclovir, valganciclovir, cidofovir and foscarnet for the treatment of CMV infections (the only antisense oligonucleotide that ever received marketing approval as an antiviral drug was fomivirsen [30]: it was approved for the topical treatment, upon intravitreal inoculation, of CMV retinitis). Off label, cidofovir and foscarnet are occasionally used for the treatment of severe HSV or VZV infections that have become resistant to acyclovir, valaciclovir or famciclovir (Table 1).
State of the art: currently approved systemic antiviral agents against herpesvirus infections

CMV, cytomegalovirus; EBV, Epstein–Barr virus; HSV, herpes simplex virus; OL, off label; VZV, varicella-zoster virus; +, approved; -, unapproved.
New anti-varicella-zoster virus drugs on the horizon: FV-100
In 2000, McGuigan et al. [33] described an exquisitely potent anti-VZV agent (Cf 1743), which was exclusively active against VZV without any antiviral activity against any other virus including HSV; the prototype of this class of bicyclic furano pyrimidine nucleosides proved active against a large array of VZV strains at picomolar concentrations [34]. As previously carried out for acyclovir and ganciclovir, Cf 1743 (Figure 12) was converted to its valine ester, and this prodrug, dubbed FV-100, can be considered as the most potent and selective anti-VZV agent reported to date [35]. Phase II clinical trials with FV-100 are in progress. It should be administered once-daily (for 7 days) at a dose of 100, 200, 400 or 800 mg (exact dosage still to be determined) [36], which compares favourably with valaciclovir that has to be given orally 3x daily at 1,000 mg.
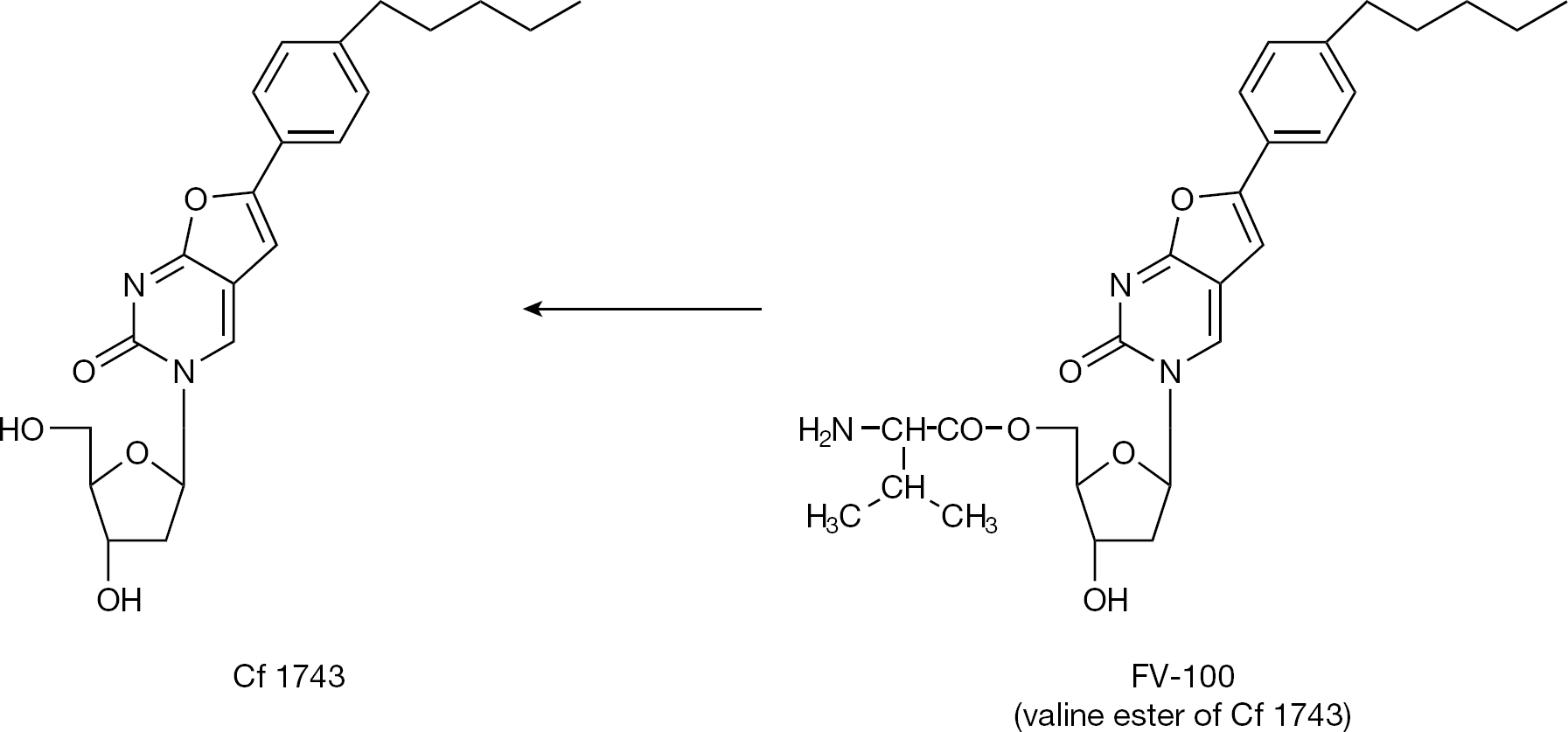
The bicyclic furanopyrimidine analogue Cf 1743 and its valine ester: exquisitely specific against varicella-zoster virus
The helicase-primase inhibitor ASP2151 (Figure 13), assumed to be effective against VZV as well as HSV has been abandoned for further development [37].
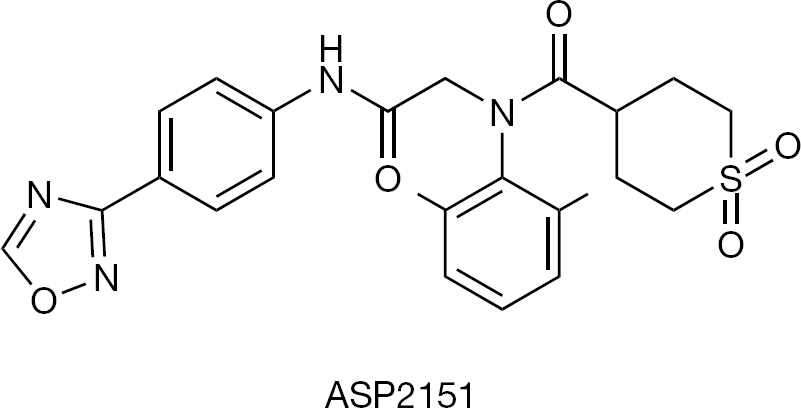
Helicase-primase inhibitor, effective against both herpes simplex virus and varicella-zoster virus Development discontinued.
New anti-herpes-simplex virus drugs on the horizon: BAY 57–1293, AIC316, pritelivir
The helicase-primase inhibitor BAY 57–1293 (Figure 14) was first described by Kleymann et al. [38,39] in 2002. This compound, together with BILS 179 BS [40], focused on a new target for new antiviral agents active against HSV. BAY 57–1293 was transferred from Bayer to AiCuris for further development. It has been extensively studied for its potential to generate drug-resistant mutants by Biswas and Field et al. [41–52]. The clinical potential of BAY 57–1293 is restricted to HSV. It remains to be determined to what extent BAY 57–1293 (AIC316) compares to the utility of acyclovir, still the ‘gold’-standard for the treatment of HSV infections.
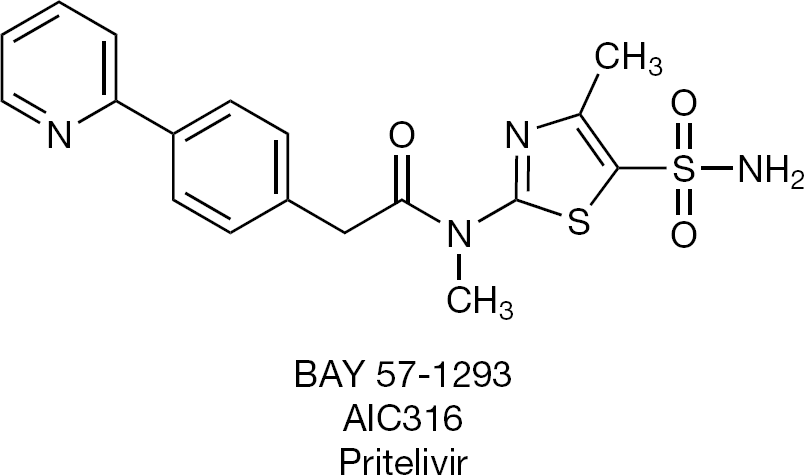
Helicase-primase inhibitor, under development for herpes simplex virus
New anti-cytomegalovirus drugs on the horizon: AIC246, letermovir
At present, there are four new candidate compounds for the treatment of HSV, VZV and CMV infections: CMX001, FV-100, BAY 57–1293 (AIC316) and AIC246 (letermovir; Table 2). For an update on new antivirals under development for the treatment of double-stranded DNA virus (including HSV) infection, see Dropulic and Cohen [53]. The development of maribavir (Figure 15) for the treatment of CMV infections has been discontinued [54].
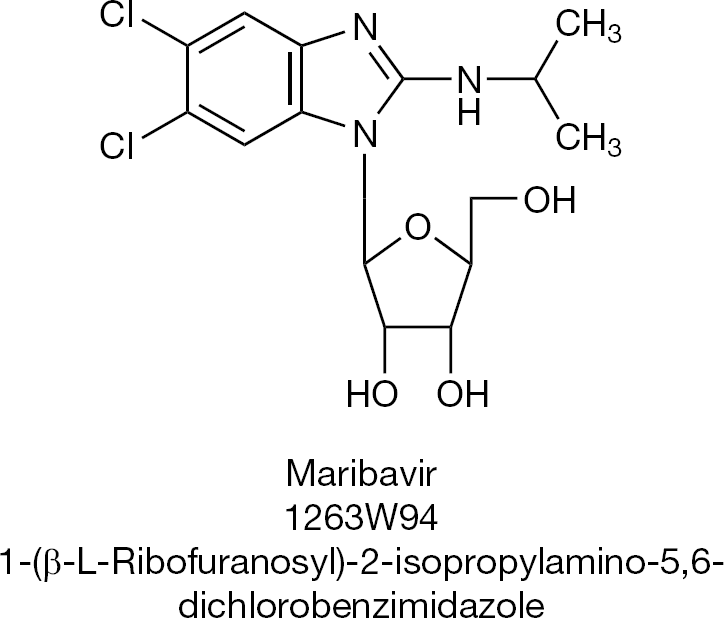
Specific inhibitor of human cytomegalovirus
New antiviral agents at the horizon: in vitro antiviral activity

Discontinued. +, antiviral activity; -, antiviral inactivity.
Instead, AIC246 (letermovir; Figure 16), which inhibits HCMV replication via a mechanism that involves the viral terminase but is distinct from that of maribavir [55], is now being pursued for its clinical potential to treat cytomegalovirus infections. It exhibits excellent in vitro inhibitory activity against HCMV laboratory strains and clinical isolates [56], but no other betaherpesviruses (that is, rat cytomegalovirus, or human herpesvirus type 6 [HHV-6]) or α- or γ-herpesviruses [57]. Initial clinical results point to the successful treatment of multidrug-resistant human cytomegalovirus (HCMV) disease with AIC246 [58].
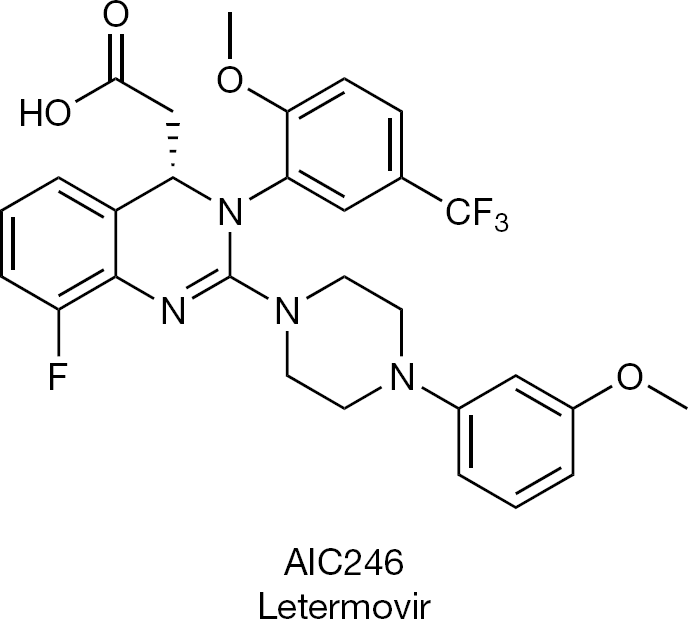
Specific inhibitor of human cytomegalovirus
Epilogue
Anti-HSV agents may indirectly affect HIV-1 virus titres: through its inhibitory effect on HSV replication, valaciclovir may help reducing plasma HIV-1 levels in HIV-1/HSV-2-coinfected individuals [59–61].
Tenofovir, a topical microbicide effective against HIV, as demonstrated by the CAPRISA study [62], was recently reported to inhibit HSV-2 replication [63]. However, tenofovir did so within the concentration range of 20–200 μg/ml, whereas adefovir inhibited HSV-2 replication within a concentration range of 0.5–50 μg/ml, and cidofovir did so within the concentration range of 0.2–50 μg/ml [63].
Footnotes
Acknowledgements
This review was originally based on the lecture presented at the Symposium on Viruses and Antiviral Therapy (in honour of Hugh J Field), Queens' College, Cambridge, UK, 23 March 2012.
The author declares no competing interests.