
Abstract
Background:
In continuation of our search for new anti-HIV and anti-HCV agents, the suggestion, synthesis and structure elucidation of a new series of 2,6-diamino-4-alkylthio- or (2-benzylhydrazinyl)-5-p-chlorophenylazopyrimidines), 2,6-diamino-4-(2-benzylhydrazinyl)-5-(aryl-[1,1′-biphenyl]-4-yl)pyrimidines, 2,6-diamino-4-(aryl)-5-(aryl[1,1′-biphenyl]-4-yl) pyrimidines), 6-(aryl)-1,3-dimethyl-5-nitro pyrimidine-2,4-dione and 6-amino-4-methoxy-N,N-dimethyl-6-arylpyrimidines were described.
Methods:
The anti-HIV-1 (strain IIIB) and HIV-2 (strain ROD) activity of the newly synthesized pyrimidine analogues was evaluated in vitro in human MT-4 cells using the MT-4/MTT assay. Similarly, the same compounds were evaluated in vitro for their selective antiviral activity against HCV in the Huh 5–2 replicon system (type 1b, Con1 strain).
Results:
None of the tested compounds exhibited inhibition of HIV-1 and HIV-2 replication in cell culture. Even though many compounds yielded a 50% effective concentration in the HCV replicon system with selectivity indexes up to 6.9, none of the compounds matched the selection criteria of a selective inhibitor of virus replication in this assay (that is, >70% inhibition at concentrations that do not elicit an anti-metabolic effect on the host cells).
Conclusions:
Structural modification of these compounds might optimize their anti-HCV activity by introducing diverse and potent functional groups at the pyrimidine backbone, like nitrile residue. Because of the nature of the molecules, these new derivatives will also be evaluated for their potential anti-HIV activity.
Introduction
Pyrimidines are compounds that in vitro possess biological activity against a wide spectrum of unrelated viruses, such as poliovirus [1], herpes virus [2] and HIV [3–5]. For the latter, two diarylpyrimidines, rilpivirine [6] (
Compounds 1, 2 and 3
Hydrazine-pyrimidine-5-carbonitrile derivatives inhibit the growth of a wide range of cancer cell lines at concentrations as low as 10−7 M [22], whereas, 2,4-diamino-N4-6-diarylpyrimidines were identified to block the proliferation of tumour cell lines in vivo, especially duodenal cancer (DU145; 50% inhibitory concentration =0.23 μM) [15]. The dynamic behaviour of this compound scaffold in multiple biological systems encouraged us to further investigate their properties within the anti-infectives field [23]. In particular, the synthesis of 4-aryl-2,6-diaminopyrimidines is still largely unexplored, despite their potentially interesting agrochemical and medicinal properties [24].
Recently, we have synthesized a series of 5-nitro-and 5-amino-6-arylsulfanyl-1-propyl-pyrimidine-2-4-dione, their 6-arylsulfanyl-1,3-dimethyl and the 2-amino analogues [25], as well as a range of substituted 1,2,4-triazolo thymidines [26], with evaluation of their anti-HIV activity.
We next aimed to explore the anti-HCV activity of the compounds. HCV belongs to the family of Flaviviridae and is an important aetiological agent causing chronic hepatitis that can progress further to hepatocellular carcinoma [27]. The current therapeutic protocol for HCV infection used to consist of interferon in combination with ribavirin that is usually accompanied by strong side effects and a moderately successful rate [28,29].
Recently, two protease inhibitors have been approved for the treatment of HCV infection and several more selective antivirals are in advanced clinical development. However, there is still a need for compounds with an alternative mechanism of action and compounds are an important tool to further unravel the exact mechanism by which the virus replicates.
In the present work, we report the synthesis of new mono- and diaryl pyrimidines via the Suzuki cross-coupling procedure [30–32] and their in vitro activity on the replication of HIV-1, HIV-2 and HVC-1b.
Materials and methods
Chemistry
Melting points are uncorrected and were measured on a Büchi melting point apparatus B-545 (Büchi Labortechnik AG, Flawil, Switzerland). NMR data were obtained on 400 and 600 MHz (1H) and 150.91 MHz (13C) spectrometers (Avance III; Bruker, Reinstetten, Germany) with TMS as internal standard and on the δ scale in ppm. Heteronuclear assignments were verified by heteronuclear single quantum coherence spectroscopy experiments. Microanalytical data were obtained with a Vario Elemental analyzer (Shimadzu; Kyoto, Japan). Analytical silica gel TLC plates 60 F254 were purchased from Merck (Darmstadt, Germany). All reagents were obtained from commercial suppliers and were used without further purification.
2,6-Diamino-4-phenylthio-5-p-chlorophenylazopyrimidine
A solution of 2,6-diamino-4-chloro-5-p-chlorophenylazopyrimidine (
2,6-Diamino-4-ethylthio-5-p-chlorophenylazopyrimidine
The method was analogous to the previous procedure, using
2,6-Diamino-4-(2-benzylhydrazinyl)-5-p-chlorophenylazopyrimidine
To a solution of
General procedure of Suzuki reaction for preparation of 9–14
A mixture of halopyrimidine and arylboronic acid in n-propanol (15 ml) was stirred for 15 min. To this mixture was added Pd(OAc)4 (650 mg, 0.19 mmol), triphenylphosphine (498 mg, 0.19 mmol) and 2M aqueous solution of Na2CO3 (3.5 ml). The reaction mixture was refluxed under nitrogen for 4–6 h and the completion of the reaction was monitored by TLC. After cooling, water was added (7 ml), followed by stirring for 5 min. The mixture was partitioned with ethyl acetate (3×10 ml) and the combined organic layers were washed subsequently with 5% Na2CO3 solution (2×10 ml), brine solution (2×10 ml) and finally with water (10 ml). The organic phase was decolourized with charcoal, filtered and the filtrate was dried (Na2SO4), filtered through Celite and evaporated to dryness to give the desired product after purification.
2,6-Diamino-4-(2-benzylhydrazinyl)-5-(4′-fluoro-[1,1′-biphenyl]-4-yl)pyrimidine
From
4,6-Diamino-4-(2-benzylhdrazinyl)-5-(3′,4′-dimethoxy[1,1′-biphenyl]-4-yl)pyrimidine
From
2,6-Diamino-4-(3,4-dimethoxyphenyl)-5-(3′,4′-dimethoxy[1,1′-biphenyl]-4-yl)pyrimidine
From
2,6-Diamino-4-(4-fluorophenyl)-5-(4′-fluoro[1,1′-biphenyl]-4-yl)pyrimidine
From
2,6-Diamino-4-(2-fluorophenyl)-5-(2′-fluoro[1,1′-biphenyl]-4-yl)pyrimidine
From
2,6-Diamino-4-(4-nitrophenyl)-5-(4′-nitro[1,1′-biphenyl]-4-yl)pyrimidine
From
2,6-Diamino-5-(4-chlorophenylazo)-3H-pyrimidin-4-one
A solution of
6-(2-Fluorophenyl)-1,3-dimethyl-5-nitropyrimidine-2,4-dione
From 6-chloro-1,3-dimethyl-5-nitropyrimidine-2,4-dione (
1,3-Dimethyl-5-nitro-6-(4-nitrophenyl)pyrimidine-2,4-dione
From
1,3-Dimethyl-6-(2-methylthiophenyl)-5-nitropyrimidine-2,4-dione
From
Virology
In vitro anti-HIV assay
Evaluation of the antiviral activity of the compounds
The MTT assay is based on the reduction of yellow coloured 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Acros Organics, Geel, Belgium) by mitochondrial dehydrogenase of metabolically active cells to a blue-purple formazan that can be measured spectrophotometrically. The absorbances were read in an eight-channel computer-controlled photometer (Safire; Tecan, Männedorf, Switzerland), at two wavelengths (540 and 690 nm). All data were calculated using the median optical density (OD) value of tree wells. The 50% cytotoxic concentration was defined as the concentration of the test compound that reduced the absorbance (OD540) of the mock-infected control sample by 50%. The concentration achieving 50% protection from the cytopathic effect of the virus in infected cells was defined as the 50% effective concentration (EC50).
The results are summarized in Table 1. Nevirapine [38] and azidothymidine [39] were included as reference compounds. All newly synthesized compounds had a selectivity index <1.
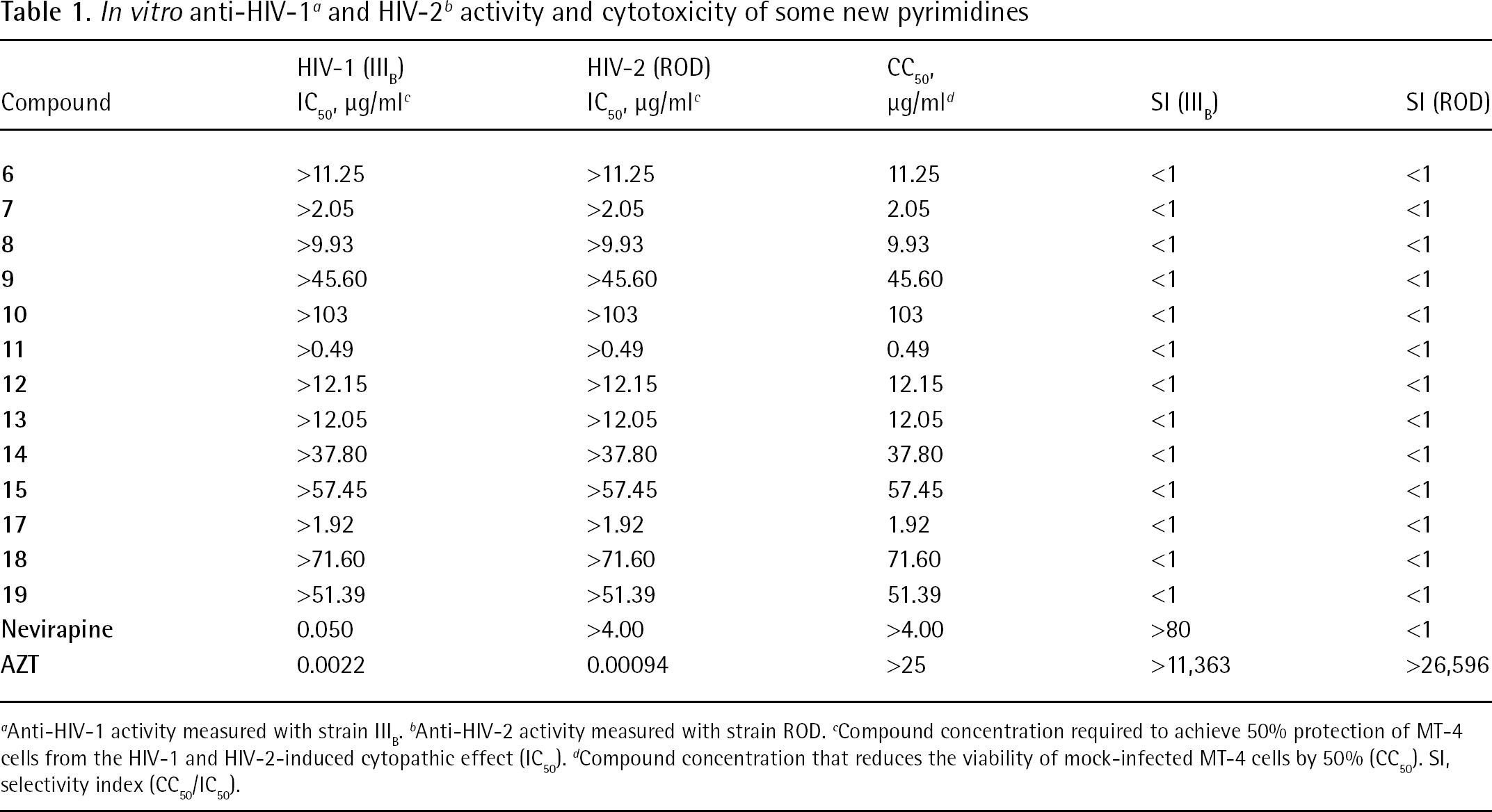
Anti-HIV-1 activity measured with strain IIIB.
Anti-HIV-2 activity measured with strain ROD.
Compound concentration required to achieve 50% protection of MT-4 cells from the HIV-1 and HIV-2-induced cytopathic effect (IC50).
Compound concentration that reduces the viability of mock-infected MT-4 cells by 50% (CC50). SI, selectivity index (CC50/IC50).
In vitro anti-HCV assay
Compounds
In vitro anti-HCV-1b activity, cytotoxicity and inhibition percentage of some pyrimidines
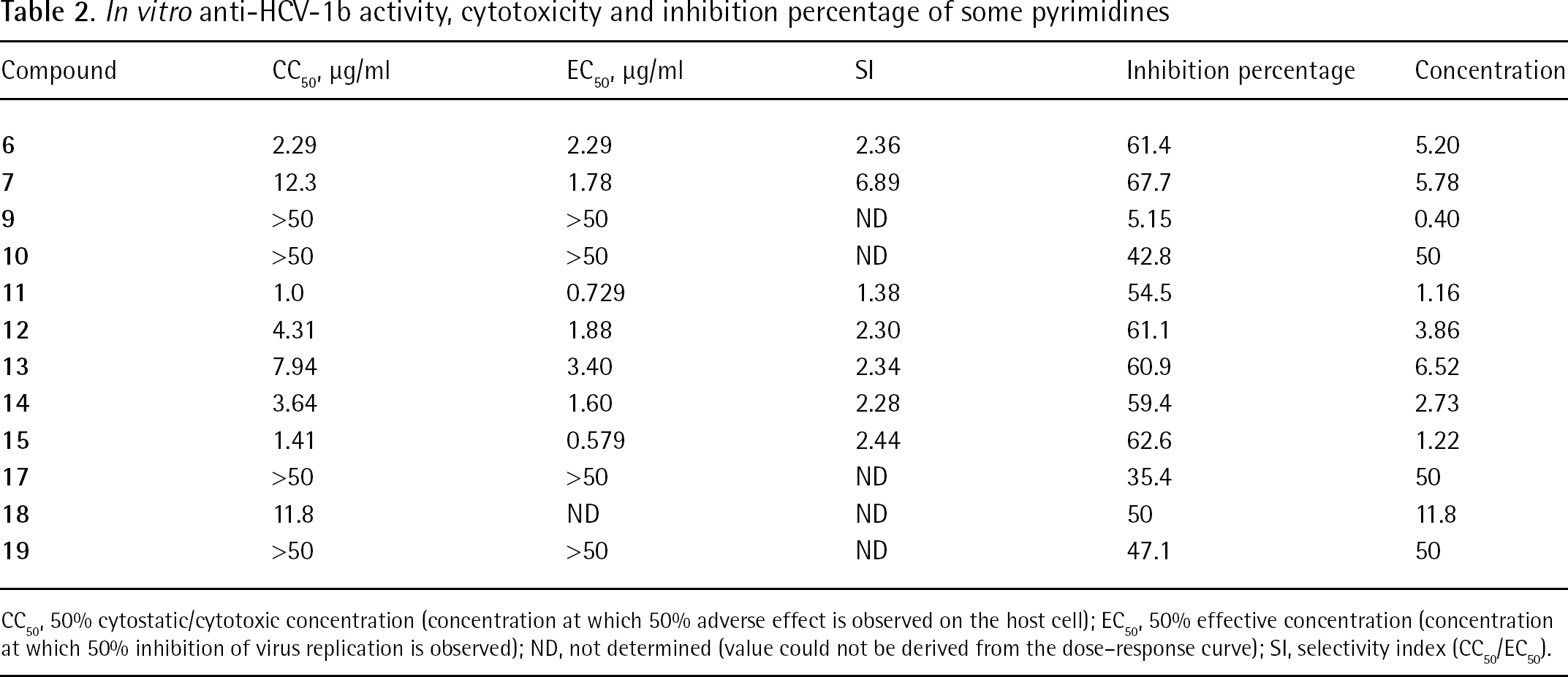
CC50′ 50% cytostatic/cytotoxic concentration (concentration at which 50% adverse effect is observed on the host cell); EC50′ 50% effective concentration (concentration at which 50% inhibition of virus replication is observed); ND, not determined (value could not be derived from the dose–response curve); SI, selectivity index (CC50/EC50).
Cells and HCV
The Huh-5-2 and Huh 9–13 HCV subgenomic replicon-containing cells were provided by R Bartenschlager (University of Heidelberg, Heidelberg, Germany).
Antiviral assays
Huh 5.2 cells, containing the HCV genotype 1b I389lucubi-neo/NS3-3′/5.1 replicon [40] were sub-cultured in Dulbecco's modified Eagle's medium supplemented with 10% FCS, 1% non-essential amino acids, 1% penicillin/streptomycin and 2% Geneticin at a ratio of 1:3 to 1:4 and grown for 3–4 days in 75 cm2 tissue culture flasks. One day before addition of the compound, cells were harvested and seeded in assay medium (Dulbecco's modified Eagle's medium, 10% FCS, 1% non-essential amino acids, 1% penicillin/streptomycin) at a density of 6,500 cells/well (100 μl/well) in 96-well tissue culture microtitre plates for evaluation of anti-metabolic effect and CulturPlate (Perkin Elmer, Waltham, MA, USA) for evaluation of the antiviral effect. The microtitre plates were incubated overnight (37°C, 5% CO2, 95–99% relative humidity), yielding a non-confluent cell monolayer.
The evaluation of the anti-metabolic as well as the antiviral effect of each compound was performed in parallel. Four-step, 1-to-5 compound dilution series were prepared for the first screen, to collect data for a more detailed dose-response curve, an eight-step, 1-to-2 dilution series was used. Following assay setup, the microtitre plates were incubated for 72 h (37°C, 5% CO2, 95–99% relative humidity). For the evaluation of anti-metabolic effects, the assay medium was aspirated, replaced with 75 μl of a 5% MTS solution in phenol red-free medium and incubated for 1.5 h (37°C, 5% CO2, 95–99% relative humidity). Absorbance was measured at a wavelength of 498 nm (Safire2; Tecan), and optical densities (OD values) were converted to percentage of untreated controls. For the evaluation of antiviral effects, assay medium was aspirated and the cell monolayers were washed with PBS. The wash buffer was aspirated and 25 μl of Glo Lysis Buffer (Promega) was added allowing for cell lysis to proceed for 5 min at room temperature. Subsequently, 50 μl of Luciferase Assay System (Promega, Madison, WI, USA) was added, and the luciferase luminescence signal was quantified immediately (1,000 ms integration time/well; Safire2; Tecan). Relative luminescence units were converted into percentage of untreated controls.
The EC50 and EC90 (values calculated from the dose–response curve) represent the concentrations at which 50% and 90% inhibition, respectively, of viral replication, is achieved. The 50% cytotoxic concentration (value calculated from the dose–response curve) represents the concentration at which the metabolic activity of the cells is reduced by 50% as compared to untreated cells.
A concentration of compound is considered to elicit a genuine antiviral effect in the HCV replicon system when the anti-replicon effect is well above the 70% threshold at concentrations where no significant anti-metabolic activity is observed [40].
Results
The azo-pyrimidine derivative
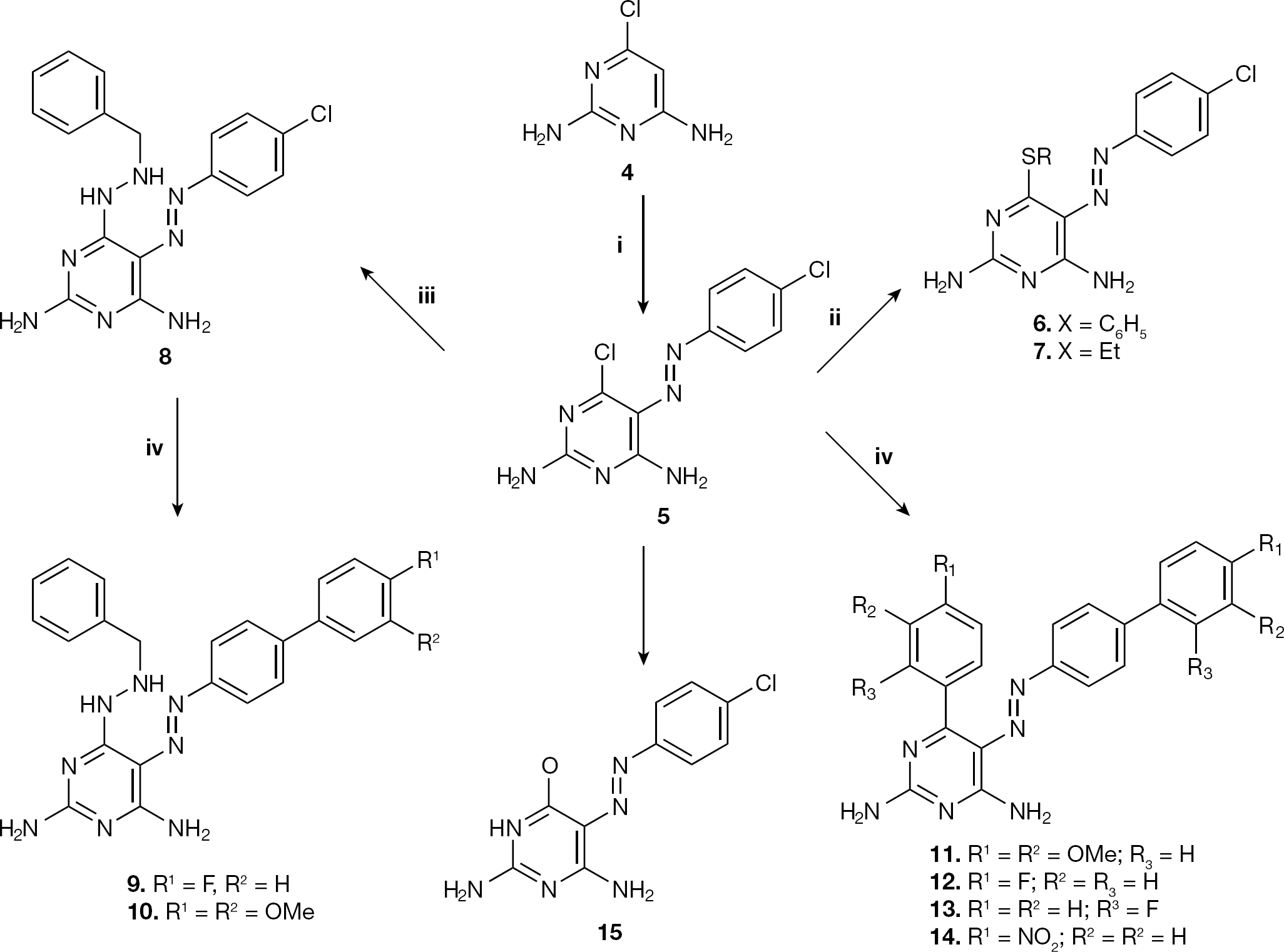
Synthesis of mono- and diarylpyrimidines and analogues
The structures of the newly synthesized compounds
Next, our efforts focused on the synthesis of new 6-aryl-pyrimidine-2,4-dione derivatives to study their effect on HIV and HCV replication in cell culture. Thus, treatment of 16 with aryl boronic acids using Suzuki methodology [32] afforded 17–19 in 68, 63 and 64% yield, respectively (Figure 3).
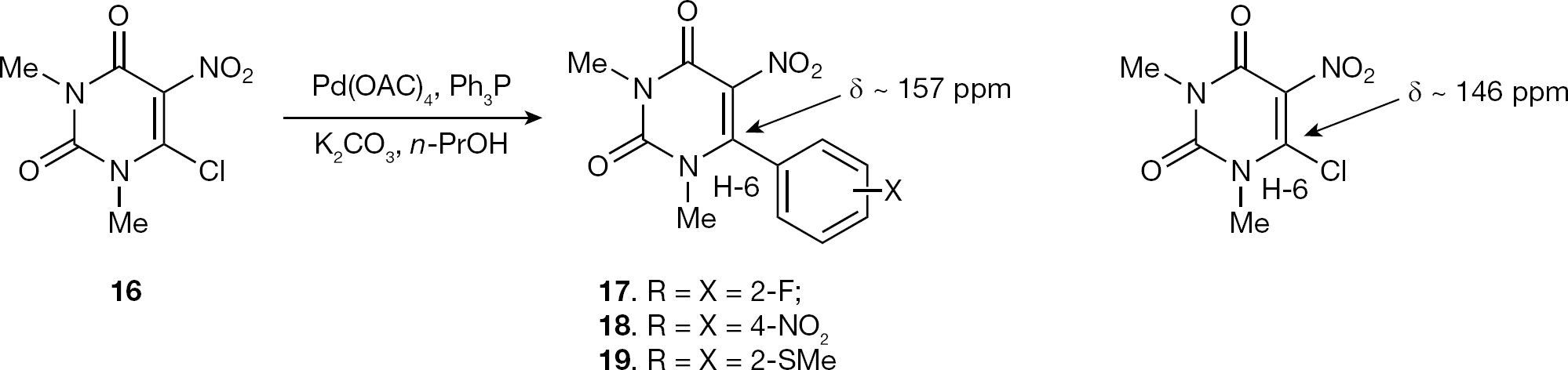
Synthesis of 6-aryl-1,3-dimethyl-5-nitropyrimidine 2,4-diones
The assignment of protons and carbons of the pyrimidine and aromatic rings were deduced in comparison to our previously reported data on 6-arylsulfanyl pyrimidines [25]. In the 1H NMR spectra of 17–19, the N-1 and N-3 methyl groups were resonated at the region (δ = 3.62–3.32 ppm) and (δ = 3.18–2.50 ppm), respectively. The 13C NMR spectra of 17–19 showed signals at higher fields (δ = 168.8–162.8 ppm), attributed to the carbonyl group whereas the resonances at (δ = 157.2–154.9 ppm) and (δ = 151.4–150.9 ppm) were assigned to C-6 and C-2, respectively. Resonances at δ = 108.6, 108.3 and 90.3 ppm were assigned to C-5 pyrimidine carbons. From the 13C NMR spectra, it is clearly shown a shift in ppm values (approximately 10 ppm) between C6-Carom and C6-Cl, indicative for substitution of a chloride group by aryl moiety.
Compounds 6–15 and 17–19 were evaluated in vitro for their selective antiviral activity against anti-HIV-1 (strain IIIB) and anti-HIV-2 (strain ROD) activity in human T-lymphocyte (MT-4) cells using the MT-4/MTT assay [33,34]. None of the compounds inhibit HIV-1 and HIV-2 replication in cell culture.
Discussion
Non-nucleoside reverse transcriptase inhibitors (NNRTIs) are notorious for rapidly leading to virus drug resistance development, primarily based on the emergence of the K103N and Y181C mutations in the HIV-1 reverse transcriptase. Newer NNRTIs, such as capravirine, dapivirine (TMC 125) and DPC 083, are resilient to these ‘NNRTI’ mutations, and, therefore, offer considerable promise as future anti-HIV-1 drugs. NNRTIs are targeted at a specific ‘pocket’ binding site within the HIV-1 RT that is distinct from, but both spatially and functionally related to, the catalytic site where the nucleoside reverse transcriptase inhibitors and nucleotide reverse transcriptase inhibitors interact [42]. However, capravirine, an imidazole derivative, is active against HIV variants with the single mutations K103N, V106A or L100I, which confer resistance to NNRTIs and form an extensive hydrogen-bond network with the RT main chain, a network that is unlikely to be disrupted by simple side-chain mutations [43]. Unfortunately, none of our pyrimidine derivatives produced any protective effect against the HIV-induced cytopathogenic effect in MT-4 cells.
The establishment of stable HCV subgenomic replicon systems [44,45] in the human hepatoblastoma cell line Huh7 has provided a useful system for the development of new antiviral approaches against HCV [46–49]. The overriding aims of the new therapeutic strategies are higher efficacy associated with shortened duration of treatment, favourable mode of administration, and thus improved tolerability and adherence.
However, we have evaluated in vitro our new pyrimidine derivatives 6, 7, 9–15 and 17–19 for selective antiviral activity against HCV in the Huh 5–2 replicon system (type 1b, Con1 strain). Even though for many compounds an EC50 was obtained with a selectivity index of up to 6.89 (for example, compound 7; Table 2), none of the compounds matched the selection criteria of a selective inhibitor of virus replication in this assay (that is, >70% inhibition at concentrations that do not elicit an anti-metabolic effect on the host cells).
Therefore, structural modification of these compounds might optimize their anti-HCV activity by introducing diverse and potent functional groups at the pyrimidine backbone, like nitrile residue. In conclusion, we explored the anti-HIV and anti-HCV activity of a novel series of pyrimidine derivatives in an attempt to identify selective inhibitors of viral replication.
Footnotes
Acknowledgements
We thank U Haunz and A Friemel of the Chemistry Department, University of Konstanz, Germany, for the NMR experiments. We also would like to acknowledge Stijn Delmotte, Tom Bellon, Annelies De Ceulaer and Caroline Collard for their excellent technical assistance in the acquisition of the anti-HCV data.
The authors declare no competing interests.