
Abstract
At the 25th International Conference on Antiviral Research, I received a special recognition for my contribution to the International Society of Antiviral Research over a period of 25 years (from 1987 until 2012). This review follows the theme of my presentation at that event, which comprised 10 reminiscences, all with a Japanese connection concerning the success, or otherwise, in the clinical development of: double- and single-stranded polynucleotides; suramin, a polysulfonate; dextran sulfate, a polysulfate; brivudin; BVaraU; 2′,3′-dideoxynucleoside analogues; HEPT; adefovir and tenofovir; CXCR4 antagonists; and elvitegravir.
Introduction
This ‘anniversary’ review lecture was written at the occasion of the 25th anniversary of the International Conference of Antiviral Research (ICAR) and International Society of Antiviral Research (ISAR) in Sapporo, Japan (16–19 April 2012). Both ISAR and ICAR were conceived in Il Ciocco, Italy, in 1987 during the NATO Advanced Study Institute/FEBS Advanced Course on Antiviral Drug Development. To commemorate this anniversary and to reflect the fact that this is the second time that ICAR has been held in Japan, I present herein a narrative account of 10 personal reminiscences with a Japanese connection. I dedicate these ‘10-in-1’ stories to the Japanese friendships I built up over a period of almost 40 years starting from the mid-1970s.
Polynucleotides
The discovery of the induction of interferon by double-stranded RNAs, such as poly(I).poly(C) (Figure 1) by Hilleman et al. [1–5] at Merck, could be considered as a major breakthrough in interferon research, as it proved that interferon, up until the mid-1960s considered as an isosteric principle, really existed and could be therapeutically useful if induced endogenously. Various 2′-modified poly(I).poly(C) analogues were synthesized, for example, poly(2′-azido-2′-deoxyinosinic acid).poly(cytidylic acid) [6] and poly(2′-fluoro-2′-deoxyinosinic acid).poly(cytidylic acid) [7]. However, these compounds, which originated from a collaborative effort with Ikehara [6,7], were not further pursued for their therapeutic potential, because with the cloning of interferon, the clinicians' attention had in the meantime shifted from the endogenous induction of interferon to exogenously applied interferon. Exogenous interferon-α and -β would later prove useful in the treatment of chronic hepatitis C and multiple sclerosis, respectively.
Double-stranded RNA and poly(I).poly(C)
However, in the 1970s, a second possible application of polynucleotides became apparent, which was their potential use as inhibitors of the reverse transcriptase. This enzyme had been discovered in 1970 by Temin and Mizutani [8] and Baltimore [9] and thus started the search for reverse transcriptase inhibitors, focusing initially on single-stranded polynucleotides as they might inhibit the enzyme by competing with the single-stranded genome of the oncoviruses, then believed to be at the origin of leukaemia and other cancers. Thus, again in collaboration with Ikehara, we described several polynucleotides, that is, poly(2-methylthioinosinic acid) [10], poly(2-azaadenylic acid) [11] and various 2-and 2′-substituted polyadenylic acids [12], as inhibitors of the oncovirus reverse transcriptase. Yet, as the interest and belief in the possible role of the reverse transcriptase in human cancer waned, so did the interest in the pursuit of polynucleotides as reverse transcriptase inhibitors. The only retrovirus that remained unequivocally associated with human cancer, that is, adult T-cell leukaemia, was human T-cell leukaemia virus (HTLV) type 1 [13], but no reverse transcriptase inhibitors ever reached significant momentum for the treatment of this disease.
Suramin
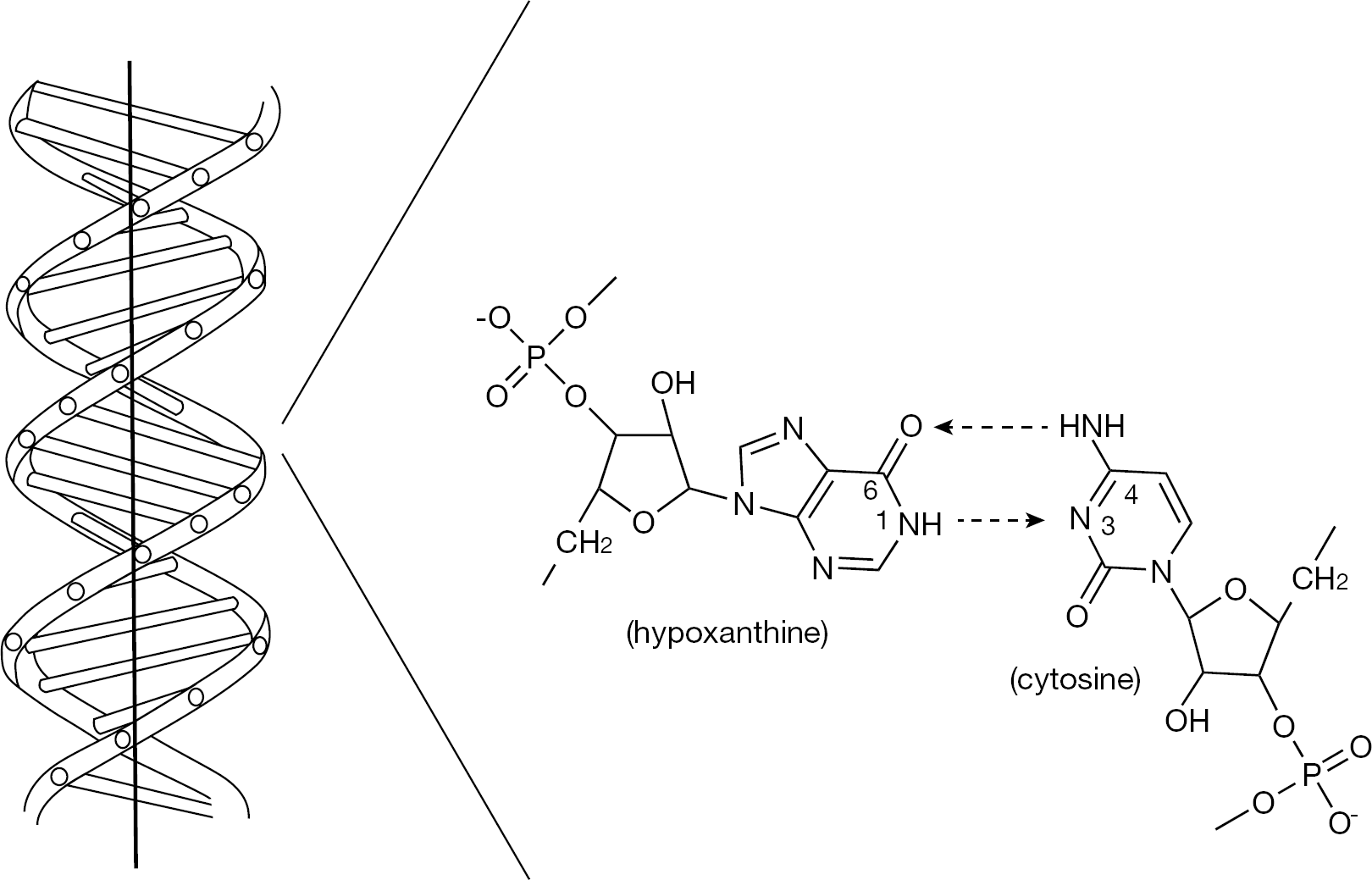
In 1975 I found serendipitously that suramin was a potent inhibitor of the reverse transcriptase; this was perhaps not surprising considering the polyanionic structure of suramin, a hexasulfonate (Figure 2). The expectation that suramin could exhibit any antitumor activity, that is, in mice bearing leukaemic cells, was not fulfilled, however, and in 1979 I published an article in Cancer Letters [14] about the potent inhibitory effect of suramin on the reverse transcriptase activity of murine retroviruses, without mentioning the failure I had encountered with suramin in inhibiting leukaemic cell growth. For the next few years, suramin, which had been used since 1920 in the treatment of sleeping sickness, languished until it was revived by Broder (at the suggestion of Gallo) as an inhibitor of HIV-1 [15]. In fact, suramin was the first reverse transcriptase inhibitor, shown by Mitsuya et al. [15], to inhibit the in vitro infectivity of HIV-1 Suramin was also the first anti-HIV agent to be shown to be effective in suppressing HIV-1 replication in vivo [16]. At about the same time, Rozenbaum et al. [17] had reported equally promising in vivo anti-HIV activity with a polyoxometalate (HPA-23) in the treatment of HIV-1 infection (prior to 1987, HIV-1 was known as HTLV-III and lymphoadenopathy-associated virus type 1 [LAV-1]).
Suramin (hexasodium salt) Commonly used brand names are Moranyl, Naganol, Antrypol, Germanin, Bayer 205.
Although suramin did not enjoy a long career as a potential anti-HIV drug, it allowed an important principle to be established, that as long as it was administered (as one intravenous injection of 1 g per week), it suppressed virus replication, which subsequently resumed, after administration of the drug was ceased [16]. Suramin was not further pursued for the treatment of AIDS (HIV infections) essentially for two reasons: it was too toxic, especially for the liver and kidneys, and in 1985 zidovudine (AZT; azidothymidine) had emerged as a more specific, less toxic and more potent HIV inhibitor.
Dextran sulfate
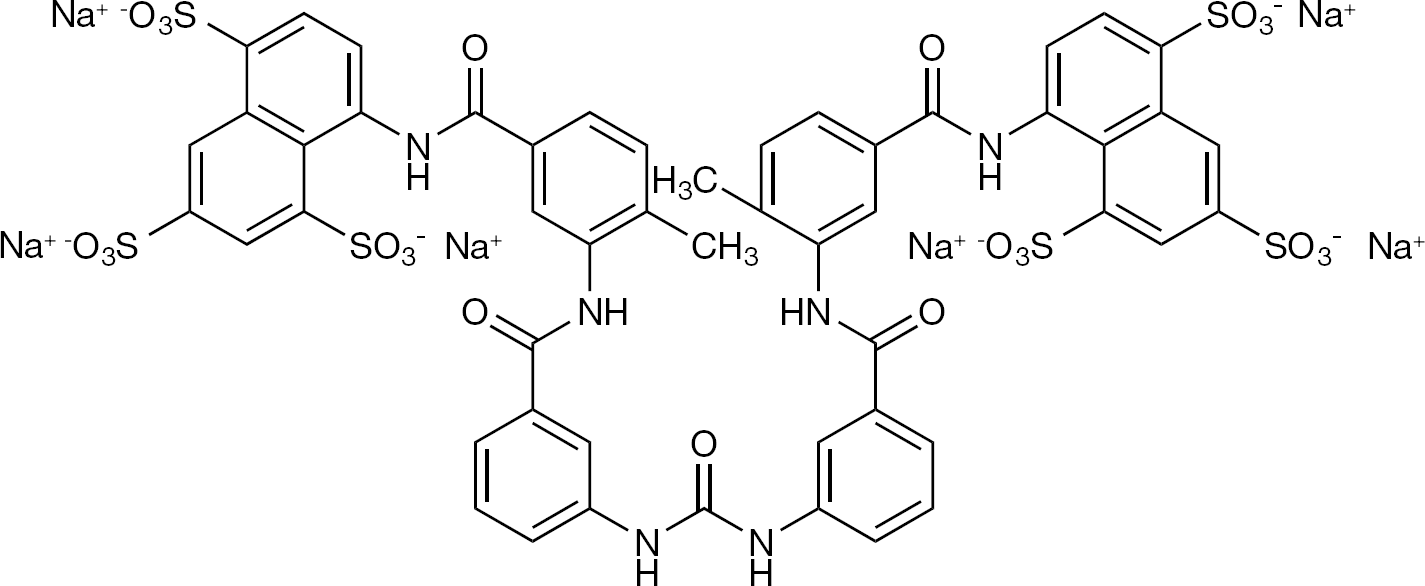
In 1987, two groups, a Japanese team [18] and a Japanese/Belgian team [19], reported simultaneously that dextran sulfate (Figure 3) exhibited a potent inhibitory effect on the replication of HIV. The potency and specificity of dextran sulfate was such that for some time it was considered an attractive candidate for the treatment of HIV infections. At this time, the requirement for parenteral (intravenous or subcutaneous) administration of the compound was not considered to be an issue. Both the groups of Ueno and Kuno, now associated with Mitsuya and our own group (led by Baba) then ascertained that dextran sulfate actually inhibited HIV virion binding to the CD4+ T-cells [20,21].
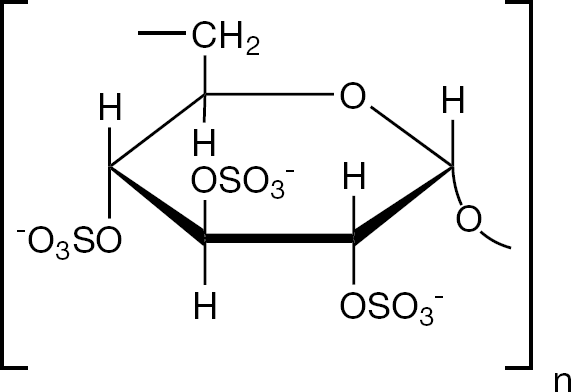
Dextran sulfate
We further showed that the anti-HIV activity of dextran sulfate extended to a variety of sulfated polysaccharides, all depending on the same principle, the inhibition of HIV binding to cells [22–25]. Despite its remarkable anti-HIV potency in vitro, dextran sulfate was not pursued further in vivo, primarily because of its poor oral bioavailability. However, polysulfates and the less biodegradable polysulfonates still have potential as topically applied microbicides, for the prevention of HIV, herpes simplex virus (HSV) and bacterial (that is, chlamydia) infections.
Brivudin
Brivudin (BVDU; (E)-5-(2-bromovinyl)-2′-deoxyuridine; Figure 4) was originally synthesized by Barr in the laboratory of Jones and Walker (Laboratory of Chemistry, Birmingham University, Birmingham, UK) as a possible radio-sensitizing agent, before it was shown by my laboratory to be an extremely potent inhibitor of HSV-1 [26,27] and varicella-zoster virus (VZV) [28]. Under the enthusiastic leadership of De Somer, we were the first to evaluate the clinical potential of BVDU in immunosuppressed patients with severe VZV infection [28]. In 1982, in collaboration with Shigeta (Fukushima Medical College, Fukushima, Japan) we ascertained the exceptionally high in vitro antiviral activity of BVDU against VZV. Although clearly less active than acyclovir against HSV-2, BVDU proved, in vitro, to be appximately 1,000-fold more potent than acyclovir against VZV. Several studies pointed to the superiority of BVDU over acyclovir in inhibiting VZV replication in vitro [29–32].
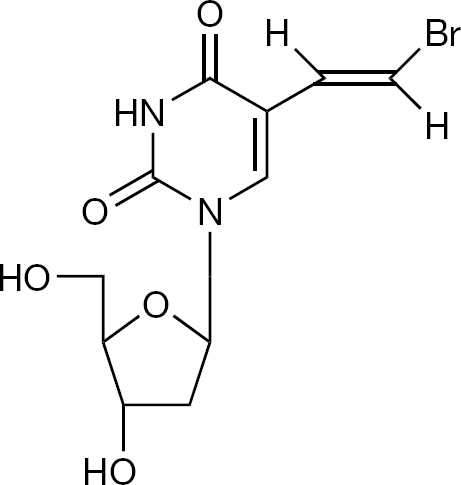
Brivudin (BVDU)
After oral BVDU was shown to be at least equally effective as intravenous acyclovir in the treatment of VZV infections [33], the compound was approved for clinical use, first, as Helpin® in the former DDR (East Germany) for the treatment of herpes zoster in immunosuppressed patients and, finally, in a multitude of countries; among others, Germany (Zostex®), Italy (Brivirac®) and Belgium (Zerpex®) for the treatment of herpes zoster in immunocompetent patients. Because of the potentially lethal interaction of (E)-5-(2-bromovinyl)uracil (BVU), the degradation product of BVDU, with 5-fluorouracil, it should not be used in combination with 5-fluorouracil. The rapid degradation by phosphorylase results in a very short half-life, but this process, which occurs at the liver, is reversible and with the help of thymidine regenerates BVDU, strengthening its antiviral activity [34].
BVaraU
Shortly after the discovery of BVDU as a specific and potent anti-HSV-1 agent, we also discovered the potent anti-HSV-1 activity of its arabinofuranosyl counterpart, BVaraU (Figure 5) [35]. At that time, we were joined by Machida et al. [36,37] at Yamasa Shoyu Company (Choshi, Japan), who had specifically focused on the anti-VZV activity of BVaraU. In fact, a comparative study of 20 anti-herpes agents against 10 different strains of VZV [30] found BVaraU as the most potent anti-VZV agent, with a 50% inhibitory concentration of approximately 1 ng/ml, slightly surpassing BVDU in potency.
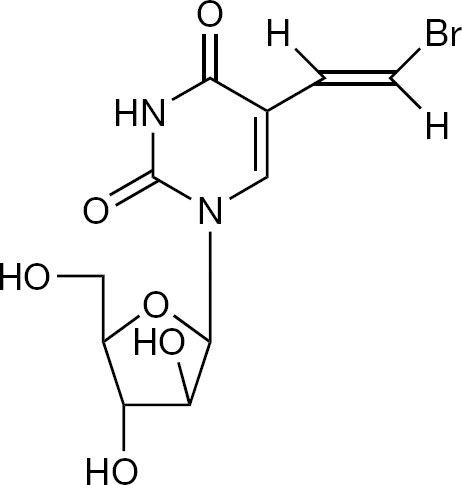
BVaraU
A problem arose with BVaraU, however: when given orally, it was rapidly deglycosylated by enteric bacteria to release BVU [38,39], and BVU, being an inhibitor of dihydropyrimidine dehydrogenase, the enzyme responsible for the first step in the degradation pathway of uracil, thymine, and 5-fluorouracil, caused an accumulation and, therewith associated toxicity, of 5-fluorouracil. The interaction of BVaraU, via BVU, proved lethal in a number of patients [40–45].
Yet, the interaction of BVU with 5-fluorouracil did not have to be necessarily deleterious: in repeated studies Iigo et al. [46–51] showed that BVU, generated from BVDU, could enhance the antitumour activity of 5-fluorouracil and its prodrugs in mice, and Keizer et al. [52,53] further confirmed this potentially favourable effect of BVDU in enhancing the antitumor activity of 5-fluorouracil in cancer patients. The release of BVU from BVDU is reversible and occurs principally in the liver; that of BVU from BVaraU occurs primarily in the gut and is irreversible. The differential pharmacodynamics in the release of BVU from BVaraU and BVDU may have far-reaching implications in that the rate and extent by which BVU is released from BVaraU may primarily aggravate the toxicity of 5-fluorouracil (and thus explain its lethality), whereas for BVDU the balance might be shifted towards a potentiation of the antitumour activity.
Nucleoside reverse transcriptase inhibitors
The era of the nucleoside reverse transcriptase inhibitors (NRTIs) started with the discovery of AZT (Figure 6), first presented under its company codename BWA509U at the AIDS Antiviral Agent Workshop, Public Health Service (National Institutes of Health, Bethesda, MD, USA, 3 June 1985), as an inhibitor of the infectivity and cytopathic effect of HTLV-III [54]. Shortly thereafter, Mitsuya and Broder [55] reported the inhibitory effect of several other 2′,3′-dideoxynucleoside analogues, 2′,3′-dideoxyinosine (didanosine; ddI) and 2′,3′-dideoxycytidine (zalcitabine; ddC; Figure 6) on the in vitro infectivity of HTLV-III. Baba et al. [56] were the first to describe the anti-HIV activity of 2′,3′-didehydro-2′,3′-dideoxythymidine (stavudine; d4T).
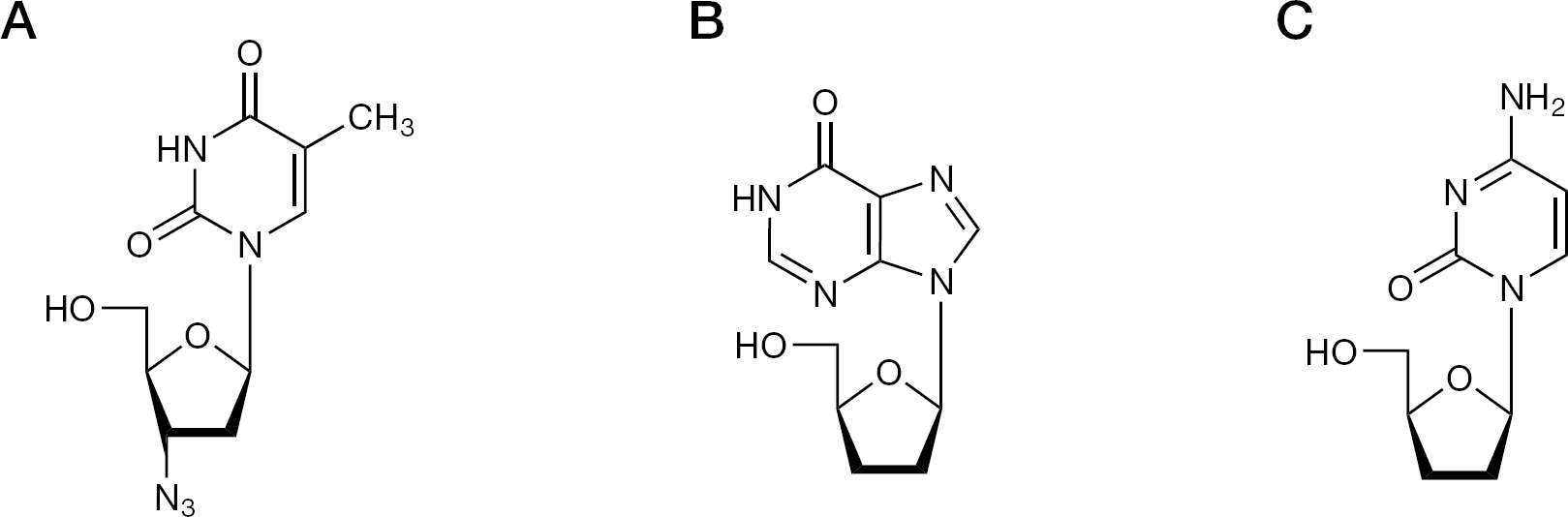
Nucleoside reverse transcriptase inhibitors
Within the same year (1987), two other groups, led by Prusoff [57] and Yamamoto [58], respectively, would describe the anti-HIV activity of d4T. The list of 2′,3′-dideoxynucleoside (ddN) analogues clinically approved and used in the treatment of HIV infections now includes three more compounds: lamivudine (3TC), abacavir (ABC) and emtricitabine ((−)FTC) [59]. The ddN analogues are collectively called NRTIs and share the same mode of action in that they all need to be phosphorylated to their 5′-triphosphate form before they serve as DNA chain terminators: they interact as alternate substrates/competitive inhibitors with the natural substrates dTTP, dATP, dCTP or dGTP. As a rule, they are used in combination with other anti-HIV drugs, particularly non-nucleoside reverse transcriptase inhibitors (NNRTIs) and nucleotide reverse transcriptase inhibitors.
Non-nucleoside reverse transcriptase inhibitors
1-([2-Hydroxyethoxy]methyl)-6-(phenylthio)thymine (HEPT; Figure 7) was the first of the NNRTIs. However, at the time of its first description, the term ‘NNRTI’ did not yet exist. Hence, HEPT was recognized as a novel lead for specific anti-HIV-1 agents. It had been synthesized at Showa University (Tokyo, Japan) by Tanaka and Miyasaka [60]. Considering the presence of a 2-hydroxyethoxymethyl side chain (a feature of acyclovir), one might conclude that the HEPT derivatives had originally been synthesized as potential anti-herpetic agents. However, they did not exhibit any anti-herpetic activity, but, unexpectedly proved active against HIV-1 [60,61]. Starting from the original HEPT, a large variety of analogues were synthesized, and from extensive mechanism-of-action studies, it became clear that they interacted specifically with the HIV-1 reverse transcriptase at an allosteric site, different from but spatially close to the catalytic site of the NRTIs [62–69]. Emivirine (MKC-442; Figure 7) [70] was the member of the HEPT series that advanced furthest, reaching Phase III clinical trials, before development was halted. Emivirine (Coactinon®) had in the meantime been transferred to Triangle Pharmaceuticals, which was later taken over by Gilead Sciences.
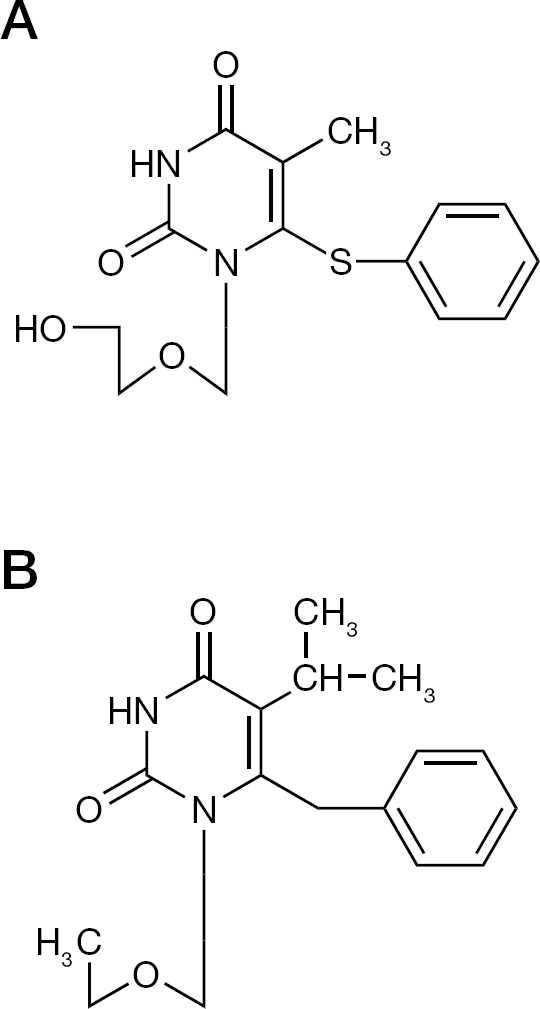
Non-nucleoside reverse transcriptase inhibitors
The TIBO derivatives were identified as NNRTIs at the same time as HEPT [71]. Preliminary assessments noted the structural similarities between the HEPT derivatives (prototype emivirine) and TIBO derivatives (prototype tivirapine) [72]. More in-depth analysis revealed the similarity in molecular conformation, upon docking of HEPT, TIBO and various other NNRTIs within their HIV-1 reverse transcriptase ‘pocket’ binding site [73–75]. Five NNRTIs have been licensed for clinical use: nevirapine, delavirdine, efavirenz, etravirine and rilpivirine [59]. Delavirdine is no longer used, whereas efavirenz and rilpivirine are now used primarily in combination with FTC and tenofovir disoproxil fumarate.
The acyclic nucleoside phosphonates
The era of the acyclic nucleoside phosphonates (or acyclic nucleotide analogues) started with the discovery of (S)-HPMPA or (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine (Figure 8) as a broad-spectrum anti-DNA virus agent [76]. Nucleoside phosphonates differ from the normal nucleotides in that they contain a phosphonate instead of a regular phosphate (Figure 8) linkage, and as a consequence thereof, exhibit a long-lasting intracellular half-life with broad-spectrum antiviral activity against a variety of DNA viruses (including HBV) and retroviruses (including HIV). The early group of the acyclic nucleoside phosphonates included three compounds that were ultimately approved for clinical use: (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine ([S]-HPMPC; cidofovir, Vistide®) [77]; 9-(2-phosphonylmethoxyethyl)adenine (PMEA, adefovir) [76,78] and (R)-9-(2-phosphonylmethoxypropyl)adenine ((R)-PMPA, tenofovir; Figure 8). Cidofovir was approved for clinical use in the (intravenous) treatment of human cytomegalovirus (HCMV) retinitis in AIDS patients; adefovir in its prodrug form (adefovir dipivoxil; Hepsera®; Figure 8) was approved for the (oral) treatment of chronic HBV infections; and tenofovir has been approved in its prodrug form (tenofovir disoproxil fumarate; Viread®; Figure 8) for the (oral) treatment of both HIV and HBV infections. For the treatment of HIV infections (AIDS), tenofovir disoproxil fumarate has also been approved in several drug combinations, that is, as Truvada® if combined with FTC [79], as Atripla®, if combined with FTC and efavirenz [80], and most recently, as Complera® (in the US; or Eviplera® in the EU), if combined with FTC and rilpivirine [81].
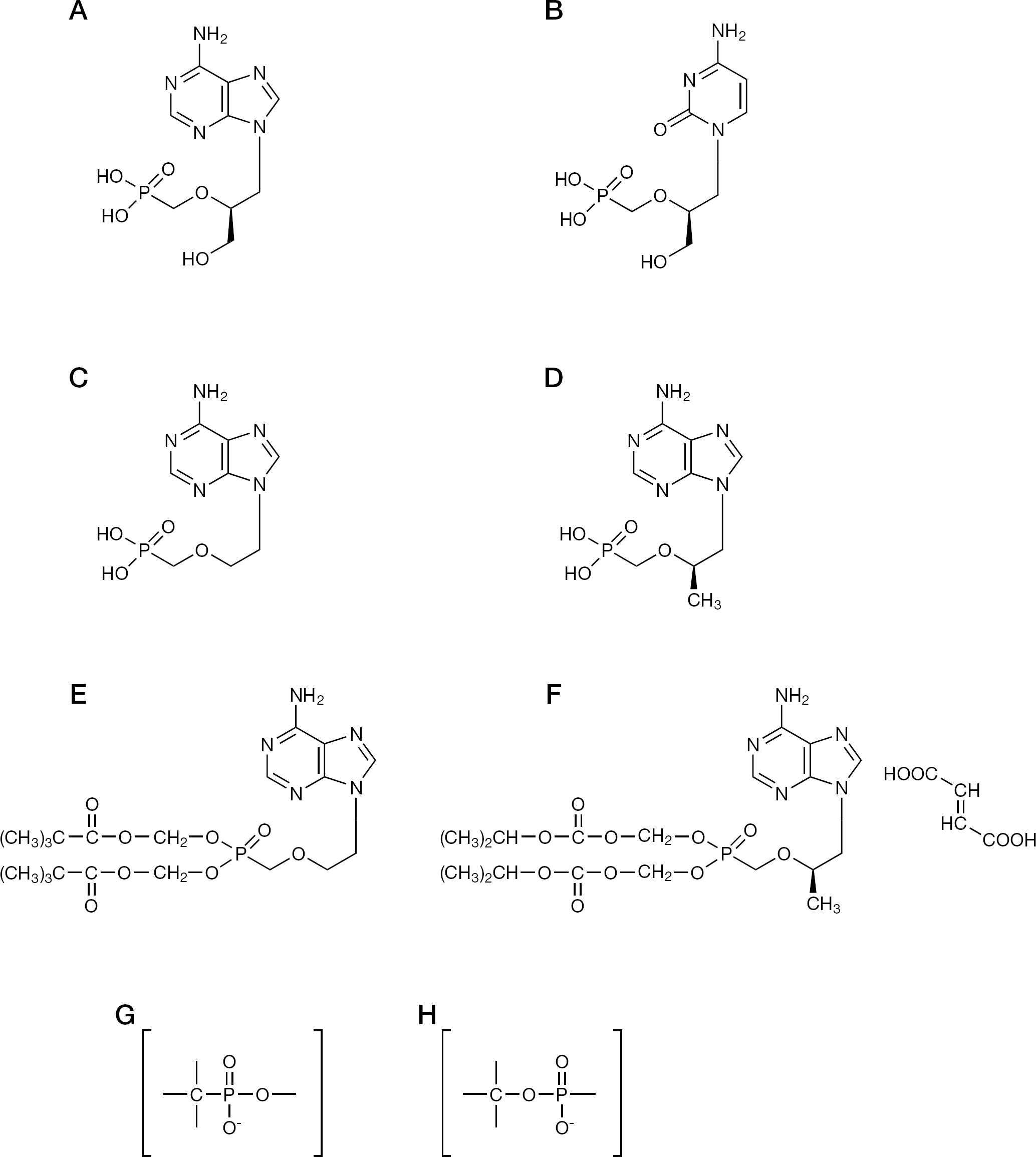
Acyclic nucleoside phosphonates
In addition to its use in the therapy of HIV and HBV, tenofovir disoproxil fumarate has also gone in the direction of the prevention (both topically, as microbicide, and systemically as Viread® or Truvada®) of HIV infections [82]. The usefulness of adefovir and tenofovir in the treatment of HBV infections was pioneered by the original work of Yokota et al. [83–85] in the early 1990s.
CXCR4 antagonists
In 1997, three papers appeared in the same issue of the Journal of Experimental Medicine [86–88] reporting three different CXCR4 antagonists as inhibitors of HIV infection: AMD3100, T22 and ALX40-4C (Figure 9). Following their report on the oligopeptide T22, Yamamoto et al. reported on two additional CXCR4 antagonists, KRH-1636 [89] and KRH-3955 [90]. These CXCR4 antagonists were originally pursued as inhibitors of HIV replication for use in the treatment of HIV infection. However, during the further exploration of the therapeutic potential of AMD3100 [91], it became evident that the compound had an interesting side effect, that is, due to its antagonization of CXCR4, it proved useful as a stem cell mobilizer (plerixafor; Mozobil™), and it was finally licensed for this purpose [92,93]. Its commercialization has been transferred from AnorMed to Genzyme, and finally, Sanofi. The compound has been formally approved for use as a stem cell mobilizer in the autologous transplantation of hematopoietic stem cells in patients with non-Hodgkin's lymphoma and multiple myeloma. It would now seem imperative to examine whether Mozobil would also be useful for stem cell mobilization in other clinical situations, and whether these indications could be extended to other CXCR4 antagonists, such as KRH-1636 or KRH-3955, as well.
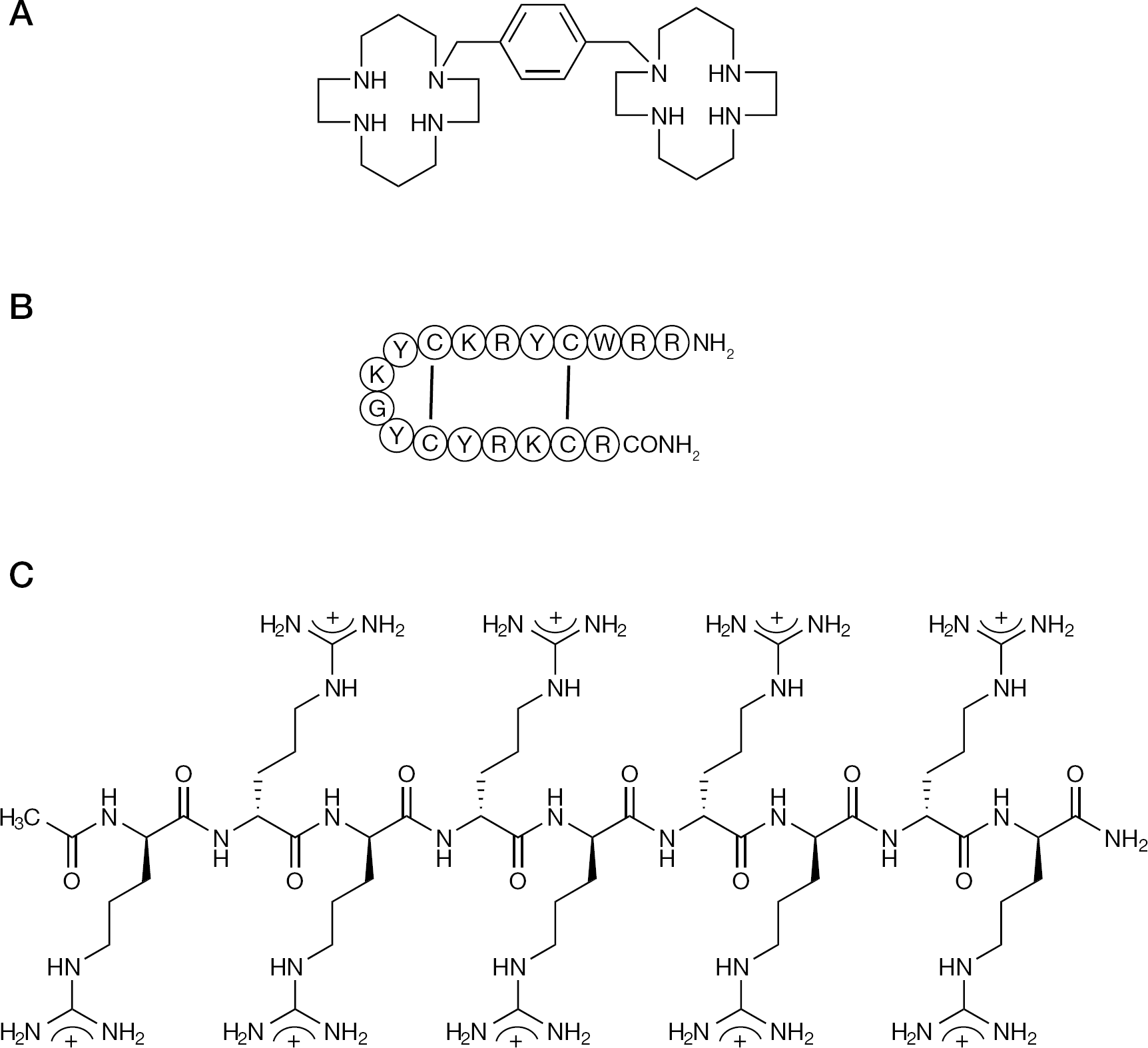
CXCR4 antagonists
Elvitegravir is scheduled to be part of the so-called Quad pill, containing, in addition to elvitegravir, a booster or pharmacoenhancer, cobicistat as well as FTC and tenofovir disoproxil fumarate. This is the first (‘all Gilead’) Quad pill that should be administered orally once-daily in the treatment of HIV infections [98].
Elvitegravir
Elvitegravir represents a novel inhibitor of HIV integrase, which originated in Japan (JTK-303; Figure 10) [94,95]. It was then licensed to Gilead Sciences (GS-9137), where it was further developed [96,97], with the aim to commercialize it for the treatment of HIV infections.
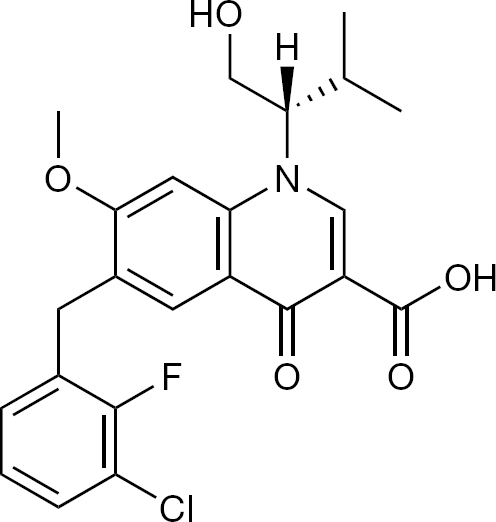
Elvitegravir
Conclusions
This review covers ten accounts of antiviral development, which, over a period of approximately 30 years, started either in Japan and/or strongly profited from a Japanese influence. These stories focus on polynucleotides (as either inducers of interferon or inhibitors of the reverse transcriptase), suramin, a reverse transcriptase inhibitor and anti-HIV agent, dextran sulfate, an inhibitor of HIV binding to the cells, BVDU, a potent inhibitor of HSV-1 and VZV, BVaraU, another potent inhibitor of HSV-1 and VZV, the NRTIs as anti-HIV agents, the NNRTIs as anti-HIV-1 agents, the acyclic nucleoside phosphonates as broad-spectrum antiviral agents, and which are also active against HBV, the CXCR4 antagonists, which were discovered as anti-HIV agents, but now evolved to stem cell mobilizers, and the HIV integrase inhibitor, elvitegravir.
Footnotes
Acknowledgements (Japanese colleagues in alphabetical order)
I am glad to dedicate this paper to my Japanese coworkers, colleagues and friends and to Christiane Callebaut for her proficient editorial assistance in preparing this presentation and paper. Masanori Baba, Toshikazu Fukui, Mitsuaki Hosoya, Masaaki Iigo, Satoru Ikeda, Morio Ikehara, Nakao Ishida, Masahiko Ito, Kunisuke Izawa, Eiichi Kodama, Tasuke Konno, Haruhiko Machida, Tokumi Maruyama, Akira Matsuda, Hiroaki Mitsuya, Tadashi Miyasaka, Shuichi Mori, Hideki Nakashima, Mika Okamoto, Katsuhiko Ono, Tohru Otake, Takashi Sakuma, Shiro Shigeta, Hiroshi Shiota, Satoshi Shuto, Hiroshi Takaku, Hiromichi Tanaka, Masaru Ubasawa, Naohiko Yamamoto, Naoki Yamamoto, Tomoyuki Yokota and Tohru Ueda.
This review is based on a lecture presented at the 25th ICAR, 16–19 April 2012, Sapporo, Japan.
The author declares no competing interests.