
Abstract
Each year, the International Society for Antiviral Research (ISAR) organises a conference covering many differing aspects of antiviral research. The 25th International Conference on Antiviral Research (ICAR) was held in Japan. This special anniversary meeting was co-sponsored by the Japanese Association for Antiviral Therapy.
This Workshop Report contains summaries of the four major lectures and each of the invited presentations in the Clinical symposium and in the three mini-symposia. Of the many interesting contributor presentations, there are brief summaries of a small selection of these. This report concludes with a few personal comments and observations.
A brief summary of this report is included within the ISAR News published in this issue of AVCC.
Introduction
This review provides an overview of the conference highlights. As this is a research conference, any references to clinical results should not be taken as a recommendation for clinical use. I wish to thank all those authors who have kindly provided me with copies of their presentations and for giving me valuable comments.
This report does not follow the chronology of the meeting, but starts with the 25th Anniversary Lecture, then the presentations by the recipients of the Society's two major awards, the keynote address, the clinical symposium, three mini-symposia and contributed presentations, the last of which gives hope of a cure for HIV. Occasionally, as an aside, I have added my own comments.
25th Anniversary Lecture: Success and failure in antiviral drug development; personal reminiscences with a Japanese connection
Erik De Clercq (KU Leuven, Rega Institute for Medical Research, Leuven, Belgium)
Just prior to this Anniversary Lecture, Joe Colacino (International Society for Antiviral Research [ISAR] President) presented the prestigious ‘Service to the Society Award’ to Erik, one of the founders of ISAR, in recognition of his continued dedication, support and service to the Society and its members. This award has been presented to only two other members of the Society, Drs George Galasso and Earl Kern (Figure 1).

Erik De Clercq giving the 25th Anniversary Special Lecture, having been presented with ISAR's Service to the Society Award
For this anniversary presentation, Erik chose to tell 10 stories, each of which contained a Japanese connection. Most of these stories were illustrated by photographs from Erik's own archives, including one showing a very young Masanori Baba who did so much to organize this 25th ICAR.
Polyl.polyC
PolyI.polyC was a known interferon inducer. Together with Morio Ikehara, Erik worked on analogues and investigated single stranded (ss) RNA as inhibitors of reverse transcriptase (RT) [1]. On 10 to 23 May 1987, there was a NATO meeting at Il Ciocco, Italy. Erik showed a photograph (Figure 2) of the attendees, pointing out several people who have been notable supporters of ISAR. This meeting can be regarded as a model for ICAR meetings. As a clarification, Williamsburg of 1988 was the second International Conference on Antiviral Research (ICAR). There was no meeting called the ‘First ICAR’ but the Rotterdam meeting of 1985 has generally been referred to as the first ICAR. Erik considers Il Ciocco as the ‘virtual first’ because ideas about both ISAR and ICAR were developed at that meeting.

Attendees at Il Ciocco, Italy in May 1987: this meeting can be regarded as a model for ICAR meetings
Suramin
This is an inhibitor of RT [2]. It was found to inhibit HIV replication by Mitsuya et al. [3]. Erik's early work on RT inhibitors was presented at Urabandai, Japan, at the 9th ICAR in 1996.
Dextran sulphate
Several papers were published by Ueno, Mitsuya and Baba but this compound did not become a clinically useful drug [4,5].
Brivudin (BVDU)
In 1980, a proof-of-concept trial, in four varicella zoster (VZV) patients, was published [6]. Comparative anti-VZV activity of various nucleoside analogues was published by Shigeta et al. [7]. Twenty years later, brivudin reached the market, first in East Germany, then in the whole of Germany, Italy and Belgium, and several other European countries.
BVaraU
Machida (Yamasa Company, Choshi, Japan) discovered that bacterial deglycosylase released BVU from BVaraU [8]. Unlike BVDU, this step was essentially irreversible. Due to the interaction between BVU and 5 fluorouracil, an anti-cancer drug, the clinical use of BVaraU was terminated.
AZT
The first paper on zidovudine (AZT) against HIV was published by Mitsuya et al. [9]. This work led to didanosine (ddI) and zalcitabine (ddC) which, in turn, led Masanori Baba, in Erik's laboratory, Bill Prusoff and Yamamoto to the drug, stavudine (d4T). A success.
HEPT
Miyasaka and Tanaka (Showa University, Tokyo) synthesized HEPT [10] and sent it to Erik for antiviral testing. This led to many HEPT analogues being tested. Emivirine progressed to Phase III trials but was then stopped.
Phosphonates
In a long collaboration with Antonin Holý (Academy of Sciences, Prague, Czech Republic) and later with Gilead Sciences, Erik evaluated (S)-HPMPA, (S)-HPMPC, PMEA which became known as adefovir, and (R)-PMPA which became known as tenofovir [11]. As Erik did not have an HBV assay available, he asked Yokota in Shigeta's laboratory to test against HBV. The prodrug of tenofovir, tenofovir disoproxyl fumarate (tenofovir DF) has become a mainstay in the therapy of HIV and HBV.
AMD 3100
AMD 3100 is active against HIV as a CXCR4 antagonist. AMD 3100 was discovered to be a stem cell mobilizer and is used as such clinically [12]. In Naoki Yamamoto's laboratory in Tokyo, several other CXCR4 antagonists were discovered.
Elvitegravir
Elvitegravir (EVG) was originally discovered at Japan Tobacco Company, the second HIV integrase inhibitor. EVG is a component of the QUAD pill.
Several failures certainly but also some successes to cherish.
Gertrude Elion Memorial Award Lecture: Using phospholipid mimicry to increase efficacy and safety of acyclic nucleoside phosphonate antivirals by Karl Hostetler (University of California, San Diego, CA, USA)
Karl started his presentation with a characteristically cheerful photograph to remind the audience of Gertrude Elion at the 1997 ICAR (Figure 3).

Gertrude Elion Memorial Award winner: Karl Hostetler
The idea of using phospholipid mimicry was stimulated by a 1967 paper on lysophosphatidylcholine [13]. In 1993, acyclovir (ACV) diphosphate diglyceride was shown to be active against ACV-resistant, thymidine kinase negative (TK−) herpes simplex virus (HSV). However, this derivative of ACV was not orally bioavailable. Hexadecyl-oxypropyl-P-ACV (HDP-P-ACV) was found to be active against HBV because of kinase bypass. This led to a request from the NIAID in 1999 to investigate the possibility of modifying cidofovir to provide oral activity against smallpox and other poxviruses.
Hexadecyloxypropyl cidofovir (HDP-CDV) had enhanced antiviral activity because of greater cellular uptake of the drug. Persistence of antiviral activity was noted due to slow wash-out of the active moiety, CDVpp (half-life 8 to 10 days). It is thought that HDP-CDV, having only one lipid chain, can easily insert into the cell membrane, flip over and leave the internal membrane of cells to undergo metabolism and anabolic phosphorylation to CDVpp, the active moiety. HDP-CDV was orally active in a lethal mousepox model. HDP-CDV was found to be very active against all double-stranded DNA viruses including adenovirus and Epstein-Barr virus (EBV).
HDP-CDV (CMX001) is in clinical development. Phase I and open-label compassionate-use trials have shown that the pharmacokinetics are dose proportional, there has been no nephrotoxicity and renal function has either stabilized or improved. The only safety signal was reversible diarrhoea. In a published case study [14], a paediatric stem-cell transplant patient had adenovirus infection, with 109 copies/ml plasma. A single dose of CMX001 much reduced the viral load and subsequent doses twice weekly cleared the virus. In February 2012, a Phase II trial, for cytomegalovirus (CMV) prophylaxis in human stem-cell transplant patients at risk of CMV disease, was completed. The positive results from this trial were given in Mini-symposium: clinical development of antiviral agents.
The hexadecyloxypropyl conjugate of tenofovir (CMX157) has been investigated and is active against HIV with a 50% effective concentration (EC50) in the low nanomolar range in peripheral blood mononuclear cells (PBMCs). It is thought that the lipid enables HDP-tenofovir to ‘hitch a ride’ by inserting itself into the lipid of the HIV virion, reducing viral infectivity. In Phase I clinical trials in normal volunteers, the active moiety, TFVpp levels in PBMCs peak at about 24 to 48 h after dosing and wash-out slowly over a week.
Octadecyloxyethyl-CDV has greater activity against HIV than CDV. Using HIV RT in an enzyme assay, CDVpp did not inhibit chain extension with either RNA or DNA as the template and so full-length chains were produced. However, when CDV was present in the template, chain extension stopped at the inserted drug and no full-length DNA was noted. Since subclinical CMV replication in well-controlled AIDS patients may cause significant morbidity, it was proposed that including HDP-CDV in an AIDS regimen may be of extra benefit.
William Prusoff Young Investigator Award Lecture - HBV and HCV: Parallels, contrasts and future directions for therapy
William E Delaney IV (Gilead Sciences, Foster City, CA, USA)
Although HBV and HCV both infect the liver, their virion structure and genetic organization differ markedly. HBV has a partly double-stranded DNA about 1/3 the length of the HCV single-stranded RNA. HBV DNA has four major open reading frames, one of which codes for the only viral enzyme, the polymerase. HCV codes for ten distinct proteins, including at least four of which are enzymes involved in viral replication. Their replication strategies within infected cells differ. During infection, HBV DNA is transported into the cell nucleus and the viral DNA forms covalently closed circles (cccDNA). In this form, the viral DNA is very stable and can persist for a long time in the infected cell nucleus. In contrast, replication of HCV takes place entirely within the cell cytoplasm and there are no known long-lived forms of the virus (continuous replication is required to sustain infection). Another marked difference between these two viruses is their genetic variability. HBV has genotypes A through to H with the viral DNA having about 7 to 8% sequence divergence between genotypes. This level of variability can be exceeded within a single genotype of HCV (which can have subtype divergence of 20%); the variability between HCV genotypes (GT1-GT7) is even greater (up to 30%). These differences have major implications for therapies.
HBV polymerase is the only clinically validated viral target for HBV antiviral therapy. Several nucleoside and nucleotide analogues are highly effective, initially. Due to the persistence of HBV, therapy has to be continued for years, maybe indefinitely, to maintain viral suppression. Although mutations generally arise far more slowly in HBV than in HCV, resistance does appear over the course of years of therapy. The newest therapies have very high barriers to resistance in treatment-naive patients (tenofovir DF 0% resistance at 5 years; entecavir <2% resistance after 5 years). Those patients who are hepatitis Be antigen-negative (HBeAg -ve) usually have good antiviral outcome: after one year therapy, about 90% of patients have undetectable viral DNA when using tenofovir DF, entecavir or telbivudine. With HBeAg +ve patients, the corresponding (cross-study) values are about 75%, 70% and 60%, respectively. The percentage of patients becoming undetectable for HBV DNA will increase with additional time on therapy, unless resistance emerges. Even though the serum viral load may be reduced dramatically after a year of therapy (4 to 5 log10), the corresponding reduction in nuclear levels of the HBV covalently closed circular DNA (cccDNA) is modest (about 0.5–1 log10). Stable hepatitis B surface antigen (HBsAg) seroconversion is believed to represent a functional cure for HBV. So far, even the best anti-HBV therapies have given low rates of HBsAg seroconversion. With tenofovir DF therapy, HBsAg loss rates have slowly increased over the years of therapy, to about 10 to 11% at five years.
With HCV having several potential antiviral targets, there are many anti-HCV compounds in the pipeline. The only approved direct-acting antivirals are two protease inhibitors (PIs), boceprevir and telaprevir, which are each used in combination with pegylated interferon-α and ribavirin (PEG-IFN/RBV). Three PIs (TMC-435, BI-201335 and BMS-650032) are in Phase III trials and at least nine others in Phases I and II. There are at least nine non-nucleoside inhibitors of the HCV polymerase in Phases I and II. There are some nucleoside analogue inhibitors of the HCV polymerase in Phase III (PSI-7977, now called GS-7977), in Phase II (RG-7128, IDX184) and in Phase I (GS-6620, INX-189, TMC649128). Of the HCV NS5A inhibitors, BMS-790052 is in Phase III and GS-5885 and BMS-824393 are in Phase II.
As an aside, HCV NS5A seems to be an interesting target. NS5A has no known classical enzymatic function but domain 1 binds viral RNA, domain 2 may interact with host proteins and domain 3 is unfolded and is essential for viral assembly. Possibly, the degree of NS5A phosphorylation guides its role towards RNA replication or of viral assembly. BMS-790052 was the first NS5A inhibitor to be evaluated in clinical trials which confirmed NS5A as a valid target. Interestingly, the NS5A protein forms dimers in crystal structures and NS5A inhibitors are also dimeric in nature. Just one molecule of BMS 790052 inhibits about 10 to 100 NS5A molecules, maybe by blocking NS5A polymerization?
Some examples of HCV clinical trials were shown by William Delaney (Figure 4). GS-9451, an NS3 protease inhibitor, was evaluated in a Phase I trial against HCV (genotype 1). During the three-day monotherapy (400 mg once daily) there was about a 3.5 log10 reduction in viral load. In a similar monotherapy trial, the NS5A inhibitor GS-5885 (10, 30 or 90 mg once daily) gave a very rapid 3 log10 reduction in viral load within the first day of therapy, and about 3.5 log10 reduction by day 3. With the nucleoside NS5B (polymerase) inhibitor GS-7977, monotherapy (400 mg once daily) reduced viral load by about 4 to 5 log10 over a seven-day period with some patients becoming undetectable for HCV RNA.

William Prusoff Young Investigator Award winner: William Delaney
Genetic variability of HCV (including both divergence between genotypes and the frequency of polymorphisms at drug-resistance sites within individual genotypes) influences the success rate of antivirals, depending on the target protein. PIs, for example, BILN-2061, can be highly active against genotype 1 but much less so against genotypes 2 and 3 due to baseline polymorphism in the protease. The PI, GS-9256, was highly effective at reducing viral load during three days of therapy but at that time most patients had resistance. In a deep sequencing analysis of patients in this study, no resistance mutations were detected at baseline (limit of detection =0.25%). However, a single representative patient had four resistant mutations identified at 24 h and eleven mutations at 84 h, one of these (R155K) representing 62% of the virus population. In contrast, nucleoside inhibitors of HCV polymerase are active against all genotypes and provide a much higher genetic barrier to resistance. With GS-7977 monotherapy for 12 weeks in GT2/3 patients, there were no viral breakthroughs and 6/10 patients attained a sustained virological response (SVR). Combination of GS-7977 with ribavirin gave 100% SVR in GT2/3 patients in Phase II pilot studies. SVR is regarded as a cure for HCV.
Looking to the future for HBV, long-term viral suppression is now achievable with nucleoside therapy, but HBsAg seroconversion remains a challenge. A substantial change in treatment approach will likely be necessary to increase HBV cures and this may require combinations of immune modulators with antivirals. As noted by William Delaney: ‘We are still one giant leap away.’
For HCV, the field is developing rapidly with promising candidates being evaluated in advanced clinical trials. Recent Phase II studies have demonstrated the ability to cure most patients without the need for interferon or ribavirin. A polymerase inhibitor, combined with another pan-genotype antiviral, could potentially become a world-wide therapy for all genotypes of HCV. It seems likely that simple, convenient therapies to cure HCV will be available within the coming years.
Keynote address - structure-guided development of AIDS therapeutics: Successes, challenges and opportunities
Hiroaki Mitsuya (National Cancer Institute, Frederick, MD, USA)
After 31 years of AIDS research, highly active antiretroviral therapy (HAART), now commonly shortened to ART (antiretroviral therapy) enables an estimated 33 million people to live with HIV, median death about 65 years. Sadly, each year, about 2.5 million are newly infected and about 2 million die. Key challenges remain: the immune system is only partially restored, long-term toxicity and cost.
The rationale for investigating CCR5 inhibitors is that the Δ32 deletion is found naturally in 1% of the Caucasian population. During 4 years at the presenter's (Figure 5) laboratories, over 100 compounds were investigated and one, GRL 09117, is very potent and looks promising. Because CXCR4-tropic HIV is present in nearly all HIV-infected patients, and especially as the CXCR4-tropic HIV becomes the dominant variant later in infection, this receptor is a good antiviral target.

Keynote lecture by Professor Hiroaki Mitsuya, President of the JAAT
New RT inhibitors are being developed, for example, festinavir (Ed4T, 4′ethynyl-d4T) and EFdA (4′ethynyl-2-fluoro-2′deoxy-adenine). Festinavir was first developed at Yale University, USA, licensed to Oncolys BioPharma, a Japanese company, which progressed festinavir to Phase I trials and is now being developed by Bristol-Myers Squibb. It is a once daily RT inhibitor which is active against viruses resistant to both tenofovir and abacavir. In an enzyme assay, it is about 100-fold less toxic to mitochondrial DNA polymerase gamma than d4T. EFdA has potent activity against HIV (EC50 in the range 0.05 nM to 0.4 nM). In a DNA extension assay, EFdA is incorporated into the DNA and the next base is added but then no further DNA chain extension is possible. Hence, this is a ‘delayed DNA chain terminator’. In monkeys with simian immunodeficiency virus (SIV), EFdA (2 mg dose daily) reduced virus load to below the limit of detection.
Darunavir (DRV) is unusual among the PIs in clinical use as resistance to DRV is much less common than against other PIs. So it was thought that DRV must have some activity other than just enzyme inhibition. Dimerization of the protease is essential for its activity. Within the interface, there are 12 hydrogen bonds which account for about 75% of the dimerization energy. DRV was found to inhibit dimerization, whereas the other PIs did not. HIV, with three mutant sites, was able to refold in the presence of DRV. A new compound, GL-0519, was 6 to 10 times more potent than DRV in this dimerization assay. In conclusion, in contrast to other PIs, multiple mutant sites are required for DRV resistance due to its mode of action in preventing dimerization. This finding has opened the way to find potentially more potent compounds.
As an aside, I felt that this presentation made a good case for being optimistic that new anti-HIV drugs will contribute to the future of HIV care.
Clinical symposium
High efficacy and excellent safety of letermovir (AIC246) in CMV prophylaxis of bone marrow transplant recipients demonstrated in Phase Iib
Holger Zimmermann (AiCuris Pharmaceutical GmbH & Co, Wuppertal, Germany)
AIC246 (letermovir) has a novel mode of action. It inhibits the HCMV terminase complex (UL56, UL89 and UL104) which cleaves the viral DNA into unit lengths for packaging into virions. As this is a viral function without a host equivalent, no mechanism-based side effects are expected. Also, there should be no cross-resistance to HCMV polymerase inhibitors, such as valganciclovir (VGCV). In cell assays, the dose response is much sharper with letermovir than for ganciclovir (GCV), the corresponding EC50/EC90 values being 4.5/6.1 nM and 2.0/14.5 μM.
Phase I studies confirmed that once daily dosing was possible (time to peak concentration [Tmax] about 1.5 h, half-life about 10 h). Elimination was mainly unchanged drug via faeces. Letermovir appeared to be well tolerated and did not interact with various drugs which transplant patients would be taking. In a proof-of-concept study, which was randomized, controlled and open-label, letermovir (40 or 80 mg daily for 14 days) was given to transplant patients as pre-emptive therapy. The primary end point was decline in HCMV DNA from baseline to day 15. The control was VGCV. All treatment groups achieved a reduction from baseline. No patient developed HCMV disease under letermovir therapy. In this small trial, the efficacies of letermovir and VGCV were comparable.
In a dose ranging Phase IIb trial, prophylactic therapy (84 days) with letermovir (60, 120 or 240 mg once daily) versus placebo was started in transplant patients who had no detectable HCMV replication in the 5 days before trial start. The two primary end points were the incidence and time of onset of HCMV prophylaxis failure. The overall failure rates, which include patients who discontinued treatment for any reason, were 49%, 32%, 29% and 64%, respectively. Considering only the viral failures, the proportions were 27%, 19%, 6% and 36%, respectively. If those patients, who had detectable HCMV on day 1 of therapy, are taken out of the analysis, then the proportions were 15%, 6%, 0% and 27%, respectively. Letermovir, at all doses, had a safety profile comparable to placebo.
So far, it seems that letermovir is highly effective against HCMV viraemia and against HCMV disease at a 240 mg once daily dose.
There was a question about the ethics of running a placebo controlled trial evaluating prophylactic efficacy in these transplant patients. There is no problem as prophylaxis is not normally used because of the potential side effects of VGCV. The current standard of care is simply to monitor patients for CMV. Only when HCMV is detected, does the patient go onto active therapy.
Quad: A new single tablet antiretroviral regimen
Tomas Cihlar (Gilead Sciences Inc., Foster City, CA, USA)
Single tablet regimens have become the mainstay of HIV therapy. Atripla, introduced in 2006, contains tenofovir DF, emtricitabine and efavirenz. Complera, introduced in 2011, contains tenofovir DF, emtricitabine and rilpivirine. Although Atripla has become one of the preferred first-line regimens, it still has some limitations: cental nervous system adverse effects, rash, elevated lipids, pregnancy category D and non-nucleoside reverse transcriptase inhibitor (NNRTI) resistance. Complera is not a first-line regimen, has lower efficacy with high viral load and NNRTI resistance. To complement the existing first-line regimens, a new combination called Quad is being developed.
Quad contains tenofovir DF (nucleoside reverse transcriptase inhibitor [NRTI]), emtricitabine (NRTI), elvitegravir (integrase inhibitor) and cobicistat (PK enhancer with no HIV activity). Two registrational Phase III studies (102 and 103) demonstrated non-inferior efficacy of Quad at week 48 compared to Atripla and truvada/atazanavir/ritonavir (TVD/ATV/r), respectively. The primary end point was the percentage of patients with HIV RNA below 50 copies/ml at 48 weeks. In study 102, Quad gave faster onset of viral suppression than Atripla but by 48 weeks, the two therapies were similar (89% and 87%, respectively). Study 103 gave a similar result, faster onset of viral suppression with Quad but similar viral suppression at 48 weeks (92% and 88%, respectively). In both trials, there were 5–7% virological non-responders. Only about 2% of Quad-treated patients developed NNRTI- and/or integrase inhibitor resistance. Frequencies of adverse events as well as grade 3–4 laboratory abnormalities were similar between Quad and the comparator arms. Elevated serum creatinine was more frequent in the Quad-treated patients, primarily due to effects of cobicistat on the active renal secretion of creatinine. In contrast, Quad showed less lipid abnormalities compared to Atripla and TVD/ATV/r.
Regulatory filings have been submitted in USA, Europe, Australia, Canada and Switzerland. The FDA decision is expected by 27 August 2012 and the European decision by January 2013.
As an aside, at ICAR 2011, Tomas reported on an amidate prodrug of TFV (GS-7340) which, in short-term monotherapy (either 50 mg or 150 mg), was more effective than TDF (300 mg) in treatment-naive HIV-infected patients. A lower dose of GS-7340 will be useful when considering drug combination. GS-7340 is well positioned to potentially replace TDF as the standard prodrug of TFV, perhaps in ‘Quad 2’?
GS-7977: A nucleotide prodrug for the treatment of HCV infections
Michael Sofia (Gilead Sciences Inc., Princeton, NJ, USA)
GS-7977, formerly known as PSI-7997 (Figure 6), gave up to 2 log10 reduction of HCV RNA in a three-day monotherapy ascending dose Phase I study. When combined with PEG-IFN and RBV, in a doseranging Phase IIa trial with dosing for 14 days, there was at least a 5 log10 reduction in the GS-7977 groups compared to 2.8 log10 reduction in the group with PEG-IFN with RBV.
Structure of the HCV polymerase inhibitor, GS-7977 (formerly PSI-7977)
In a Phase IIb trial (ELECTRON) with treatment-naive patients, there were initially four groups, with a GS-7977 monotherapy group added later: GS-7997 +RBV for 12 weeks; GS-7977 +RBV+PEG-IFN to week 4, then GS-7997 +RBV to week 12; GS-7977 +RBV+PEG-IFN to week 8, then GS-7997 +RBV to week 12; GS-7977 +RBV+PEG-IFN to week 12; GS-7977 monotherapy for 12 weeks.
The first four groups all gave 100% SVR, the monotherapy gave 60% SVR. There were no virological breakthroughs. There were no safety issues.
In another Phase IIb trial, GS-7977/RBV therapy was for 12 weeks but comparing treatment-naive patients to those previously non-responders to PEG-IFN/RBV. Both groups had similar reductions in HCV RNA load, 100% of patients reaching the limit of detection (LOD) by 4 weeks and remaining below LOD to week 12. At four weeks after therapy, the proportions of patients achieving SVR were 10/10 and 1/9, respectively. Future studies in patients, who were previously non-responders, will evaluate longer treatment times and combinations with other antivirals.
Phase III studies underway are comparing GS-7977/RBV (12 weeks) with PEG-IFN/RBV (24 weeks).
Breaking news on the development of a new drug for Hepatitis C to tackle its threat in Japan
Shoichiro Goto (Janssen Pharmaceutical K K, Tokyo, Japan)
Japan has a long history of HCV infections, now known to stretch back to World War II. The age distribution in Japan and USA differ: generally >60 years old versus 30–40 years old, respectively.
In Japan, a Phase I study of TMC435, a HCV protease inhibitor, showed that the drug exposure was higher in Japanese subjects than in Caucasians. Therefore, it was necessary to adjust the dose for Phase II studies. In the DRAGON (C215) Phase II study, TMC435 (50 and 100 mg doses) with PEG-IFN/RBV were compared with PEG-IFN/RBV. The duration of therapy was 24 weeks and the primary end point was the SVR at 24 weeks after the end of therapy (SVR24). In the 100 mg group, the SVR24 was 82% (32/39) compared to 46% in the control group. There were no safety issues. Phase III trials in Japan are ongoing and are running in parallel to those in USA/Europe.
Proof of concept trial of novel p7 inhibitor BIT225 in combination with interferon-alpha and ribavirin
Michelle Miller (Biotron Ltd, Sydney, Australia)
Viroporins are a class of proteins encoded by various viruses. Viroporins form ion channels, a well-known example being the M2 protein of influenza virus. HCV p7 is a 63 aa protein with ion channel activity and, in chimpanzees, it is essential for virus assembly and release. BIT225 was selected as a lead compound targeting HCV p7. For preclinical studies, the HCV replicon assay is not suitable for this type of compound and so bovine viral diarrhoea virus (BVDV) was used as a model virus. In a Phase I study, ascending doses (35 to 600 mg) were well tolerated. The drug half-life was 7 h. In a Phase Ib study with HCV patients having any genotype, there was about a 0.5 log10 reduction in viral load.
In a Phase IIa trial, BIT225 (400 mg, 200 mg or placebo) was added to PEG-IFN/RBV for 12 weeks therapy. At the end of treatment, the proportion of patients achieving complete viral response (<50 IU/ml) were 86%, 88% and 63%, respectively. The mean reduction in HCV load was about 4 log10 for the drug groups versus about 3 log10 for the control.
Mini-symposium - therapy of infections endemic to Japan and Asia
Kouichi Morita (Nagasaki University, Nagasaki, Japan) gave a good overview of Japanese encephalitis virus (JEV), starting with its history in Japan. In the 1870s, there were episode of ‘summer encephalitis’. In 1924, there was a great epidemic with 6,125 cases of which 3,797 died. JEV was first isolated from a fatal case in 1934, reported in 1935, and isolated from a mosquito in 1938. More recently, JEV has spread to Australia. There are five genotypes, each seems to have mutated in the tropical regions and then spread to other regions. In many cases, the infection is essentially asymptomatic but if the virus survives the initial immune response, then it can spread to the brain to cause serious disease. There is no treatment available but the disease can be prevented by live attenuated or inactivated vaccines.
There are two viral proteins which are potential targets for antivirals, NS3 (helicase and protease) NS5 (polymerase). At the Tokyo Drug Centre, their database was used for in silico screening. From 207,239 molecules screened, 1,878 were initial hits, 100 bound to the receptor, 8 suitable for testing in Vero cells, 2 being toxic, giving 6 showing activity. Of these active compounds, three were shown to be active also against West Nile virus (WNV).
HTLV-1 was the first human retrovirus to be discovered over 30 years ago. Jun-ichi Fujisawa (Kansai Medical University, Osaka, Japan) summarised the current situation. HTLV-1 has an incubation period of 40 to 60 years, it can be spread via breast milk, sex or blood transfusion. Clinical treatments, with either chemotherapy or transplantation, have poor outcomes. Antiviral treatment gives good five-year survival rates for patients with chronic disease but these are poor (28%) in acute disease. The development of an HTLV-1-infected, humanized mouse model was described. This opens the way to investigate new compounds with potential for therapy.
Interest in influenza virus has been greatly stimulated by the emergence of H5N2 avian and H1N1 pandemic viruses. Simon Tucker (Biota Holdings Ltd, Notting Hill, Victoria, Australia) reviewed the recent occurrence of influenza strains and their therapy. The South East Asia region seems to be the original source of many new strains of influenza but these often return later. The H1N1 pandemic strain is notable by its absence in Japan where the H3N2 is the most common strain. Resistance to adamantine is virtually 100% but there is little resistance to the neuraminidase inhibitors (NIs). The avian H5N1 strain is still causing concern although much less common than at its peak in 2006. Up to February 2012, there had been about 500 cases and many deaths.
Asia has a rich pipeline of anti-influenza compounds. Japan has been the highest user of NIs, mostly oseltamivir (oral) but also zanamivir (inhaled) and peramivir (intravenous). Together with routine diagnostic testing, health and cost benefits are emerging. More recently, laninamivir octanoate (single dose inhaled) was approved in Japan. In the 2011 season, the most used drugs were oseltamivir (50%) and laninamivir octanoate (35%). As elsewhere in the world, the oseltamivir-resistant mutant, H274Y, has been detected but remains infrequent. Looking forward, favipivavir (T705) NDA was filed first in Japan and is in Phase III trials in the USA. In answer to a question about resistance, Don Smee has tried to find favipivavir resistance in cell culture but so far without success. Another promising compound, dimeric zanamivir, is in preclinical studies.
The meeting was fortunate in having Takaji Wakita (National Institute of Health, Tokyo, Japan) speak about HCV replication models. Although a sub-genomic HCV replicon system was published in 1999 [15], an infectious HCV system was not available until Takaji published his work on the JFH-1 HCV in HuH7 cells [16]. Having established a successful method with HCV genotype 1 (GT-1), Takaji tried to find similar systems for other genotypes. A replicon system with GT-2 was successful but no infectious virus was obtained. Mutations in the NS5A gene were introduced to make it similar to that in GT-1. Some infectious virus was detected but with low infectivity. More mutations led to improved infectivity but, after 25 days in culture, the infectivity dropped markedly. Working with GT-3, it was possible to get a replicon system after introducing some mutations.
A replicon system is fine for testing compounds acting on the earlier stages of virus replication but not for compounds targeting the late stages of replication. Takaji introduced JFH-1 virus RNA into cells by electroporation. Virions, albeit non-infectious, were released into the medium and this system enabled compounds to be tested.
Pei Yong Shi (Novartis Institute for Tropical Diseases, Singapore) presented three examples to illustrate different approaches to anti-dengue drug discovery. The virus has two proteins with multiple enzymatic activities. NS5 codes for a methyltransferase and the polymerase, NS3 codes for a protease, helicase, NTPase and 5′-RTPase.
An adenosine analogue, NITD-008, inhibits several flaviviruses, including dengue fever virus. It has good pharmacokinetic properties, is well absorbed orally (bioavailability about 50%) and is metabolized inside cells to the mono-, di- and tri-phosphate. In a dengue mouse model, either a single dose of 75 mg/kg or 10 mg/kg twice daily for 3 days gave a good effect with 100% survival versus 0% in the placebo group. Unfortunately, a two-week toxicity study in rats showed severe side effects. The reason for this is being investigated.
The second approach was to target the viral methyl transferase activity which is essential for dengue virus replication. Examination of the crystal structure revealed a hydrophobic pocket not present in human transferases. A compound was found which binds selectively to the viral transferase. To obtain proof of concept, drug and transferase were co-crystallised. This is an area for further work. The third approach was to inhibit pyrimidine synthesis. Although a model compound gave apparent antiviral activity in cell culture, reversible by added excess uridine, it was ineffective in vivo as there is too much uridine in blood.
Hepatitis B: Is a cure possible, is it necessary?
These questions were posed by Timothy Block (Hepatitis B Foundation and Drexel University, Philadelphia, PA, USA).
Currently, only about half of patients with chronic hepatitis B fall within the guidelines for treatment.
For patients 50 years old, the ten-year mortality risk is between 3 and 10%. Drug treatment halves this risk. Hepatitis B cccDNA serves as a long-lived viral reservoir, surviving in the infected hepatocytes for their lifetime, days to weeks. It was reported recently (2012) that cccDNA can be reduced by IFN treatment [17]. Therapy with current nucleoside and nucleotide analogues can lead to HBsAg seroconversion which is seen as a functional cure. However, the proportions of patients are very low, about 2% per year on therapy. Another approach is needed. Therapeutic vaccines have been tested with disappointing results but these need to be evaluated in combination with antivirals. Several vaccine trials are ongoing.
Mini-symposium - clinical development of antiviral agents
The progression of CMX001 through clinical trials was summarised in the Elion Award Lecture. Scott Foster (Chimerix, Inc., Durham, NC, USA) described a Phase II prophylaxis trial in stem cell transplant patients, for about ten weeks (range 9 to 11 weeks) to week 13 after transplantation. CMX001 was administered either once (100 and 200 mg) or twice (100 and 200 mg) weekly versus placebo. At baseline, patients had to be CMV DNA-negative. Proportions of patients with CMV viraemia (>1,000 copies/ml) at any time were 6, 7, 4 and 2 versus 25%, respectively.
Considering only those patients who did not have viraemia within 24 h after the start of treatment, the proportions were 2, 2, 0 and 0 versus 15%, respectively (P<0.002 for twice weekly dosing).
In an investigation of viral resistance, a mutation (R1052C) was detected at baseline in one patient and in two at viral breakthrough. As position 1052 is beyond the usual region for resistance mutations, recombinant virus will be used to test if this is a true resistance mutation. Six other changes were detected but their significance is, as yet, unknown. One other mutation, known from cell culture studies, was not detected.
MiR-122 is a micro-RNA present in liver cells. There are two six-nucleotide regions within the 5′UTR of HCV, known as seed sites 1 and 2 (S1 & S2), which bind to miR-122. This step is essential to protect the viral RNA from degradation and for HCV replication. The sequence of S1 and S2 are highly conserved through all genotypes of HCV. Miravirsen (SPC3649) is a 15-olignucleotide which binds to miR-122 and so blocks the binding to HCV. In cell culture studies, HCV resistance has not been detected. In vivo, the terminal plasma half-life is about 4 weeks. Miravirsen is the first in class to reach clinical trials.
Amy Patick (Santaris Pharma, San Diego, CA, USA) reported on a Phase II proof-of-concept, dose-escalating trial in treatment-naive patients with genotype 1 chronic HCV infection. Miravirsen (3, 5 or 7 mg) was given weekly for 4 weeks (5 doses) and the patients were followed to week 18. There was a clear dose response and treatment was well tolerated. With the 7 mg dose, 4/9 patients became HCV RNA undetectable. The miR-122 binding region of HCV RNA from every patient was sequenced at baseline, at 5 weeks and at 4 weeks if the virus levels had risen above the nadir. No changes were detected in the miR-122 binding region. Further trials, in combination with other antiviral compounds, are planned.
Some pyrimidinediones have been evaluated as potential microbicides to prevent the transmission of HIV-1. Bob Buckheit (ImQuest BioSciences, Inc., Frederick, MD, USA) summarised the work with IQP-0528 as the lead candidate and with IQP-0532 as back-up candidate. These NNRTIs have been combined with tenofovir and formulated in various forms, as gels, as vaginal film and in rings, the rings staying in place for 30 days. These have been evaluated in monkeys. The vaginal film was particularly good in delivering high drug concentrations quickly, within 30 min.
As an aside, developing really effective topical microbicides has proved to be a huge challenge but it is good to hear of further work in this area – the potential to limit the spread of HIV is such a worthwhile aim.
Mini-symposium - building a better clinical candidate: Issues, strategies and tools
The concept of prodrugs is well accepted and exploited within the antiviral field but less so in other areas. Reza Oliyai (Gilead Sciences, Foster City, CA, USA) summarised the role of prodrug design in drug discovery and lead optimization. Initially, a typical aim for using a prodrug was to increase bioavailability from oral administration. More recently, the prodrug approach has had a variety of aims, including: aqueous solubility, permeability, chemical or physical stability, targeting a particular tissue, by-passing an inefficient enzymatic step.
Some examples were given. Fosamprenavir, a prodrug of amprenavir, increases aqueous solubility and reduces the number of tablets from 16 to 2. Because the conversion back to the parent drug is so quick, the oral absorption is unaltered. Tenofovir disoproxil fumarate (TDF) gives improved bioavailability of tenofovir (TFV). Even better, the drug can be delivered into cells. An amidate prodrug, GS-7340, was selected due to its stability while in circulation but is converted to the active drug inside the cell (Clinical Symposium, Quad: a new single tablet antiretroviral regimen).
As an alternative to the prodrug approach, novel formulation options may be useful, for example, the use of liquid crystals. The bioavailability and trough concentrations of omeprazole have been improved by a new formulation.
Although high throughput screens give hits, many of these will never become useful drugs. Robert Zahler (PharmD Consulting, New York, NY, USA) explained why this is so in his presentation on reactive metabolites in drug discovery: implications, anticipation and management. There are well-recognised chemical entities which are associated with toxicity. Many examples were given: cyclopropyramines give radical cations which attack proteins; hydrazines are carcinogenic in rats – as always, there may be exceptions such as atazanavir, a PI for HIV; anilines and nitro-aromatics may be mutagenic; thiomides and thioureas (often seen in screening hits) form a complex with iodine which reacts with proteins and may be hepatoxic; olefins and alkynes are subject to a ‘health warning’ but may be alright – entecavir is a useful drug as it is not subject to oxidative metabolism; electron-rich heterocycles, for example thiophenes, are hepatoxic to 1% of patients; quinones and iminoquinone may be hepatoxic – for example, troglitazone (for diabetes) was withdrawn due to hepatoxicity.
Risk mitigation: be aware of the potential problems and evaluate the candidate compound in the appropriate tests. For instance use the Ames test for potential genotoxic compounds. Irreversible binding to proteins should be taken as a warning sign.
Paul Scola (Bristol–Myers Squibb, New York, NY, USA) illustrated the process for improving an initial compound into a drug candidate using, by way of example, the discovery of asunaprevir (BMS-650032), a PI for the treatment of HCV.
The initial chemical design was to obtain a good fit into the HCV protease. This was guided by information from X-ray crystal determinations. Then, carefully chosen parts of the molecule were modified to improve the physical properties. This led to BMS-781 but its oral bioavailability was <10%. A rat model, testing the blood drug concentration at 4 h, was used to guide further modifications. The best candidates were tested for trough concentrations greater than ten times the EC50 value. This led to BMS-605339. Previously, in clinical trials, there had been some ECG changes and mild bradychardia. Looking back at the preclinical studies, such effects had been seen in the dog but only at >50-fold the human dose. An isolated rabbit heart model allowed direct assessment of pacemaker function and was as sensitive as in the clinic. This led to BMS-650032 (asunaprevir).
Asunaprevir, a PI, is active against HCV genotype 1, less so against other genotypes. Having shown activity in a Phase II trial, it is being progressed in combination with daclatasvir, an NS5A inhibitor.
As an aside, immediately after the conference, on the 19 April 2012, BMS announced a mid-stage study result of the combination trial. At 24 weeks after the end of treatment, 77% of patients were cured.
The development of another HCV PI, ACH1625 (sovaprevir), was reported by Avinash Phadka (Achillion Pharmaceuticals, New Haven, CT, USA). As in the previous presentation, the first step was to optimise the drug-protease interaction. Eventually, this led to sovaprevir: EC50 11 nM, EC90 31 nM, 50% cytotoxic concentration (CC50)>31,000 nM, metabolically stable in human subjects, although much less so in monkeys. After intravenous administration to rats, the plasma half-life was only 0.3 h but this was due to the drug going to the liver. The area under the curve (AUC) for liver was about 130 times that for plasma. With a dose of 10 mg/kg, the drug concentration in the liver at 24 h was over the EC50 value.
In a Phase Ia trial, doses of sovaprevir from 200 mg twice daily to 600 mg once daily were tested and HCV RNA levels were reduced by up to 3 log10. In the first segment of a Phase IIa trial, sovaprevir (200, 400 and 800 mg) was combined with PEG-IFN/RBV for 28 days followed by PEG-IFN/RBV only for 44 weeks. The highest dose gave 5 log10 reduction in HCV RNA. The proportions of patients with undetectable (<25 IU/ml) early viral levels at 12 weeks (EVR12) were 56%, 75%, 47% and 27% (PEG-IFN/RBV), respectively. A second segment of the trial was initiated in which sovaprevir and PEG-IFN/RBV were given for 12 weeks, followed by PEG-IFN/RBV only for 12 weeks if the patient had achieved EVR12, otherwise for 36 weeks. Although this segment is still ongoing, the initial results are more encouraging. The proportions of patients achieving EVR12 were 95%, 75%, 100% and 27%, respectively. No safety issues have been identified.
In a follow-up programme, an aim was to have a PI active against all genotypes and against known resistant viruses. ACH2684 has good activity against genotype 1 (EC50 0.18 nM, EC90 0.28 nM), genotype 3 (EC50 4.8 nM) and the other genotypes (EC50<1 nM). It does not inhibit CYP enzymes (IC50>5 μM). In a Phase I trial, ACH2684 seems to be at least as active as sovaprevir.
Dominique Surleraux (Idenix, Cambridge, MA, USA) gave an overview of how their anti-HCV programme has developed recently, starting with IDX184, a 2′-C-methylguanosine nucleotide liver-targeting prodrug. In a Phase IIb trial, two doses of IDX184 (50 and 100 mg) were coadministered with PEG-IFN/RBV for 12 weeks with response-guided extension of PEG-IFN/RBV. The EVRs were 81% and 93%, respectively. There were no safety issues.
As an aside, in an HCV replicon system, resistance to IDX184 has been seen after 25 days, the mutation being S282T. The mutant virus has about 5% replication capacity. Although investigated in samples from clinical trials, no resistance has been detected, nor has the known S282T mutant been found. This suggests that IDX184, in common with other nucleoside/nucleotide polymerase inhibitors, presents a high genetic barrier to HCV.
Choosing another HCV target, IDX719 is an NS5A inhibitor (see Prusoff Lecture above). A dose-ranging (5 to 100 mg once daily) Phase I study in patients with genotypes 1, 2 or 3 was started in January 2012. At the 100 mg dose, there was a 3.7 log10 reduction in HCV RNA. Just starting, a proof-of-concept study will evaluate IDX719 (100 mg dose once daily for 3 days) in patients with genotypes 1, 2, 3 or 4. It is planned to start a combination trial of IDX184 and IDX719 in late 2012.
Since January 2011, the group has synthesised 1,500 nucleotides with a focus on prodrugs containing a single diastereoisomer. The lead candidate is IDX19368. It is expected that the IND will be submitted in mid-2012.
Contributor presentations
Immune response to Fluzone™ and FluMist™ vaccines in BABL/c mice and efficacy against A/CA/04/2009 (pandemic H1N1) virus challenge
Bart Tarbet (Utah State University, Logan, UT, USA)
Two trivalent vaccines for seasonal influenza, Fluzone™ (inactivated) and FluMist™ (attenuated), were evaluated for efficacy in mice. Three concentrations of each vaccine were tested but this summary will focus on just the full dose. This study compared the efficacy against a virus challenge and investigated the immune responses following vaccination (Table 1).
Summary of test results with Fluzone™ and FluMist™
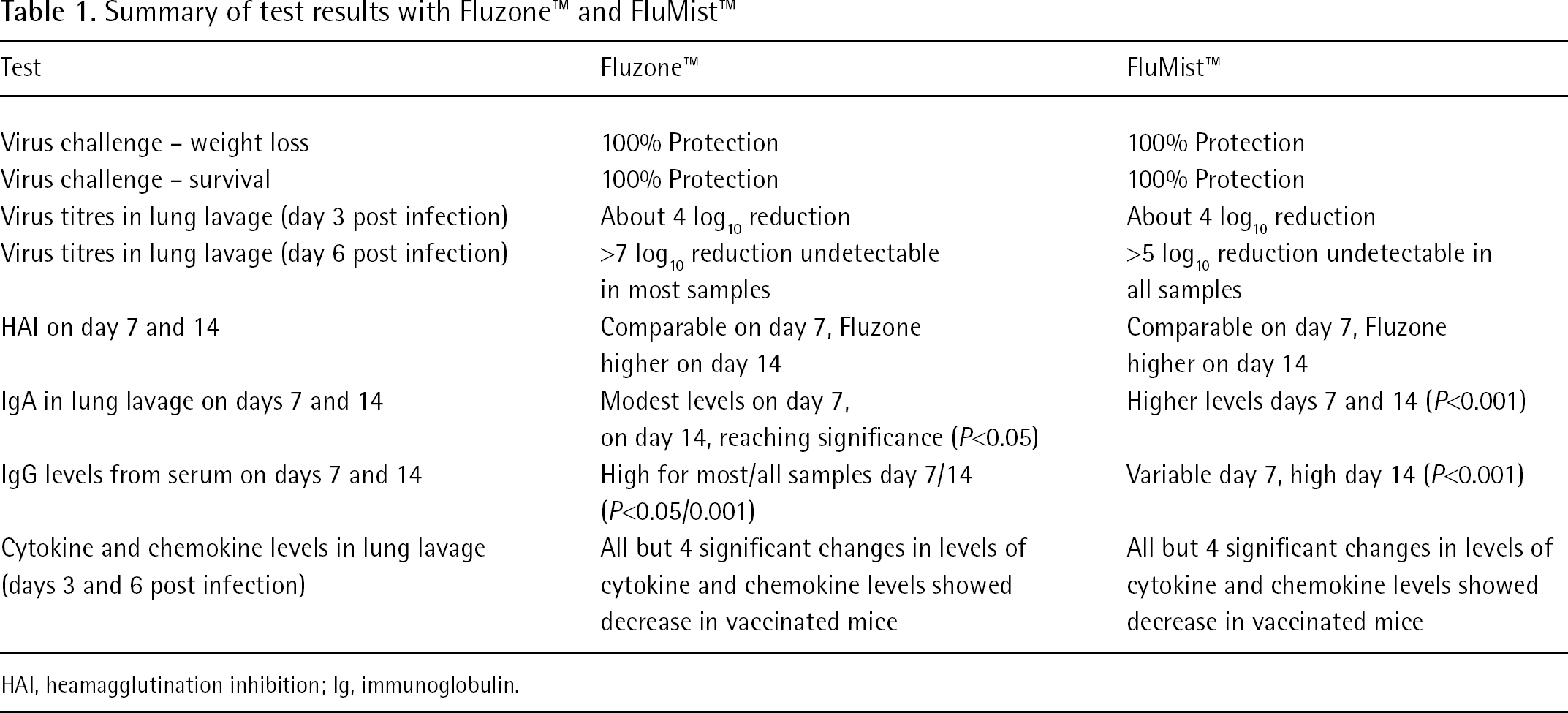
HAI, heamagglutination inhibition; Ig, immunoglobulin.
Both these vaccines had good efficacy against an influenza strain which was included in the vaccines. Influenza virus-specific antibodies are currently being evaluated by virus neutralization against homologous and heterologous influenza subtypes.
Mechanism of Ivermectin-mediated flaviviral helicase
Eloise Mastrangelo (University of Milan, Milan, Italy)
Flaviviruses are ssRNA viruses such as dengue (DENV, 4 serotypes), yellow fever (YFV), WNV, JEV and tick borne encephalitis (TBEV) viruses. At ICAR 2011, Eloise reported that Ivermectin, which has been used clinically for the treatment of various parasitic diseases for more than 20 years, was active against flaviviruses. Based on the crystal structure of Kunjin viral helicase, the authors hypothesized that the ssRNA entrance site (α-helical gate) could be a drug target. An in silico docking search led to the identification of some candidate compounds, including Ivermectin. This presentation described the studies investigating the mode of action of Ivermectin.
Double mutants of YFV (Thr413Ile and Asp414Glu), DENV (Thr408Ile and Asp409Glu) and WNV (Thr409Ile and Asp410Glu) were prepared and tested in comparison with the corresponding wild-type viruses. This work confirmed that these mutations abolished the activity of Ivermectin. Enzymatic assays using helicases from YFV, DENV and WNV demonstrated that Ivermectin acts as an uncompetitive inhibitor (inhibition constant [Ki] values were 0.019, 0.35 and 0.18 μ M, respectively).
It would be interesting to obtain any anecdotal evidence for Ivermectin having activity against flaviviruses during its clinical use as an anti-helmintic drug.
Sensitivity of clinical isolates to helicase primase inhibitors and no detection of resistance mutations above background frequency
Hugh Field (University of Cambridge, Cambridge, UK)
The helicase primase inhibitor, AIC316 (BAY 57–1293), is being evaluated in clinical trials for efficacy against herpes simplex virus (HSV)-1 and HSV-2 (Excellent efficacy and pharmacokinetics of AIC316, a novel drug against herpes simplex virus (HSV) types 1 and 2 in preclinical and Phase I/II studies by Alexander Birkmann). The purpose of this study was to seek evidence of drug-resistance in clinical isolates of HSV-1 and HSV-2 obtained from the USA prior to the clinical use of this class of inhibitors. Clinical isolates of HSV-1 (27) and HSV-2 (32) were obtained, passaged once in Vero cells and stored at −70°C. In a standard plaque reduction assay, all the isolates would be regarded as sensitive but it was notable that the spread of EC50 values, and especially the spread of EC90 values, was greater for HSV-2 isolates than HSV-1 isolates (mean EC90 values for HSV-1 and HSV-2 were 0.045 μM [sd 0.015] and 0.16 μM [sd 0.10], respectively).
All isolates (about 106 PFU in Vero cells) were screened in the presence of AIC316 to detect drug-resistant mutants that may be present at low frequency. All plaques were recorded and single plaques were passaged once in the absence of AIC316. Following confirmation of AIC316-resistance, those regions, within the UL5 (helicase) and UL52 (primase) genes which are associated with resistant mutations, were sequenced. Among the HSV-1 isolates, 19/27 (70%) yielded at least one resistant plaque. The most common mutations were at position K356 in the helicase gene. In addition, three plaques had a mutation (A899T) in the primase gene. It was noted that all these mutations occurred at low frequency (approximately 10−6). Somewhat similar results were obtained with the HSV-2 isolates, 8/31 (25%) yielding at least one plaque, the most common mutation site was in the corresponding position (K355) of the helicase gene and only one plaque had resistance in the primase gene, again at the corresponding position, A905T. The one remarkable difference was that one of the HSV-2 plaques had the helicase gene mutant, G351R, at relatively high frequency (approximately10−4; Table 2).
Pre-existing mutations giving resistance to AIC316, a helicase primase inhibitor
Bold represents presence at high frequency (10−4); mutant isolated from US2/22 which was from an immunocompromised patient. All others at about 10−6 frequency.
Although the clinical significance of these pre-existing mutations is unknown, one may speculate. Perhaps, in immunocompetent patients, the small amount of drug-resistant virus will be adequately controlled by the immune system. In immunocompromised patients, such pre-existing resistant mutants may lead to early appearance of resistant virus causing clinical problems.
Excellent efficacy and pharmacokinetics of AIC316, a novel drug against herpes simplex virus (HSV) types 1 and 2 in preclinical and Phase I/II studies
Alexander Birkmann (AiCuris GmbH & Co, Wuppertal, Germany)
When introduced over a decade ago, acyclovir (then its prodrug, valaciclovir) and famciclovir made a major impact on HSV disease, however, there has been a notable absence of new therapeutic options since that time. In contrast to acyclovir and famciclovir, which inhibit the viral polymerase, AIC316 inhibits the helicase/primase complex, as confirmed by resistance studies. In cell culture studies, all mutations locate in the UL5 (helicase gene) or less commonly in UL52 (primase gene).
In a Phase I dose-escalation study, doses of 5, 25, 100 and 200 mg were administered. The terminal half-life was long, up to 80 h. There were no safety issues. A Phase II trial investigated HSV-2 mucocutaneous shedding. HSV shedding is thought to be of high relevance for patients. There may be a brief, low copy episode (⩽104 HSV DNA copies), prolonged subclinical episode (104 to 106 HSV DNA copies) assumed to be mainly associated with transmissions, and clinical disease (peak copy number ⩾106). The treatment was for 28 days with 5 mg once daily, 25 mg once daily, 75 mg once daily or 400 mg once weekly versus placebo. There was a dose-dependent reduction in virus shedding rates (21.2%, 9.2%, 2.0% and 5.2% and 16.5%, respectively). The corresponding lesion rates were 13.3%, 4.6%, 1.1%, 1.3% and 9.1%, respectively. The total numbers of recurrences were 18%, 12%, 3%, 6% and 12%, respectively. No safety issues have been identified. There has been no emergence of resistant virus in subjects in the Phase II trial, an encouraging result albeit the number of subjects is small.
Therapeutic efficacy of ST-246 in cynomolgous macaques challenged with monkeypox virus via aerosol
Andrew Russo (Lovelace Respiratory Research Institute, Albuquerque, NM, USA)
As yet, there are no compounds approved by the FDA for general use against orthopoxvirus infections. ST-246 is being progressed through the ‘animal rule’ process. This presentation summarized two studies investigating how late treatment could start after infection with monkeypox and still be effective. In the first study, ST-246 treatment (10 mg/kg once daily for 14 days) was initiated on days 1, 2, 3 and 4 post infection. Having achieved essentially full protection in all treated groups, a second study looked at treatments starting day 4 (to give an overlap), days 5, 6, 7 and 8. The study was continued to day 45 post infection for observation.
Various parameters were followed. In the placebo group, serum C-reactive protein (CRP; parameter for liver inflammation) rises at day 3 and weight loss was mainly from days 4 to 6. Early treatments abolished CRP rise and later treatments reduced the levels quickly. When treatment was started on day 4 (D4), there was only slight weight loss. D5 gave quick recovery from the weight loss already present. Viral loads were measured. Early treatments, D1 and D2, prevented virus from reaching detectable levels. D3 gave a large reduction in viral load although virus was detected. D4 gave about 1 log10 reduction in peak viral load. D5 had a high viral load but this reduced quickly. All animals in groups D1 to D5 survived, D6 4/6 (67%) survived, D7 100% survived and D8 3/6 (50%) survived. Even D8 had a better survival than placebo, 2/8 (20%). This is yet another example in which ST-246 has shown excellent therapeutic activity.
Towards virus eradication: Excision of HIV-1 proviral DNA using LTR-specific recombinase
Joachim Hauber (Heinrich Pette Institute - Leibniz Institute for Experimental Virology, Hamburg, Germany)
The ‘Berlin Patient’ gives hope that a cure for HIV may one day be possible. This HIV-infected patient had a bone marrow transplant to treat myeloid leukaemia. Importantly, the transplanted cells were also homozygous with respect to the HIV-1 CCR5 coreceptor inactivating Δ32 allele. Remarkably, for more than four years, the patient has remained free of detectable HIV without any HAART. A functional cure, in which the immune system is able to fully control HIV replication, would be a great step forward but even better would be an approach which eliminates HIV proviral DNA.
It is possible to use molecular evolution technology to make a Tre-recombinase highly specific for the long terminal repeats (LTR) of HIV. In a culture of CEM T-cells, infected with HIV and treated with recombinant Tre, the excised HIV DNA can be detected. Moreover, a novel lentiviral SIN-vector gives conditional expression of Tre-recombinase (LV+Tre). First, LV+Tre safety was checked in vitro over a 15-week period. Then LV+Tre and LV-control were used to genetically alter primary human CD4+ T-cells. Mice were transplanted with these engineered T-cells, subsequently infected with HIV, and virus replication was followed over time. The active treatment was better than the control (P<0.001), significantly suppressing viral load. Interestingly, one mouse in the treated group appeared to be free of HIV (LOD=20 copies HIV RNA/ml) at 24 weeks post infection. This mouse has been given the title ‘Hamburg Mouse’!
It may be possible to use drugs which activate resting, HIV-infected cells, thereby making those cells susceptible to antiviral therapy and immune attack. Maybe an improved approach is its combination with lentiviral vector-mediated conditional expression of Tre, which targets the LTR sequences of HIV. These LTRs are about 30 bases long and the Tre target sequence is highly conserved. A very important advantage with this Tre-enzyme is that the host DNA is not targeted and therefore no loose DNA ends exist. Instead, Tre excises specifically the HIV DNA and accurately rejoins the ends at the chromosomal integration site.
Conclusion
This ICAR meeting had many interesting presentations. Perhaps more than in recent years, reports on clinical results and on chemistry-related topics were well represented. It is impossible to record all these in this review, but it is hoped that this report is indicative of the range of topics covered.
Personally, I found the major award lectures, the keynote address, the clinical symposium and the mini-symposia gave me a good overview of important areas of antiviral research. Those presentations, reporting clinical results, proved without doubt that antiviral chemotherapy is continuing to make important new progress. For HBV therapy, resistance to tenofovir has not yet been seen, even after 5 years of treatment, but the proportion of patients having a ‘cure’ (becoming HBsAg seropositive) is still low (about 10%) although slowly increasing. The outlook for HCV is most promising. Inhibitors of the HCV polymerase (GS-7977 and IDX184) and the HCV NS5A inhibitors (BMS-790052, GS-5885 and BMS-624393) are active against all genotypes and likely, when combined, to offer effective cures. I am not so sure of a future role for the HCV PIs. Generally, the PIs have activity against only one genotype and they present a lower genetic barrier to HCV than do the HCV polymerase inhibitors. A new approach, using a micro-RNA, miravirsen, has shown encouraging positive results in a Phase II trial. For over a decade, the mainstays of HSV and VZV therapies have been acyclovir, its prodrug valaciclovir and famciclovir. A helicase/primase inhibitor, AIC-316, may be the first new alternative for HSV infections. Similarly, valganciclovir has been the key therapy for CMV infections but now CMX001 and AIC246 are showing promise in Phase II trials. New advances in HIV treatment were illustrated by festinavir (in early clinical trials) and Quad, a new convenient combination pill. A contributor presentation gives hope that a cure for HIV infection may one day be possible. Influenza virus therapy is set to change with an NDA for favipivavir, filed first in Japan. To protect against bio-terrorism, ST-246 has again shown excellent therapeutic activity, this time in a monkey pox model of smallpox.
A notable absence, this year, was a presentation on apricitabine (ATC). Over several years, Susan Cox (formerly Avexa, Richmond, Victoria, Australia) has described the progress of this nucleoside HIV RT inhibitor from preclinical studies through to long-term clinical studies. Since ICAR 2011, Avexa have been looking for a partner to progress ATC through to approval but little progress has been made. In a company statement dated 23 November 2011, Avexa announced that two new patents had been filed following the full analysis of the Phase II/III data which showed that ATC, with two marketed drugs, gave greater synergistic benefits than were expected. Previously, ATC had shown a remarkably high genetic barrier to HIV resistance and a good safety profile. Using the synergy recently reported, ATC could be included in a combination pill to provide a valuable alternative to current therapies. I hope to hear another presentation on ATC at ICAR 2013.
A great strength of ICAR meetings is the potential for cross-fertilization between differing areas. This ICAR had three mini-symposia covering diverse topics such as history of virus outbreaks, prodrugs and which chemical entities not to include in a candidate drug. I would like to add my thanks to the ISAR Officers and Conference Committee, and to our co-sponsors JAAT, for organizing another successful ICAR meeting, and to Masanori Baba, our local host, for such a warm welcome to Sapporo.
Footnotes
The author declares no competing interests.