
Abstract
In the search for new anti-influenza agents, the viral polymerase has often been targeted due to the involvement of multiple conserved proteins and their distinct activities. Polymerase associates with each of the eight singled-stranded negative-sense viral RNA segments. These transcriptionally competent segments are coated with multiple copies of nucleoprotein (NP) to form the ribonucleoprotein. NP is an abundant essential protein, possessing operative and structural functions, and participating in genome organization, nuclear trafficking and RNA transcription and replication. This review examines the NP structure and function, and explores NP as an emerging target for anti-influenza drug development, focusing on recently discovered aryl piperazine amide inhibitor chemotypes.
Introduction
Influenza virus is a single-stranded negative-sense segmented RNA virus and a member of the Orthomyxovirus family. Influenza virus causes an acute transmittable infection of the respiratory tract mucosa. Global epidemics are responsible for an estimated 250,000–500,000 deaths annually [1]. Cyclical emergence of new strains with increased pathogenicity can occur via accumulation of mutations in a process called ‘antigenic drift’. Alternatively, a new strain may arise from a human virus acquiring gene segments from swine or avian strains during coinfection, referred to as the ‘antigenic shift’. These pandemic human strains usually have a higher potential for morbidity and mortality due to the lack of a pre-existing immune response. The 1918 flu pandemic was extraordinarily virulent, with death estimates exceeding 50 million [2]. In 1997, several people died in Hong Kong after contracting highly pathogenic H5N1 avian influenza [3–5]. Additional cases surfaced throughout South Asia in 2003–2005, with mortality rates approaching 60% [6]. With the potential threat of an H5N1 pandemic more severe than the 1918 Spanish flu, both the search for new influenza targets and the screening for novel chemotypes as inhibitors of influenza replication have escalated [7].
Two antiviral classes have been approved for the treatment of influenza infection: M2 ion channel blockers (amantadine and rimantadine) and neuraminidase inhibitors (zanamivir, oseltamivir, peramivir and laninamivir, with the latter two currently approved only in Japan). Adamantanes were the first anti-influenza drugs approved. Their mechanism of action interferes with the ion channel activity of the viral M2 protein that is required for uncoating [8,9]. Deficiencies of adamantanes include lack of activity against influenza B [10], central nervous system side effects [11] and rapid resistance development [12,13]. In fact, close to 90% of all circulating H3N2 viruses and initially 100% of swine-origin H1N1 subtype were resistant to amantadine or rimantadine, while 30–50% of other H1N1 isolates remained susceptible [14,15].
The neuraminidase inhibitors halt nascent virus release (budding) from infected cells and reduce influenza mobility within the airway mucosa, via inhibition of the viral enzyme [16–20]. The two neuraminidase inhibitors available worldwide are zanamivir (Relenza®) and oseltamivir (Tamiflu®). A shortcoming of zanamivir is its poor oral bioavailability [21,22], requiring it to be administered as an inhaled aerosolized powder [23,24]. Oseltamivir is formulated as an oral prodrug [25,26] and is the more widely used agent. However, resistance to oseltamivir has been generated through clinical use, which began to appear in Europe in 2007 [27]. By 2008, the circulating H1N1 viruses were essentially resistant due to an H274Y mutation [28] and, although rare, some H5N1 cases were resistant as well [29]. In 2009, the swine flu pandemic strain of H1N1 (HI1NI pdm09) essentially replaced the circulating oseltamivir-resistant H1N1 viruses. This emergent virus strain is sensitive to oseltamivir, but with its use, resistance continues to develop [30,31].
One way to combat resistance would be through the use of combinations of antivirals against different targets, a strategy that has worked well with viruses such as HIV [32–36]. Thus, the search for inhibitors of other targets in influenza virus has intensified. The viral polymerase, whose proteins comprise more than half of the coding sequence of the viral genome, and which has many potential targets for intervention, has been a key part of this effort. One potential route revolves around inhibitors targeting the influenza nucleoprotein (NP) [37–40]. NP is involved in viral transcription and replication, which entails binding to viral RNA (vRNA) and complementary RNA (cRNA). NP possesses many viral and host binding partners during various stages of the virus life cycle. An overall examination of NP structure and function reveals a versatile protein with multiple functions for antiviral targeting. This article will initially review what is known about the NP protein, and then discuss the identification of novel inhibitors targeting NP and their potential mechanisms of action.
Nucleoprotein structure and function
NP association with viral RNA and polymerase Influenza viruses have their genomic RNA associated with multiple copies of NP and a heterotrimer polymerase complex comprising the PB2, PB1 and PA protein subunits to form stable ribonucleoproteins (RNPs) [41]. Four out of the eight influenza genomic segments encode proteins essential for messenger RNA (mRNA) and vRNA synthesis [42]. Segment 5 encodes the RNA-binding protein NP [43], while the three largest segments encode the proteins of the heterotrimeric polymerase [44]. The polymerase is associated with the ends of the vRNA [45], while the remainder of the vRNA is encapsidated by NP. These transcriptionally competent segments are called RNPs [46].
NP is the structural unit of RNPs [47]. The NP monomer is a 56 kDa basic protein [48,49] with inherent single-strand RNA-binding properties, while possessing no sequence specificity [50]. RNA segments are encapsidated by NP with a periodicity of one NP for each 24 bases of RNA [47,51]. NP binds to vRNA via the ribose-phosphate backbone, leaving the bases exposed to solvation, ribonuclease digestion and, most importantly, accessibility to the polymerase as a template for transcription [50].
Electron micrographs of RNP reveal double-helical rod-like bodies [47,52]. Thin sections confirm that virions generally contain eight segments, organized with seven surrounding a central eighth [53,54]. The twisted RNP rods are folded and coiled back on themselves [53]. The self-oligomerization property of NP alone may give rise to overall shape and organization of RNPs. No discernable differences in higher order structuring of RNPs was observed upon RNA removal [55] or after RNA substitution with negative-charged polymers [56].
The first X-ray structure of NP indicated that H1N1 NP–NP oligomerization is mediated via a flexible tail loop that can insert into a neighbouring NP molecule [57]. The subsequently solved H5N1 NP structure has a majority of features in common with the H1N1 structure [58]. Both crystal structures revealed that NP is crescent-shaped with head, body, RNA-binding and tail domains (Figure 1A). The putative RNA-binding site is a groove situated between the body and head regions, encompassing 16 basic residues, 11 of which are conserved in representatives of influenza A, B and C strains [58].
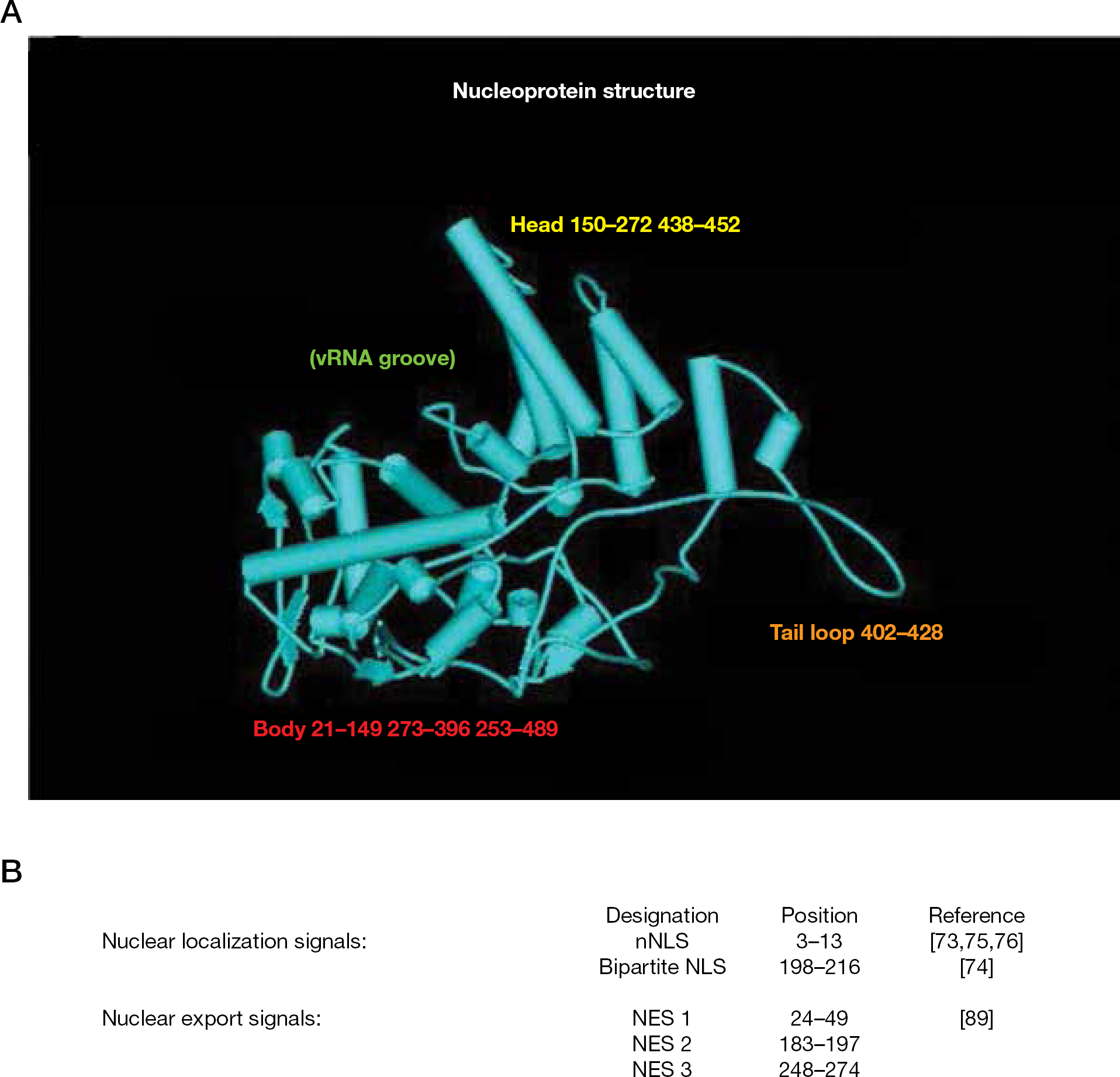
Influenza nucleoprotein structure
Purified NP preparations resolve into monomer, trimer and larger oligomer fractions on a size exclusion column, but H1N1 and H5N1 NPs crystallize exclusively as trimers [57,58]. Two single mutations in the tail loop (R416A and E339A) or the deletion Δ402–428 failed to form trimers, indicating that the tail is essential for oligomerization [57]. R416 and E339 form a critical salt bridge that stabilizes tail insertion of one NP into the body of a neighbour. Mutating them to alanine prevents this critical association. R416 was initially shown to be involved in both NP oligomerization and RNA replication, as R416A does not support efficient vRNA synthesis [59], emphasizing that NP self-oligomerization is crucial to function. An NP peptide comprising residues 402–428 inhibited both replication and NP oligomerization [60]. Furthermore, virtual screening identified small molecules that could disrupt the E339–R416 salt bridge [60] as predicted by the NP crystal structure [57].
While crystalline NP is trimeric, cryogenic electron microscopy (cryo-EM) data for recombinant RNPs, and models derived from them, indicate that NP associates as monomers in RNPs [51,61–63]. A minimal replicative RNP formed a circular structure comprising 9 NP monomers, where two NPs associate with polymerase (Figure 2) [63]. NP–polymerase association occurs via PB1 and PB2 [64–66], with the PB1 interaction being much stronger [63]. NP–NP associations within the replicative nonamer RNP are likely to result from tail loop insertion into an adjacent NP. This is consistent with the crystal structure, with the exception that the angle between monomers shifts from 120° in the trimer to 40° in the nonamer [57,63]. Mutant analysis was also consistent with NP–NP association via tail insertion, as R416A and F412A mutants exhibited drastically reduced replication and NP–NP oligomerization [63].
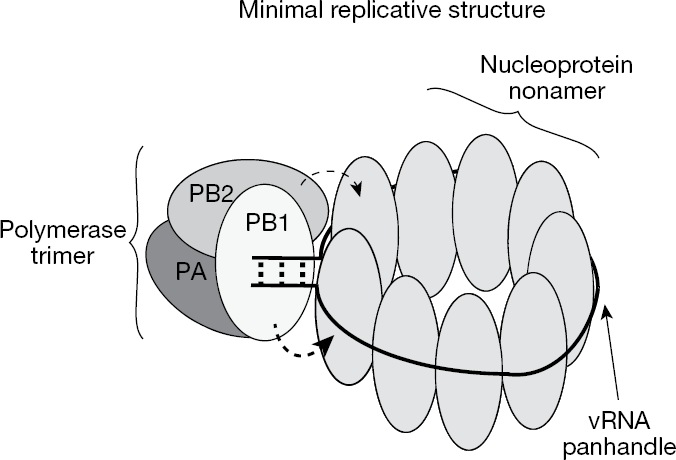
Influenza minimal replicative or RNP structure
NP function in nuclear and cytoplasmic trafficking of RNPs
NP participates in RNP translocation between the cytoplasm and nucleus [67–70]. Following adsorption, fusion and uncoating, RNPs must enter the nucleus for viral transcription [71,72]. At 100–200 Å, RNPs are too large to passively diffuse through the 9 Å nuclear pores [73] and they have been shown to be actively transported into nuclei [67]. NP has two nuclear localization signals (NLS; Figure 1B) [74–77]. Cellular importin-α (or karyopherin-α) mediates nucleus import of RNPs via NLS recognition [68,78]. The strongest and most characterized signal is the N-terminal non-conventional NLS (nNLS) [74,75,77,79]. Crystallography reveals that the nNLS is disordered and flexible enough to bind within the importin-α recognition site [57]. A weaker classical bipartite NLS resides between residues 198 and 216 [75], but the crystal structure does not support this as a functional NLS because the ‘KR’ and ‘RKTR’ motifs are too close to satisfy the minimal distance of 28 Å for an active bipartite NLS [57]. A nuclear accumulation signal identified in oocyte studies was purported to be located between residues 327–345 [80]. However, others showed that it functioned as a cytoplasmic accumulation signal (CAS) [70,75].
RNPs localize to nuclei early in infection, but with time they translocate back to the cytoplasm, illustrating that RNP trafficking occurs throughout the virus lifecycle. Matrix protein (M1) inhibits RNP nuclear import, whilst promoting export [81,82]. This M1 property is consistent with the requirement for M1–RNP dissociation prior to nuclear import and transcription and for re-association of M1 with nascent RNPs for nuclear export and packaging into virions. Interestingly, the cellular heat-shock response to influenza infection results in the inhibition of RNP export due to the failure of M1 to bind NP [83,84]. Alternatively, this result could be due to the inhibition of vRNA replication and therefore RNA assembly at high temperature [85]. The M1–NP interaction is another potential antiviral target.
Nuclear export of RNPs presents the same dilemma as with import, that is, conveying large structures through small pores. The mechanism was elucidated upon observing that leptomycin B inhibited RNP export [86–88]. Leptomycin B inhibits signal-mediated nuclear export by binding to the chromosome region maintenance 1 receptor (CRM1) [89]. Three nuclear export signals (NES) have been identified in NP, with NES 3 being CRM1-dependent (Figure 1B) [90]. The NS2 gene product, denoted NEP for nuclear export protein, is involved in RNP transport from nuclei to the cytoplasm [68]. M1 is also essential for nuclear export. Current models embrace cellular CRM1 binding to a viral NEP–M1–RNP ‘daisy-chain’ complex to expedite RNP nuclear export [87,88,91–94].
Late in infection, NP associates with the cytoskeleton [69,95,96]. In vitro studies demonstrated that NP binds to filamentous-actin (f-actin) and in cellulo experiments reveal a co-localization of actin and NP [70]. Actin has been implicated in RNP cytoplasmic retention [70] and influenza virion budding [95,97]. Mutational analysis maps a putative NP CAS to the C-terminal [70]. It should be noted that certain mutations in the C-terminal region, E339A for example, abrogate NP oligomerization and therefore may affect NP–actin affinity indirectly. Current studies indicate that cytoplasmic trafficking of RNPs involves the same microtubule machinery responsible for endosome recycling [98–100].
In order to shuttle RNPs in and out of nuclei and from the cytoplasm to the cell periphery, NP makes multiple viral and cellular protein associations involving distinct NP recognition sequences. Screening for compounds that disrupt these interactions directly or allosterically is a logical antiviral strategy.
NP role in viral mRNA transcription and vRNA replication
The influenza polymerase must synthesize three distinct RNAs (mRNA, cRNA and vRNA), all requiring NP participation and occurring in the host nucleus [71,101,102]. Viral mRNA transcription is primed with 5′-capped mRNAs scavenged from the host [103–105]. Cellular cap-structures are recognized by PB2 [106,107] and endonucleolytically cleaved 9–15 nucleotides downstream by the polymerase protein (PA) [108–110]. PB1 catalyses elongation, while mRNA termination and polyadenylation occur when polymerase reaches a uridine stretch near the vRNA 5′ terminus, but does not progress due to the constraint of the vRNA 5′-end remaining polymerase-associated [111,112]. The end result is transcriptional stuttering to yield the poly-A tail [113–117].
Viral genome (vRNA) replication involves two unprimed transcriptional events, requiring a cRNA intermediate to serve as a second template [118–120]. This entails synthesis of full-length positive-sense cRNA from negative-sense vRNA. The cRNA product must be coated with NP and associate with polymerase to be transcribed into new vRNA, therefore new viral protein synthesis is a prerequisite for replication [120]. Initiation of both cRNA and vRNA synthesis requires no primer and is reputed to occur directly with ATP, as their 5′-ends terminate in triphosphate [114,121,122]. However, NP is not required for unprimed initiation in vitro [123]. Additionally, cRNA and vRNA species are full-length, with no premature termination or polyadenylation [124,125].
The importance of NP in virus replication is demonstrated by NP temperature-sensitive mutations, which are defective for replication at non-permissive temperatures [125,126]. RNPs stripped of NP can bind and cleave capped-RNA, but conduct only limited elongation [122], while RNPs depleted of NP cannot synthesize cRNA and vRNA, proving NP is essential to genome replication [124,125]. Comprehensive mutagenesis of A/Victoria/3/75 NP has defined roles for NP in mRNA, cRNA and vRNA synthesis [127]. Three mutations and one truncation had significantly impaired cRNA synthesis, but were competent for vRNA and mRNA synthesis. Three other mutations and another truncation failed to support significant synthesis of any RNA species and induced NP aggregation [127]. Although one mutation that induced NP aggregation did not accumulate in the nucleus, other mutations induced large NP aggregates without preventing their nuclear import.
Precise NP roles during transcription and replication have been difficult to assign and details of NP mechanism of action remains elusive. NP-mediated transcription product encapsidation, transcriptional template modification and polymerase activation have all been proposed as mechanisms for switching from mRNA transcription to cRNA synthesis [128]. However, recent in vitro experiments have indicated that relative concentrations of nucleotides, capped-RNA primers, vRNA 5′ termini and polymerase can regulate the ratio of transcription verses replication [129,130]. A more precise understanding of the functions of the NP should allow for improved targeted efforts to identify novel inhibitors.
Nucleoprotein as a target for antiviral development
NP vaccine approaches
Early notions of targeting influenza NP came during vaccine development investigations. NP is a major antigenic determinant for cross-reactive cytotoxic T-lymphocytes (CTLs) recognizing epitopes common to all influenza A subtypes [131,132]. Accordingly, NP has been considered an attractive candidate for DNA or recombinant protein-mediated vaccination. NP is relatively conserved amongst influenza A strains, with less than an 11% variance [133,134]. The multitude of NP functions leaves little leeway for broad variation. Some efforts continue towards the development of a universal vaccine based on NP and are discussed elsewhere [135–139].
Small molecule inhibitor discovery: Aryl piperazine amides
Recently, several groups independently identified a series of aryl piperazine amides that inhibit influenza replication in cell culture [37–40]. These were identified through ‘black box’ antiviral screens searching for any type of inhibitor of virus replication. Once identified, the target of this new class of inhibitors was mapped to the NP through a number of studies. Compound
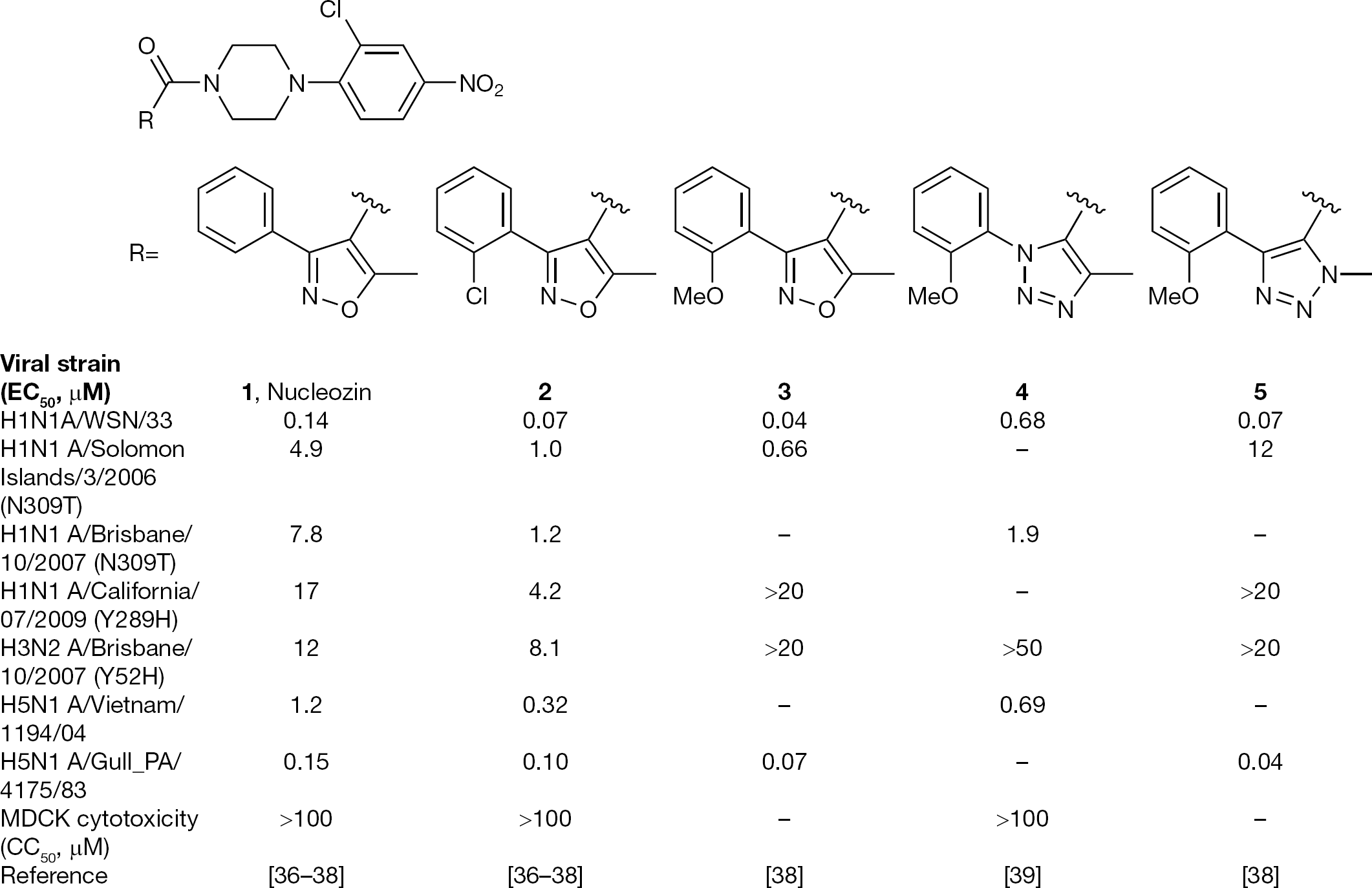
Representative small molecules that inhibit viral replication via binding to NP
Resistance was raised against a close analogue of
Virtual docking studies using the published A/WSN/33 NP structure [57] identified three prospective binding sites for nucleozin: (1) a small groove in the back of the body domain; (2) the arginine-rich RNA-binding groove; and (3) another groove in the tail-loop domain [37]. However, the only proposed site containing the Y289H and N309K resistant residues was binding site 1. Moreover, nucleozin did not inhibit RNA-binding or NP self-oligomerization that could be attributed to site 2 or 3 binding, respectively.
Using a similar cell-based infection assay employing H1N1 influenza A/WSN/33, compounds
Neither of these first two binding models for piperazine NP inhibitors incorporated all three resistant mutations observed (Y289H, N309K and Y52H). Although Y289 and N309 are relatively close to one another, the molecular distance between these residues and Y52 is greater than 17 Å [57], a span that exceeds compound length [39]. Our laboratory also discovered compound
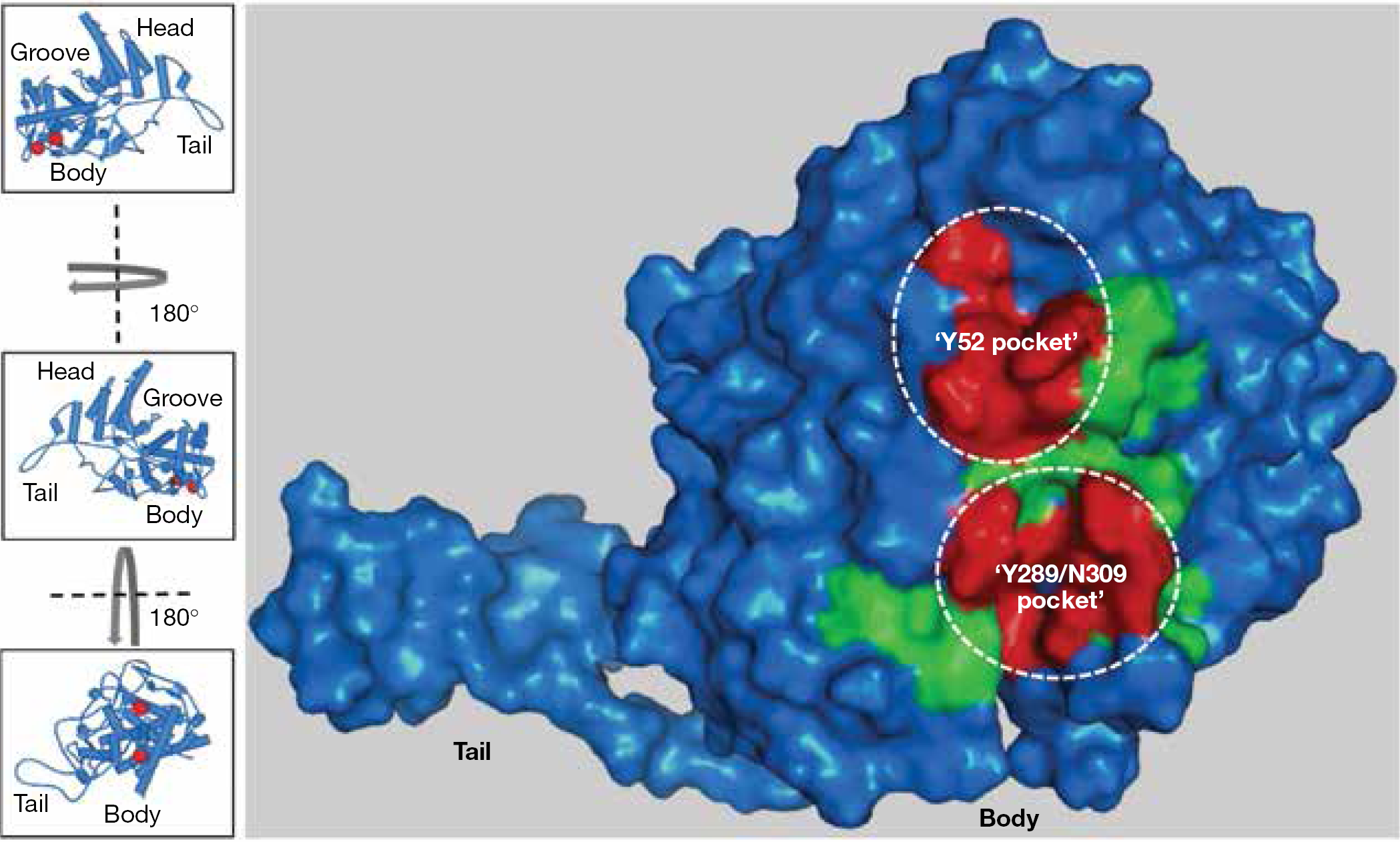
Nucleoprotein surface map of residues affecting aryl piperazine amide efficacy
Apparent discrepancies in the proposed binding site were resolved once the X-ray co-crystal structure of compound
Natural viruses bearing Y52H, Y289H or N309K mutations in their NP were significantly less sensitive to this series of inhibitors. As shown in Figure 3, two H1N1 strains with an N309T NP polymorphism were highly resistant to the series. Site-directed mutagenesis confirmed that N309K and N309T substitutions were equally resistant (unpublished results). H1N1 A/California/07/2009 virus possesses Y289H and is insensitive to inhibition by this series, and the H3N2 A/Brisbane/10/2007 virus, with the Y52H resistant signature, was refractory to compounds
Aryl piperazine amide NP inhibitor proof-of-concept in the mouse model of influenza infection
Compounds
Compounds
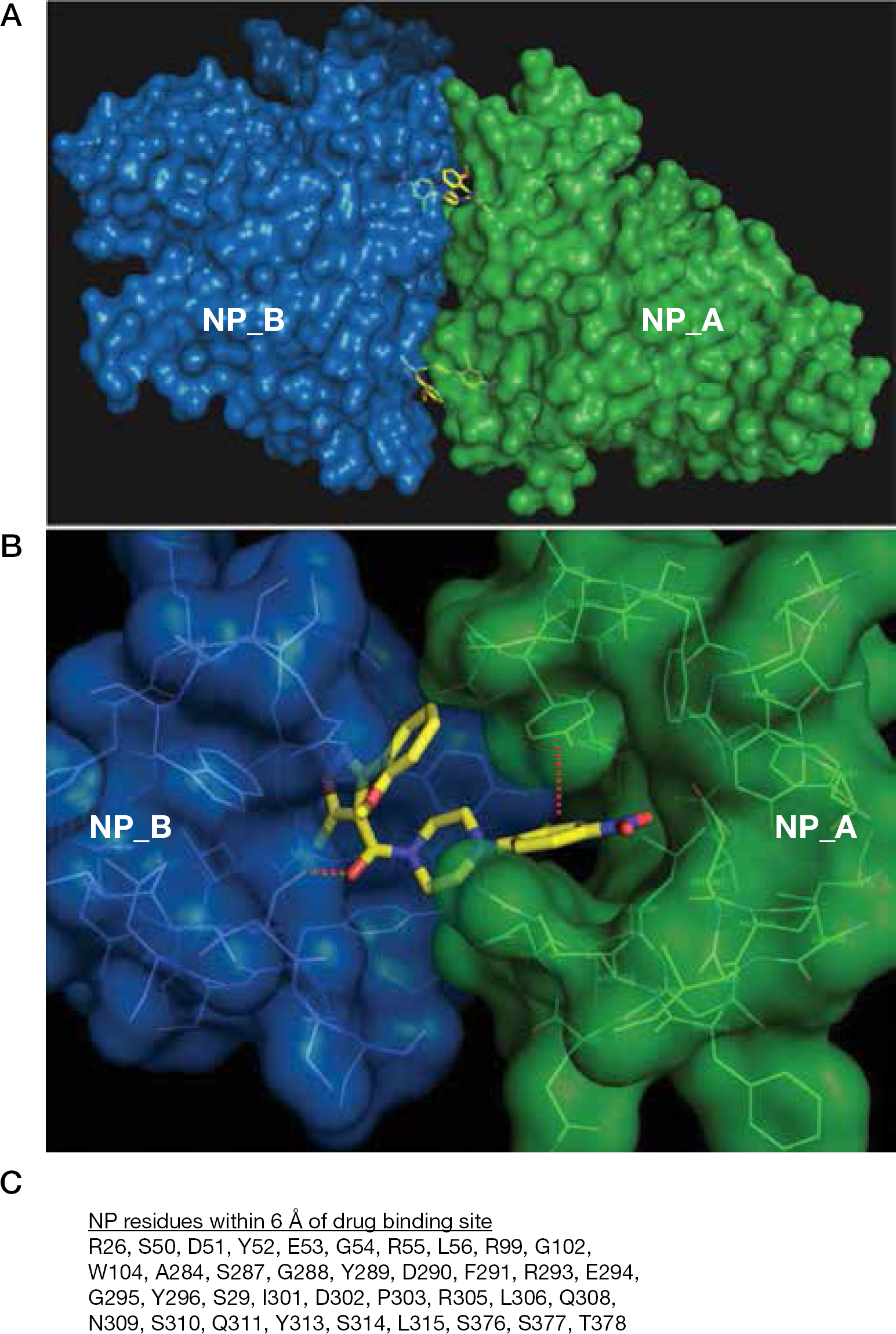
Aryl piperazine amide-induced nucleoprotein dimerization
Aryl piperazine amide NP inhibitor mechanism of action
Resistance selection to substituted aryl piperazine amides invariably yielded point mutations in the NP protein [37–39]. Reconstitution of suspected NP resistant residues into recombinant virus reproduced resistance phenotypes indistinguishable from the original compound-selected viruses, thereby unambiguously demonstrating that influenza NP is the molecular target of nucleozin and its analogues [37–39].
Nucleozin and related piperazine inhibitors have been reported to prevent nuclear accumulation of incoming infectious RNPs. Using immunofluorescence and confocal microscopy, it was observed that the addition of
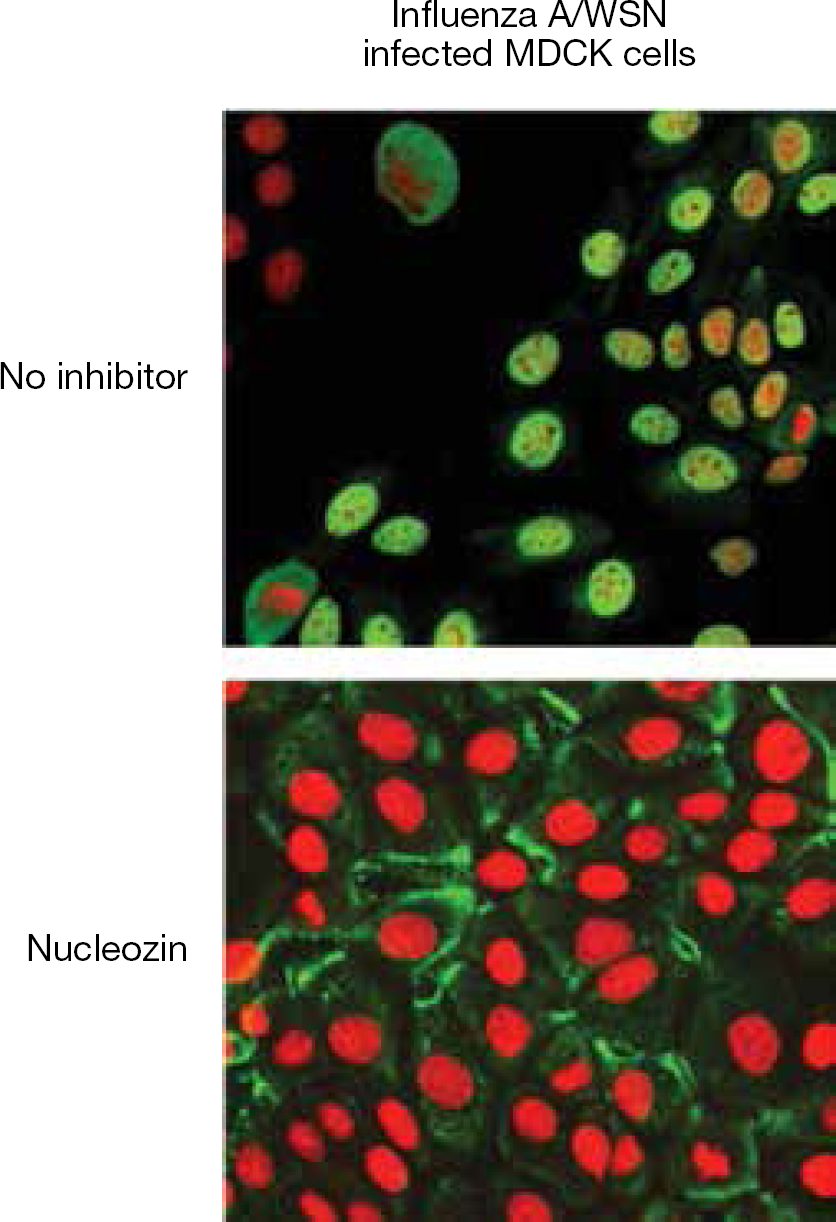
Aryl piperazine amides inhibit nucleoprotein nuclear accumulation
Further evidence of compound-induced NP aggregation was observed via gel-shift and native PAGE [37], dynamic light-scattering (DLS) [39] and analytical ultracentrifugation techniques [38]. Synthesis of compound
Taken together, these experiments suggest that this compound series exerts its antiviral effects by inducing the formation of higher-order NP oligomers that interfere with RNP and NP nuclear import, and possibly other NP functions in the virus lifecycle. It was initially speculated that compound binding to NP caused an allosteric change, stimulating NP self-oligomerization. The X-ray crystal structure of compound
These inhibitors could prevent exposure of NLS motifs to abrogate nuclear import. However, there is no overlap when comparing the NLS domains (Figure 1B) to residues within 6 Å of the binding site (Figure 5C). It is unlikely that compound-induced oligomerization would block the nNLS or bipartite NLS domains. Nonetheless, one cannot rule out that inhibitor-induced oligomerization does not perturb nuclear localization signalling via an allosteric mechanism. Amino acid R26 is in common to NES1 and the inhibitor binding pocket (Figure 1B; Figure 5C), but an effect on nuclear export has yet to be demonstrated for this series.
Nothing similar to the inhibitor-induced anti-parallel association of two NP molecules via residues within their body domains has been revealed in other X-ray crystals or cryo-EM structures obtained in the absence of inhibitor. This association is in geometric opposition to the natural tail-to-body insertions observed in NP crystal structures or found to be consistent with cryo-EM models [57,58,63]. However, one could envision an anti-parallel NP–NP interaction serving a purpose in maintaining RNP structure when vRNAs coil back upon themselves. Any inhibitor-coordinated association could interfere with a number of NP functions, binding partner associations or essential structures. Indeed, our laboratory reported nucleozin inhibition of primary mRNA transcription in vitro (Figure 7A) [39]. However, the 50% inhibitory concentration for inhibition of transcription is approximately 10-fold higher than the EC50 for overall inhibition of virus replication in tissue culture. Moreover, the slope of this in vitro polymerase inhibition curve is relatively gradual (Figure 7A; slope = 1.5) when contrasted with the steep inhibitory slopes associated with antiviral or viral yield reduction assays that encompass all stages of NP involvement (Figure 7B; Figure 7C; slopes >5). We speculate that many stages in the influenza virus life cycle employing NP could be affected by these aryl piperazine amide inhibitors and that the steep inhibitory slopes observed when analysing multiple cycle replication may reflect the summation of multiple inhibition steps.
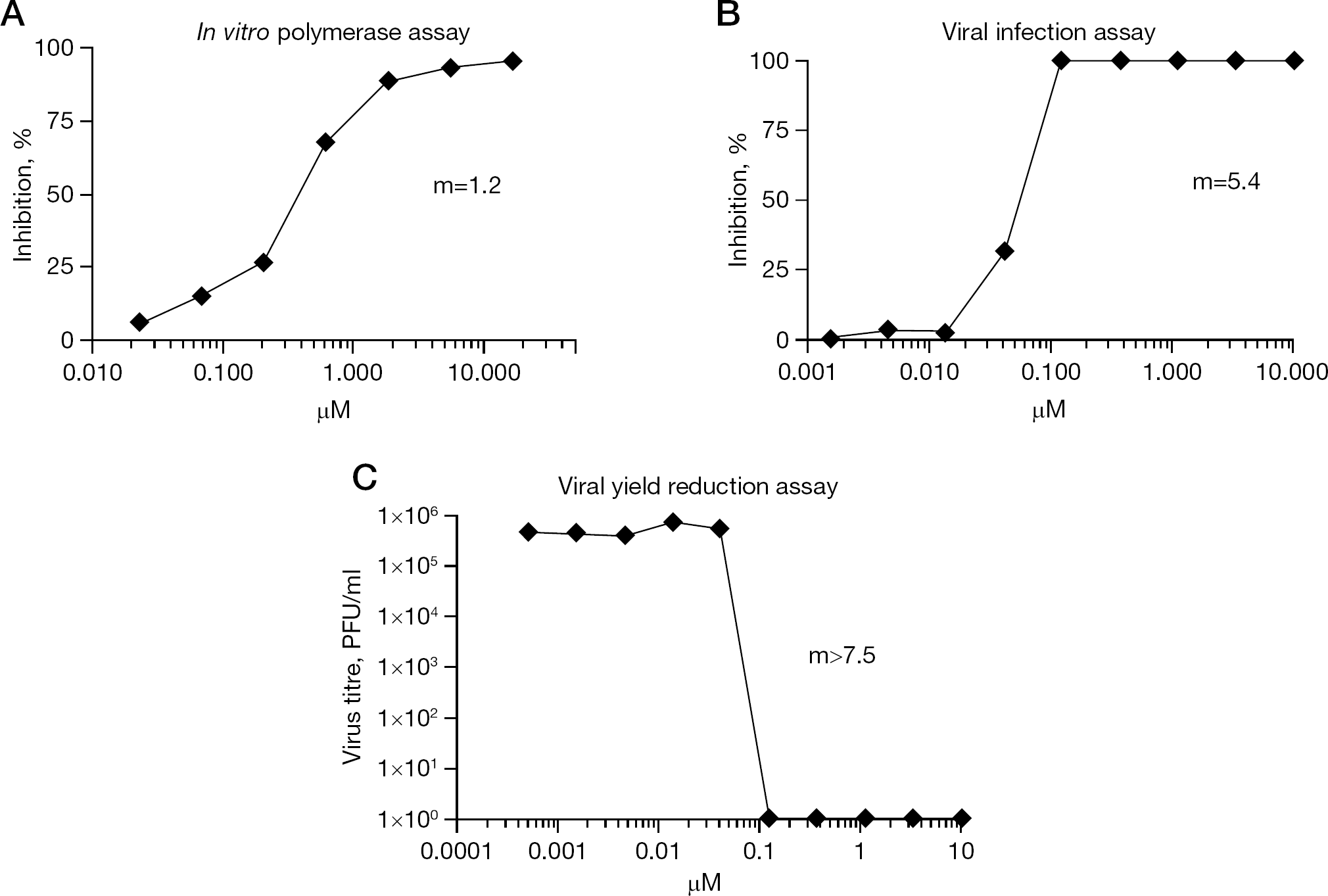
Nucleozin inhibitory profiles in various influenza assays
Other influenza virus NP inhibitors
With the crystal structure solved for H1N1 [57], H5N1 [58] and B [140] NPs and the precise interactions for NP self-oligomerization revealed, novel approaches to develop alternate inhibitors have been investigated with some early success. A peptide encompassing NP tail domain residues 402–428 inhibits oligomerization and replication [60]. Another proline-rich constrained peptide was shown to bind to NP and inhibit influenza replicons [141]. In silico screening of virtual libraries for docking to CTL epitope sites within NP identified 3-mercapto-1,2,4-triazole derivatives that inhibited virus replication [142]. Making use of photo-cross-linked chemical arrays to screen for NP-binding compounds resulted in the isolation of mycalamide-A-related structures from a natural products depository. These were shown to bind within the N-terminal amino acids 1–13, which corresponds to the nNLS domain [143]. All these inhibitors are very early in the discovery process and it remains to be seen if these leads can evolve into clinical candidates.
Respiratory syncytial virus nucleocapsid inhibitor
It is worth noting that inhibitors targeting the NP have also been identified for respiratory syncytial virus (RSV). Recently, a small molecule inhibitor was identified as a clinical RSV drug candidate. RSV604 is a benzodiazepine derivative developed from a hit identified in an antiviral cell protection assay [144,145]. RSV604 had an EC50 of 0.9 μM against RSV and was shown to target the RSV nucleocapsid (N) through resistance selection and reconstitution using reverse genetics [144]. Resistance was mapped to residues N105D, K107N with I129L, and I129L with L139I. With respect to the crystal structure of the RSV N–RNA complex, these resistant mutations reside in the most distal end of a long β-hairpin that projects away from the RNA-binding groove [146]. Intriguingly, this is analogous to the location of the piperazine influenza inhibitor resistant substitutions, which are situated on the outer surface of the body domain of the NP pointing away from the RNA groove [39,57]. In both examples, these inhibitor resistant residues are distal to the RNA-binding sites, in regions that are proposed to interact with their respective viral polymerases [57,146].
Conclusions
The development of new anti-influenza antivirals to augment the current antiviral armamentarium remains a high priority. A recent meta-analysis of vaccine effectiveness concluded that vaccines decrease both the risk of the symptoms of influenza and time off work, but their effects are minimal, with no evidence of a reduction of complications or transmission [147]. Similarly, there was inconclusive evidence to demonstrate a reduction of complications that required antibiotics after antiviral treatment with oseltamivir [148]. Moreover, the emergence of resistance to current inhibitors remains problematic, due to the high mutation rate of the vRNA-dependent RNA polymerase and the ability to select resistance during the course of a single infection. As a result, there remains an unmet medical need for new influenza antivirals and a push to use them in combination therapy.
The influenza NP is an appealing antiviral target, being a relatively conserved and crucial viral protein with both structural and functional roles. Aryl piperazine amide inhibitors directed against NP have been discovered by several laboratories in parallel efforts [37–40]. These are synthetically tractable chemotypes that can be optimized for in vivo efficacy in the mouse model of influenza infection [39]. Further analogue synthesis and SAR could expand inhibitor coverage of naturally occurring polymorphisms at amino acid residues 52, 289 and 309 of NP that reduce the efficacy of the current series.
NP serves as the protein building-block of influenza RNPs. It is engaged in RNP translocation in and out of the nucleus and is intimately involved in viral transcription and replication. It has multiple viral and host binding partners during various phases of the influenza virus life cycle. The exact mechanism of action for all of its roles has not yet been ascertained, but many functions have proven to be dependent on the self-oligomerization property of NP. Certain small molecule compounds can interfere with NP–NP association, and perhaps effect other key NP associations, resulting in antiviral efficacy. We are just beginning to reveal the significance of this multifaceted viral protein in the development of new anti-influenza agents.
Footnotes
The authors are employees of Bristol–Myers Squibb.