
Abstract
Background:
The discovery of novel influenza virus inhibitors remains an important priority in light of the emergence of drug-resistant viruses. Toward this end, a library of over 6,000 compounds was tested for antiviral activity.
Methods:
Strains of influenza virus were evaluated by cytopathic effect (CPE) inhibition and virus yield reduction assays. Intracellular nucleoside triphosphate pools were analysed by strong anion exchange HPLC. Dihydroorotate dehydrogenase inhibition assays were conducted. Influenza virus-infected mice were treated for 5 days with D282. Results: A non-nucleoside, 4-[(4-butylphenyl)amino]-2-methylene-4-oxo-butanoic acid (D282), was discovered that inhibited influenza A and B virus CPE by 50% at 6–31 μM (giving selectivity indices of >13 to >67, based on cytotoxicity of >400 μM in stationary cell cultures). Ribavirin (positive control) was active at 14–44 μM (yielding selectivity indices of >9 to >29, with >400 μM toxicity). D282 and ribavirin inhibited virus yield by 90% at 9.5 ±3.3 and 10.8 ±3.2 μM, respectively. The antiviral activity of D282 in vitro was reversed by addition of uridine, cytidine and orotic acid. D282 exhibited an uncompetitive inhibition of mouse liver dihydroorotate dehydrogenase (inhibitor constant [Ki] of 2.3 ±0.9 μM, Michaelis constant [Km] of 150 ±16 μM). Because cellular pyrimidine biosynthesis was inhibited, D282-treated cells had decreased uridine triphosphate and cytidine triphosphate levels. D282 (≤100 mg/kg/day) failed to prevent death of mice infected with influenza.
Conclusions:
D282 was active against influenza A and B viruses by inhibiting de novo pyrimidine biosynthesis. Although effective in vitro, the compound, like others in its class, was devoid of antiviral activity in infected mice.
Introduction
The spread of influenza viruses resistant to approved antiviral drugs [1–3], the appearance of pandemic influenza A (H1N1) virus in 2009 [4], and the emergence of highly virulent avian influenza A (H5N1) viruses capable of infecting and killing humans [5] underscore the need for improved antiviral therapies. Of the four approved anti-influenza drugs (amantadine, rimantadine, oseltamivir and zanamivir), only oseltamivir is currently in widespread use. Progress is being made, however, in identifying other clinical candidates. One of these compounds is laninamivir, a long-acting neuraminidase inhibitor [6] that has recently been approved for use in Japanese clinics. Laninamivir has the same mode of action as oseltamivir and zanamivir, and is similar to zanamivir in requiring inhalation for activity. Like zanamivir, laninamivir inhibits oseltamivir-resistant H275Y influenza viruses [6]. Another compound, favipiravir, is a heterocycle that is metabolized in cells to a nucleoside triphosphate analogue that inhibits influenza virus RNA polymerase activity [7]. Favipiravir has shown considerable efficacy in mouse models of influenza virus infection [8–10]. Other known anti-influenza compounds that have not been approved for use in humans are peramivir (a neuraminidase inhibitor) [11] and ribavirin (used in combination with amantadine and oseltamivir [12]). Besides these inhibitors, the number of disclosed compounds under investigation is limited. There are efforts to design adamantane derivatives that will be effective against S31N mutant viruses resistant to amantadine and rimantadine [13].
Recently a novel inhibitor of pyrimidine biosynthesis was identified, compound A3, that was effective against influenza and other RNA and DNA virus infections in cell culture [14]. By high throughput screening, Maddry et al. [15] discovered novel benzoquinazolinones and thiazoloimidiazoles inhibitory to influenza H1N1 and H5N1 viruses, but not to H3N2 or influenza B viruses. Other investigators have identified novel structures through high throughput screening efforts followed by structural optimization [16,17]. Our own efforts have involved screening a library of over 6,000 compounds from Chemtura Corporation (Guelph, ON, Canada) for inhibitors that might be broadly active against influenza A (H1N1, H3N2 and H5N1) and influenza B viruses in cell culture. From this effort we discovered 4-[(4-butylphenyl)amino]-2-methylene-4-oxo-butanoic acid (D282; Figure 1) that is a non-nucleoside with a core structure unlike others that have been published. The antiviral activity and mode of action of D282 in vitro and in vivo are the subject of this report.
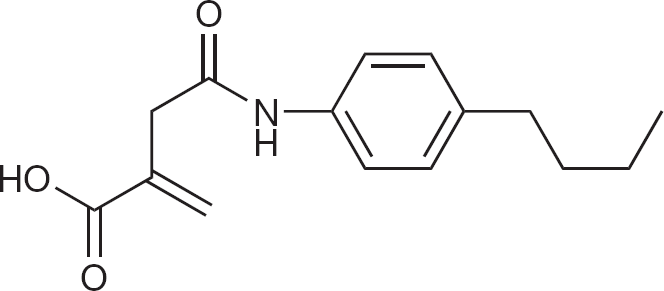
Structure of 4-[(4-butylphenyl)amino]-2-methylene-4-oxo-butanoic acid (D282)
Materials and methods
Compounds
D282 was provided for antiviral testing by Chemtura Corporation. Ribavirin was originally obtained from the former ICN Pharmaceuticals (Costa Mesa, CA, USA). D282 was not readily soluble in water or cell culture medium, but was found to be soluble at ≤16 mM in 2% sodium bicarbonate solution. Natural nucleosides (adenosine, guanosine, inosine, xanthosine, cytidine and uridine) and pyrimidine biosynthetic precursors (dihydroorotic acid and orotic acid) were purchased from Sigma (St Louis, MO, USA).
Viruses
A total of 14 different strains of influenza A and B viruses were used for antiviral testing. The following were from the Centers for Disease Control and Prevention (Atlanta, GA, USA): A/California/07/2009 (H1N1), A/New Caledonia/20/99 (H1N1), A/Solomon Islands/03/2006 (H1N1), A/Wisconsin/67/2005 (H3N2), A/Brisbane/10/2007 (H3N2), A/Perth/16/2009 (H3N2), B/Sichuan/379/99, B/Shanghai/361/02, B/Malaysia/2506/2004 and B/Florida/4/2006. A/NWS/33 (H1N1), A/Duck/MN/1525/81 (H5N1) and A/Gull/PA/4173/83 (H5N1) were obtained from sources indicated previously [18]. The H5N1 viruses are low pathogenic avian strains from North America. Pools of all of the viruses were made in Madin-Darby canine kidney (MDCK) cells.
Viral cytopathic effect (CPE) inhibition assay
Confluent monolayers of MDCK cells in 96-well microplates were infected with approximately 50–100 cell culture infectious doses (CCID50) of virus. The medium used for assays was MEM, 10 units/ml of trypsin, and 1 μg/ml of EDTA. Compounds in various concentrations were applied to cells just prior to adding virus (50–100 50% cell culture infectious doses [CCID50] per well). Three microwells at each concentration of compound were infected. Two microwells per dilution of compound were uninfected and served as toxicity controls. After 72 h of incubation in 5% CO2 at 37°C, CPE was quantified by neutral red dye uptake [18,19] using the dye at a 0.011% final concentration for 2 h. Unincorporated dye was removed from cells by aspiration, then the absorbed dye was eluted from the cells for 30 min with 0.1 ml of 50% Sörensen's citrate buffer (pH 4.2)/50% ethanol. Plates were read for optical density determination at 540 nm with 405 nm reference. Readings were converted to percentages of uninfected control and normalized to the virus control. Fifty percent virus-inhibitory concentrations (EC50 values) and 50% cytotoxic concentrations (CC50 values) were determined by linear regression analysis. Selectivity index (SI) values were calculated as CC50/EC50.
Virus yield reduction assay
Virus was replicated in 96-well plates in the presence of inhibitor in 96-well microplates as described above. After 3 days of incubation, supernatant was removed and frozen at −80°C. Later, the samples were thawed and titrated for virus on fresh monolayers of MDCK cells in 96-well plates by end point dilution method using three or four microwells for each of 10−1 to 10−8 dilutions. After 6 days' incubation the viral CPE in each well was determined by microscopic examination. Virus titre calculations of CCID50 of virus per 0.1 ml were then made using the Reed-Muench method [20]. Concentrations of compound inhibiting virus yield by 90% (one log10) were determined by regression analysis. Each of the 14 virus strains was assayed by virus yield reduction only once. In order to obtain mean values ±sd, the combined results of each virus type (H1N1, H3N2, H5N1, or B) were used. For example, the graph of data for H1N1 are results from influenza A/California/07/2009, A/New Caledonia/20/99, A/NWS/33 and A/Solomon Islands/03/2006 averaged together.
Cell proliferation toxicity assay
MDCK cells in 24-well microplates were seeded so as to be approximately 30% confluent 18 h later. D282 and ribavirin were prepared in half-log10 dilutions in medium containing 5% fetal bovine serum (FBS) to allow for cell replication, then applied to the plates and incubated for 72 h. At that time, neutral red dye was applied for 2 h, the dye was removed, the plates were rinsed, and the dye eluted as described above for the CPE inhibition assay. Media from the wells were transferred to a 96-well microplate and read for optical density as described above. The CC50 was calculated by the method used in the CPE test.
Mode-of-action studies
Initially, we performed timing experiments designed to rule out certain modes of action involved in early and late steps in virus replication. D282 was added to cells either at 1, 6 or 12 h after infection in an 18 h virus replication experiment. For these experiments, influenza A/Duck/MN/1525/81 virus was used at a multiplicity of infection of approximately one virus per cell. This virus was selected because it replicates rapidly and sufficient virus could be detected at 18 h. Virus yield from treated wells was determined by the same methods described above for virus yield reduction. Another method was to treat cells with D282 at various concentrations concurrently with natural nucleosides (each at 200 μM) during an influenza A/Victoria/3/75 infection using a low multiplicity of infection (50–100 CCID50/well, about 1 virus per 1,000 cells). Quantitation of percent viral CPE at each concentration of inhibitor was made after 72 h of incubation of cells with virus.
HPLC analysis of intracellular nucleotides
The effects of D282 on intracellular adenosine triphosphate (ATP), guanosine triphosphate (GTP), cytidine triphosphate (CTP), and uridine triphosphate (UTP) levels were assessed. T-25 flasks containing 6 to 8×106 cells of uninfected MDCK cells were treated with D282 at varying half-log10 concentrations in MEM containing 50 μg/ml gentamicin (no FBS) and incubated for 24 h. The medium was then aspirated from the flasks. Each flask was treated with 540 μl of 3.5% perchloric acid at 4°C for 5 min. Next, 270 μl of 1 N KOH/1 M imidazole was added to neutralize the sample. Finally, the liquid contents of each flask were collected, centrifuged at high speed for 1 min to pellet the perchlorate precipitate, and frozen at −80°C prior to thawing and analysis of nucleoside triphosphate concentrations.
High-performance liquid chromatography (HPLC) analysis of the samples was conducted as previously published [21–23]. Briefly, chromatographic separations were performed using an HPLC apparatus (Waters Corp., Milford, MA, USA) fitted with a 10×250 mm column packed with Whatman partisil SAX resin (Phenomenex Inc., Torrance, CA, USA). A linear gradient from 10 mM to 1 M potassium phosphate (Sigma) at pH 3.5 was run over 30 min at 1 ml/min. The 1 M buffer ran an additional 10 min before re-equilibrating the column in low salt buffer. Potassium phosphate buffer was found to be better for resolving the natural nucleoside triphosphate peaks than previously used ammonium phosphate. Detection of nucleotides was made at 260 nm with a Waters LC spectrophotometer and the peaks analysed using an integrator (Shimadzu Scientific Instruments, Columbia, MD, USA). Relative peak areas of D282-treated cells were compared to untreated cells and expressed as percentages of control levels.
Dihydroorotate dehydrogenase isolation and enzymatic assay
The procedures for performing these studies were essentially those of Peters et al. [24] with minor modifications. Mouse livers from three 20 g BALB/c mice were excised and homogenized in 100 mM Tris-Cl buffer, pH 8.0. A low speed 600× gravity spin was performed and the pellet was discarded. The supernatant fluid was subjected to an 8,000× gravity centrifugation for 30 min. The supinate fluid was discarded and the pellet was suspended in 6 ml of the above Tris-Cl buffer. Enzyme assays were performed by incubating 25 μl of crude enzyme (mitochondrial fraction) in a 200-μl reaction volume of Tris-Cl buffer for 1 h at 37°C. Reactions were stopped by the addition of 50 μl of 16% trichloroacetic acid. After chilling on ice for 15 min, a 10,000× gravity spin for 2 min was performed to pellet the protein. The clear liquid was transferred to a separate tube and partially neutralized (to about pH 5) with 40 μl of 1 N KOH. Analysis of orotic acid found in 100 μl of reaction mixture was made by Whatman partisil SAX HPLC (Waters instrument described above) using an isocratic buffer of 8 mM KH2P04, 8 mM KCl, pH 4.0 at a 1 ml/min flow rate. Elution time for orotic acid was about 7.8–8.0 min, similar to the published value [24]. Peak areas of orotic acid were plotted at 280 nm using a Shimadzu integrator. The assay relies on the fact that dihydroorotic acid has an ultraviolet absorbance of 220 nm and is invisible at 280 nm, and the formation of orotic acid is separable by HPLC with no interfering peaks. The Michaelis constant (Km value) was estimated from double reciprocal Lineweaver-Burk plots after testing two-fold dilutions of dihydroorotic acid in the enzyme assay from 1.6 to 800 μM. The 50% inhibitory concentration (I50 value) of D282 was determined by evaluating two-fold dilutions of D282 (0 μM, then 1 to 256 μM) versus a constant 200 μM concentration of dihydroorotic acid. The dissociation constant of the enzyme-inhibitor complex, also referred to as the inhibition constant (Ki value), was derived by the published equation [25] that relates I50 to Km and substrate concentration (S) as follows: I50=Ki(1+[S/Km]). Km and I50 determinations were each made from three independently performed enzyme assays. Another assay was performed to determine the type of inhibition (competitive, non-competitive or uncompetitive) exhibited by D282 by varying concentrations of dihydroorotic acid (25, 50, 100 and 200 μM) versus fixed concentrations of D282 (0, 2 or 8 μM) in the reaction mixture, with results presented as a double reciprocal (1/S versus 1/v [initial reaction velocity]) Lineweaver-Burk plot.
Animal infection experiments
Female BALB/c mice (18–20 g) were obtained from Charles River Laboratories (Wilmington, MA, USA). They were anaesthetized by intraperitoneal injection of ketamine/xylazine (at 50 and 5 mg/kg, respectively) followed by infection intranasally with a 90-μl suspension of influenza virus in cell culture medium. The infectious inoculations equated to three to four 50% mouse lethal challenge doses (MLD50). The compounds were administered once a day for 5 days starting 4 h before virus exposure. Ten drug-treated infected mice and 20 placebo-treated controls were observed daily for death through 21 days. Survivor numbers were analysed by the Fisher's exact test. Mean day of death differences were analysed by the Mann-Whitney U test. Analyses were two-tailed and made using Instat® (GraphPad Software, San Diego, CA, USA).
Results
Cell culture antiviral activity
Virus-inhibitory effects of D282 and ribavirin were determined in MDCK cell culture against 14 influenza strains (Table 1). D282 inhibited viral CPE by 50% at concentrations ranging from 6 to 31 μM. Ribavirin was inhibitory at 14 to 44 μM. In these assays, D282 had a lower EC50 value against each of the viruses compared to ribavirin. Average inhibition was determined from all viruses combined, giving values of 13 μM for D282 and 24 μM for ribavirin. Selectivity index (SI) values calculated from the data gave values of >13 to >67 for D282 and >9 to >29 for ribavirin. Mean SI values were >31 and >17 for the two compounds, respectively.
Antiviral activities of D282 and ribavirin against strains of influenza A and B viruses in MDCK cells
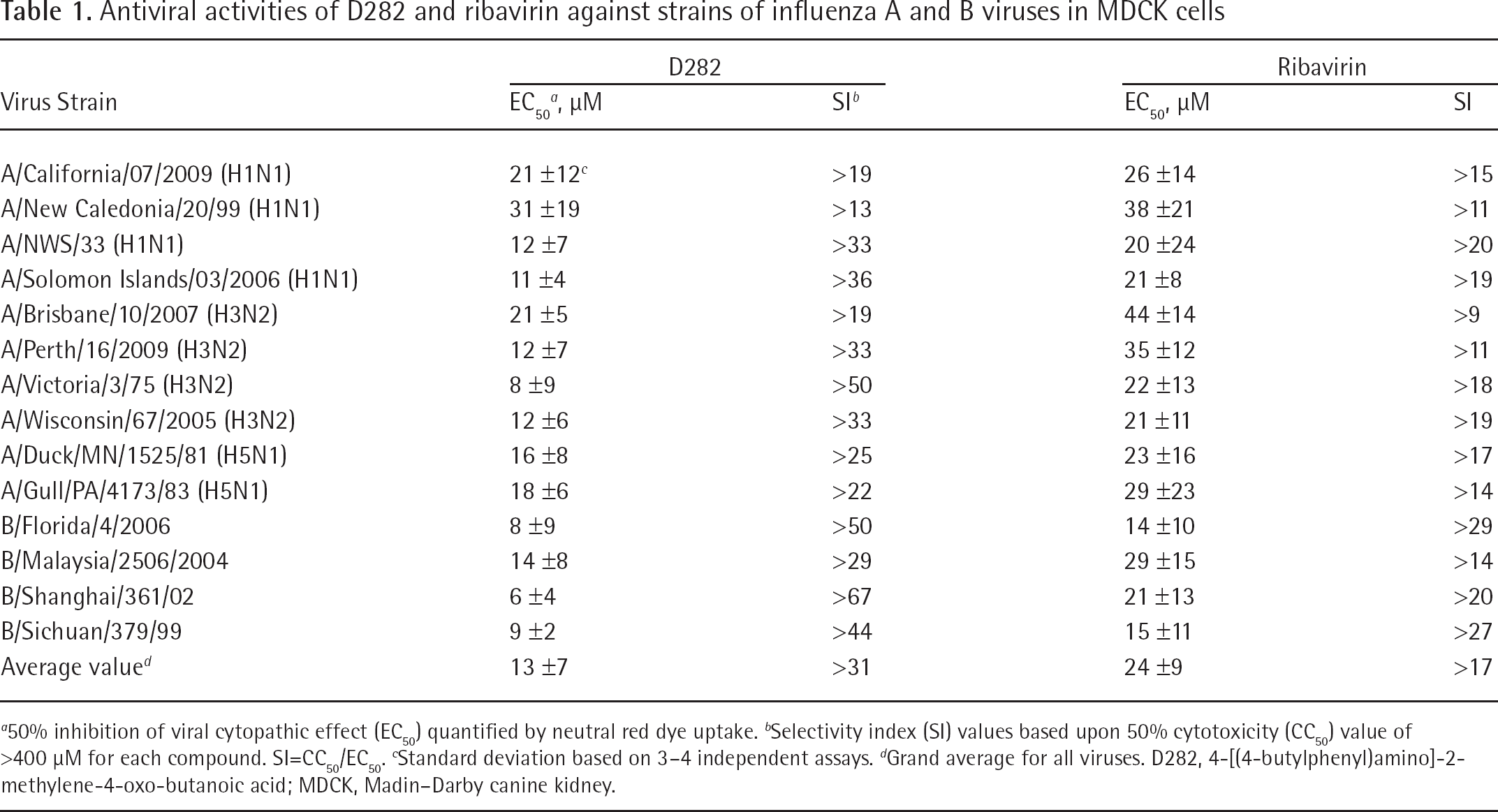
50% inhibition of viral cytopathic effect (EC50) quantified by neutral red dye uptake.
Selectivity index (SI) values based upon 50% cytotoxicity (CC50) value of >400 μM for each compound. SI=CC50/EC50.
Standard deviation based on 3–4 independent assays.
Grand average for all viruses. D282, 4-[(4-butylphenyl)amino]-2-methylene-4-oxo-butanoic acid; MDCK, Madin-Darby canine kidney.
The virus yield results for influenza A and B viruses are shown in Figure 2. From the curves, 90% virus-inhibitory concentrations (EC90 values) were calculated. EC90 values for D282 ranged from 7 to 14 μM, with a mean value (for all viruses) of 9.5 ±3.3 μM. Ribavirin was 90% inhibitory at 8–14 μM, with a mean of 10.8 ±3.2 μM. EC90 values for individual viruses are not shown. At 130 μM, all viruses were completely inhibited by D282 and ribavirin (Figure 2). The inhibition curves for D282 and ribavirin were quite similar.
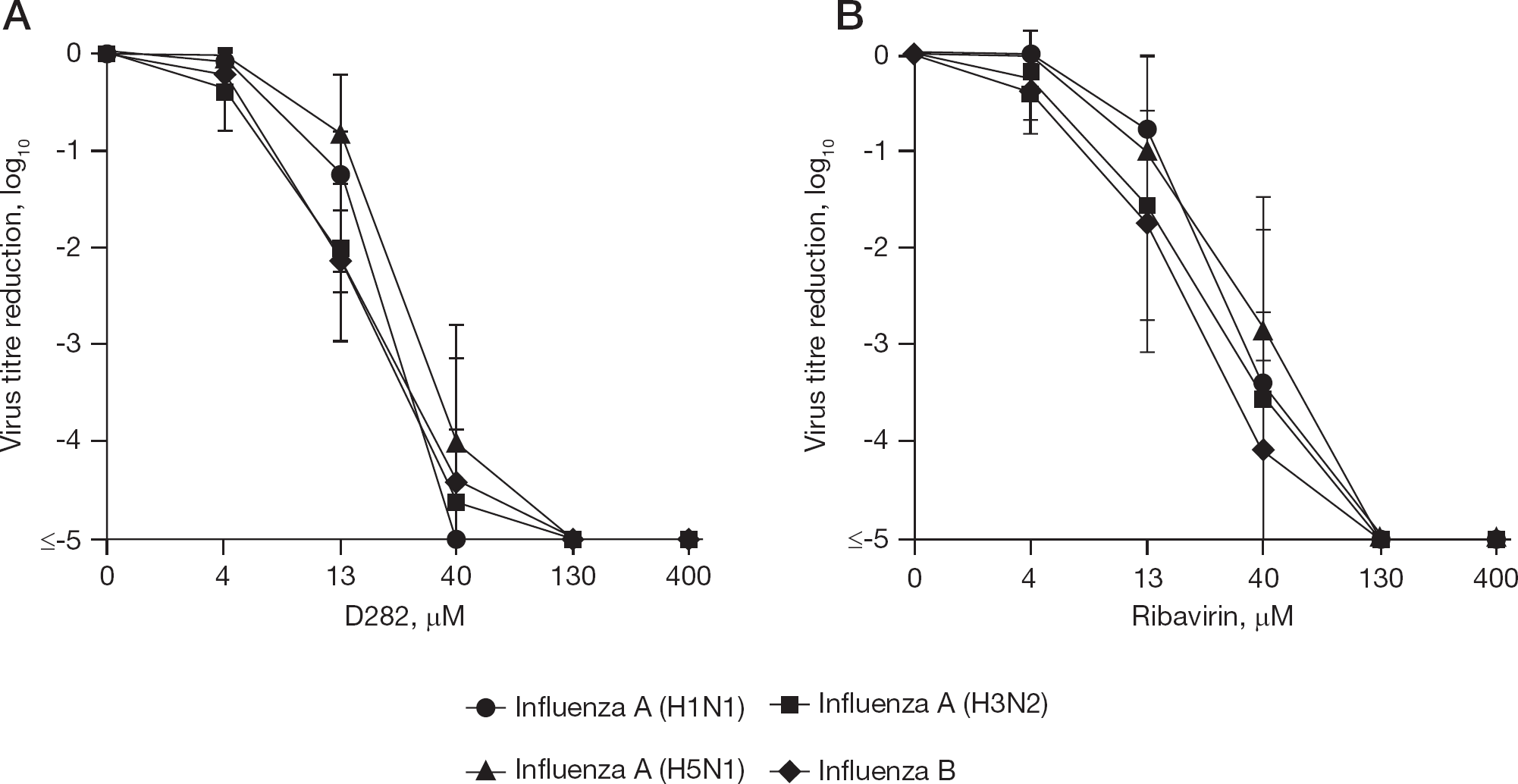
Effects of D282 and ribavirin on virus yields from MDCK cells
Cytotoxicity determinations
Tests for cytotoxicity of D282 and ribavirin were performed concurrently with the antiviral CPE studies in confluent MDCK cell monolayers. Neither compound was cytotoxic at ≤400 μM to stationary confluent cell monolayers. However, compounds that are not toxic to stationary phase cells may inhibit actively dividing cells. Indeed, in MDCK cell proliferation assays, D282 and ribavirin were inhibitory at 19 ±9 and 81 ±43 μM, respectively, in three independent experiments.
Mode of antiviral action of D282
D282 represented a new chemical entity for which the mode of action was not intuitively predicted by its structure. Thus, initially all anti-influenza virus targets were possibilities. We performed timing experiments quantified by virus yield to rule out certain modes of action involved in early and late steps in virus replication. D282 was added to cells either at 1, 6 or 12 h after infection in an 18 h virus replication experiment (Figure 3). The compound proved to be inhibitory when added as late as 6 h, but lost activity when added at 12 h. From these results we could rule out early antiviral targets, such as the prevention of virus adsorption, attachment or uncoating. Late events such as inhibition of viral neuraminidase could also be ruled out. From these results we hypothesized that the inhibitor acts on a virus replication step, such as involving viral RNA synthesis. However, not having a viral RNA polymerase assay established in our laboratory, we first looked at alternative methods to investigate this possibility.
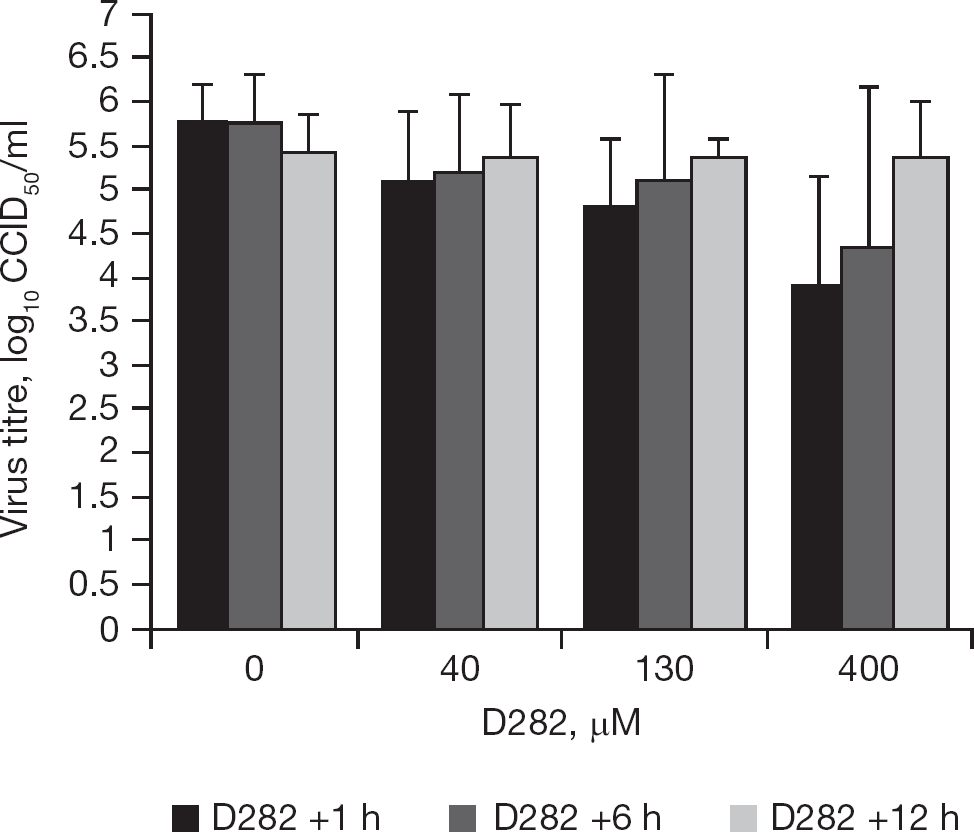
Time of addition of D282 to cells affects antiviral activity
Reversal of antiviral activity with natural nucleosides
We tested whether natural nucleosides could reverse the antiviral activity of D282, which would point to a cellular target of virus inhibition. MDCK cells were treated simultaneously with natural nucleosides and D282 starting just before infection, with viral CPE measured after 72 h (Figure 4A). Treatment of cells with uridine reversed the antiviral activity of D282. Cytidine treatment also caused reversal of activity, but to a lesser degree than uridine. This suggested that D282 might be suppressing intracellular pyrimidine pools. Adenosine treatment increased antiviral activity, as did guanosine and xanthosine treatments to a lesser degree.
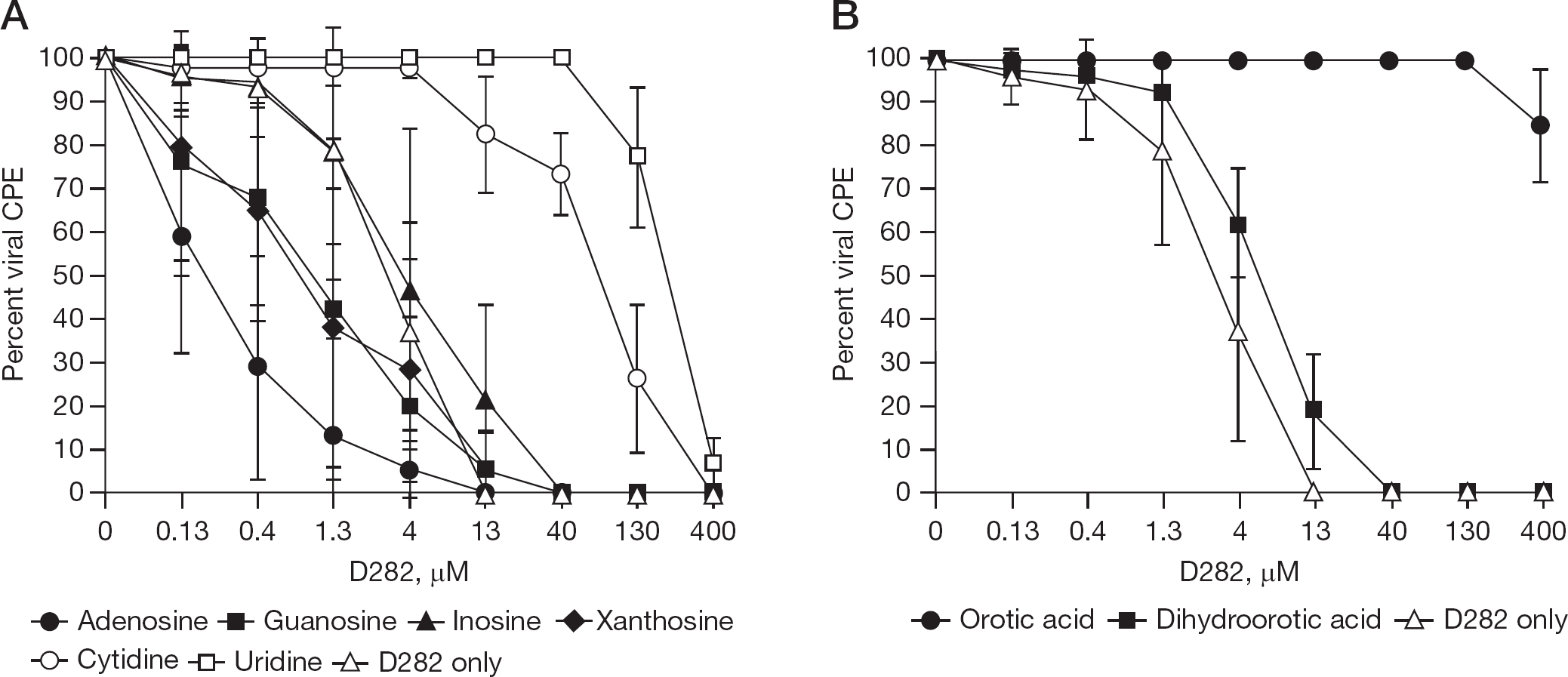
Ability of natural nucleosides, dihydroorotic acid and orotic acid, to reverse the anti-influenza virus activity of D282 in cell culture
To explore this possibility, uninfected cells treated with the D282 for 24 h were analysed for relative nucleoside triphosphate levels (Figure 5). It was found that treated MDCK cells had severe depletion in UTP, and to a lesser extent CTP. Interestingly, CTP levels treated with 400 μM D282 were actually higher than cells receiving no treatment in repeated experiments. ATP and GTP levels were relatively unaffected by treatment of cells with D282. Addition of uridine to the cell culture medium restored both UTP and CTP levels in cells (Figure 6A). Cytidine treatment partially restored UTP levels and hyper-elevated CTP levels (Figure 6B).
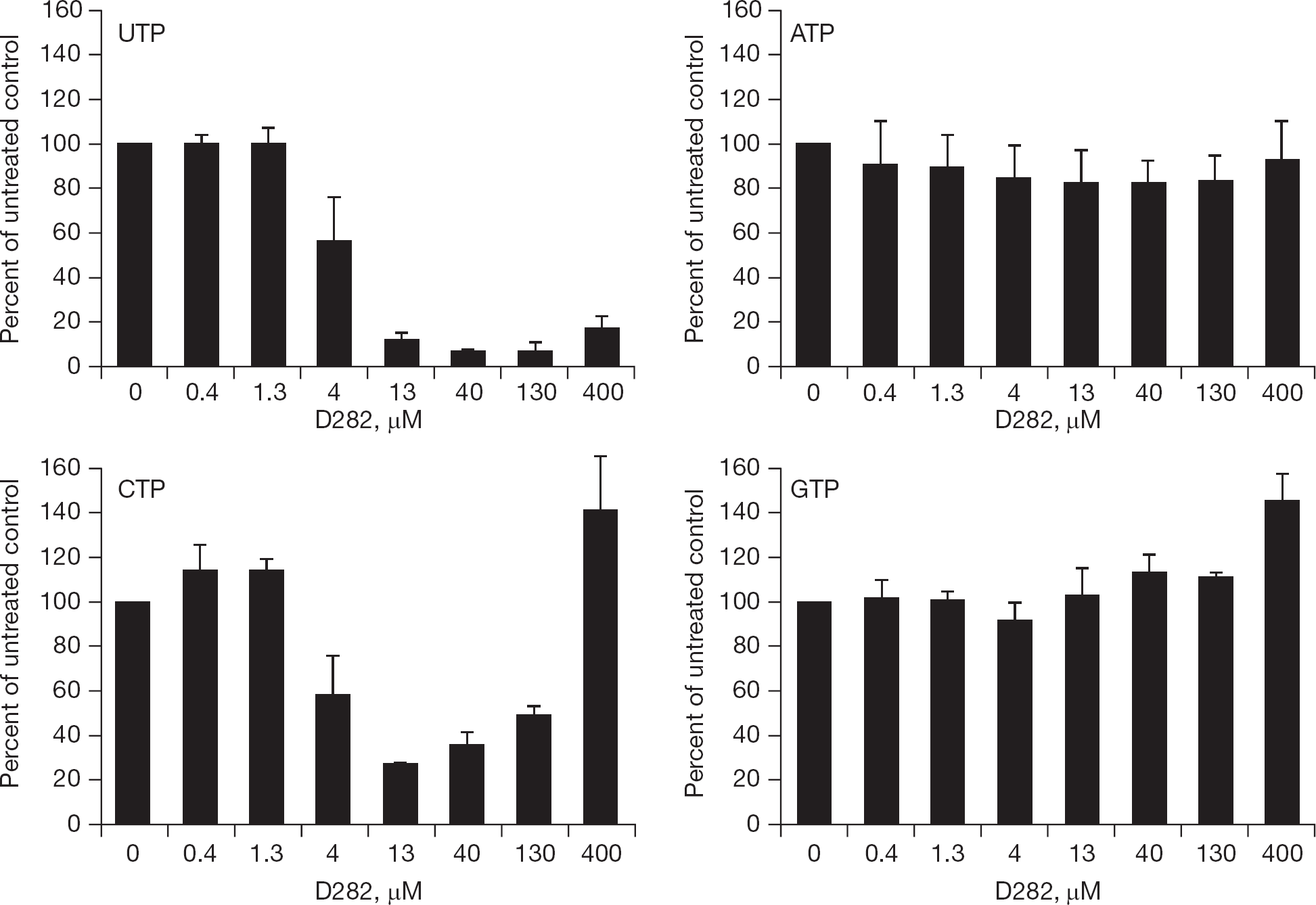
D282 suppresses intracellular nucleoside triphosphate pools in uninfected MDCK cells
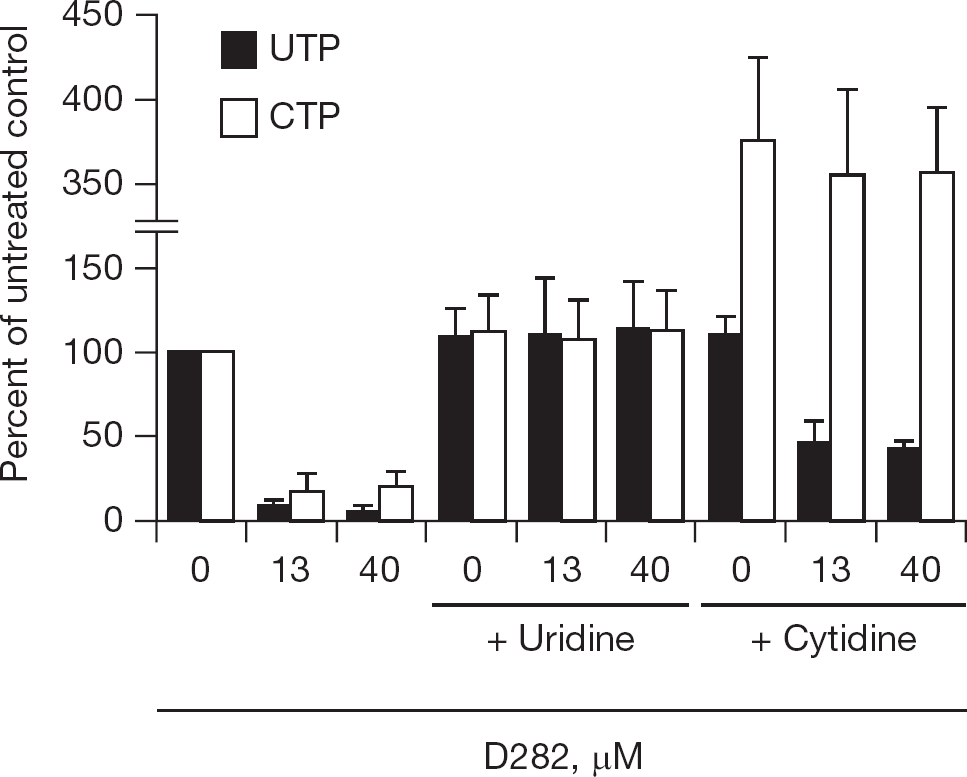
Effects of extracellular uridine and cytidine, each at 200 μM, treatment to restore nucleotide pools in uninfected MDCK cells treated with D282
Reversal of antiviral activity with orotic acid
Pyrimidine biosynthesis occurs in a multi-step pathway involving five enzymes and many substrates. This allowed for testing of compounds other than uridine or cytidine to determine whether they reversed the antiviral activity of D282. Treatment of cells with orotic acid caused reversal of antiviral activity, whereas treatment with dihydroorotic acid did not (Figure 4B). These results implicated the cellular enzyme dihydroorotate dehydrogenase as the target of action of D282.
Inhibition of dihydroorotate dehydrogenase from mouse liver
Dihydroorotate dehydrogenase was found to be inhibited by D282 in an uncompetitive manner (Figure 7), based upon the type of inhibition represented by the Lineweaver-Burk plot (parallel lines). The apparent Km value of enzymatic activity for dihydroorotic acid as substrate was 150 ±16 μM, and the Ki value for enzyme inhibition by D282 was 2.3 ±0.9 μM. The inhibition of this enzyme by D282 is consistent with the data showing reversal of antiviral activity by orotic acid (Figure 4B), since orotic acid is the product of the enzyme reaction that would be suppressed in cells treated with D282.
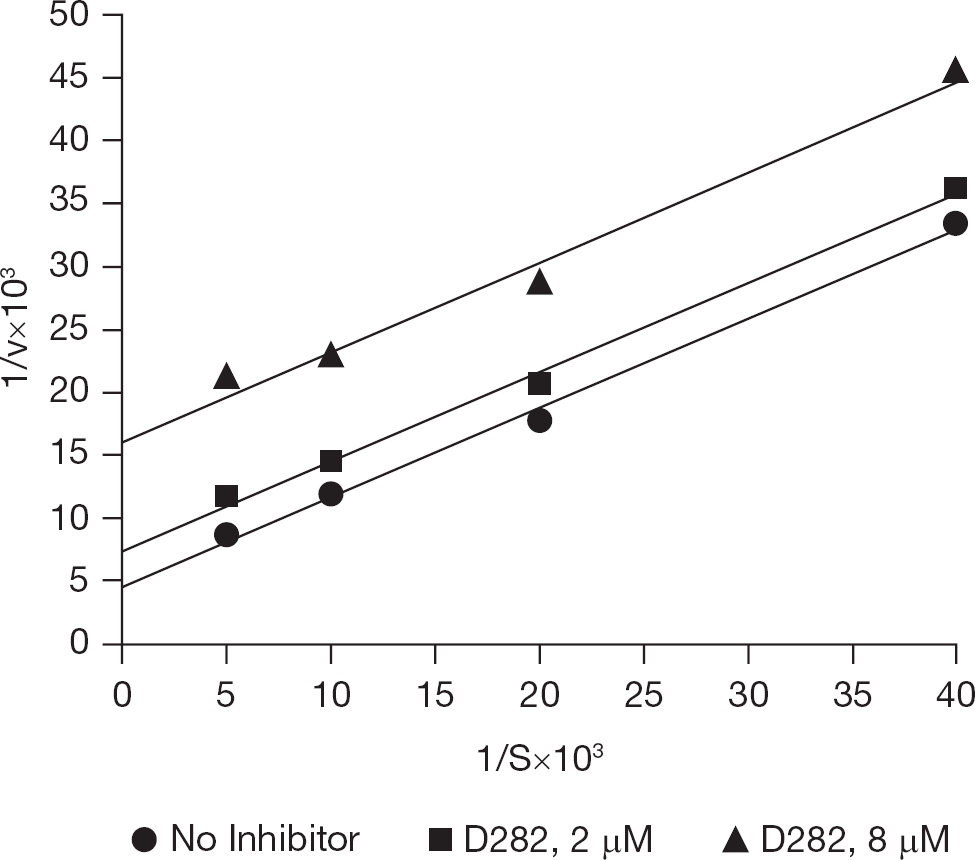
Double-reciprocal plot of the interaction of dihydroorotic acid and D282 with dihydroorotate dehydrogenase from mouse liver extracts
Antiviral activity of D282 in influenza-virus-infected mice
Mice were infected with influenza virus by intranasal route and treated twice a day for 5 days with D282 or ribavirin by three different routes of administration (Table 2). A lower concentration of D282 needed to be used for intranasal treatment compared to other routes of administration, due to causing excessive body weight loss. D282 provided no antiviral protection at the highest dose tested by each treatment route, whereas ribavirin exhibited protective activity. Data for lower (ineffective) doses of D282 are not shown.
Summary of antiviral activity of D282 compared to ribavirin in influenza (H1N1) virus-infected mice
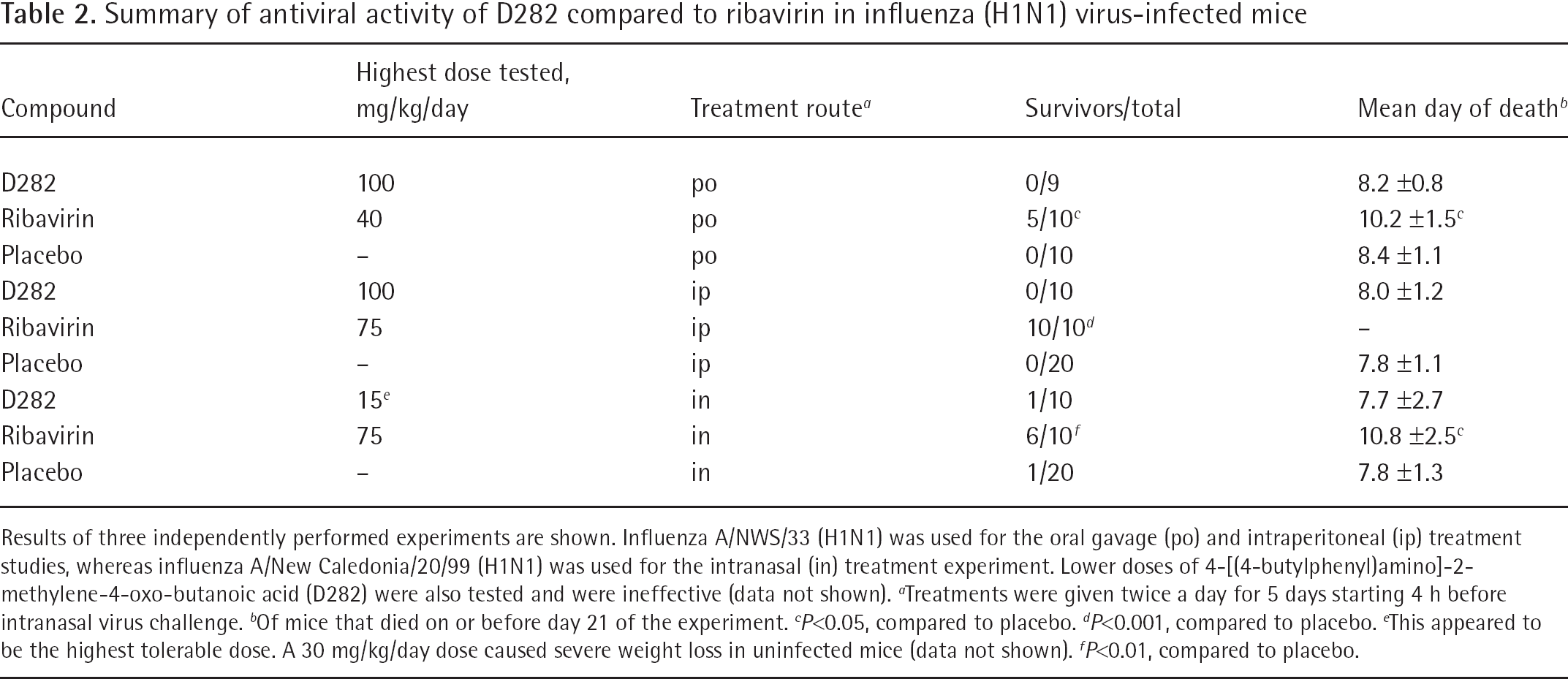
Results of three independently performed experiments are shown. Influenza A/NWS/33 (H1N1) was used for the oral gavage (po) and intraperitoneal (ip) treatment studies, whereas influenza A/New Caledonia/20/99 (H1N1) was used for the intranasal (in) treatment experiment. Lower doses of 4-[(4-butylphenyl)amino]-2-methylene-4-oxo-butanoic acid (D282) were also tested and were ineffective (data not shown).
Treatments were given twice a day for 5 days starting 4 h before intranasal virus challenge.
Of mice that died on or before day 21 of the experiment.
P<0.05, compared to placebo.
P<0.001, compared to placebo.
This appeared to be the highest tolerable dose. A 30 mg/kg/day dose caused severe weight loss in uninfected mice (data not shown).
P<0.01, compared to placebo.
Discussion
D282 was found to exhibit anti-influenza virus activity against a number of virus strains, with potency of the compound being similar against each virus. This activity was attributed to the inhibition of the cellular enzyme, dihydroorotate dehydrogenase, which leads to decreases in intracellular UTP and CTP concentrations. This was first hypothesized based upon the observation that treatment of cells with orotic acid overcomes (reverses) the antiviral activity of D282 (Figure 4B). Later we demonstrated by enzyme assays that D282 inhibited the conversion of dihydroorotic acid to orotic acid by a crude mitochondrial preparation of mouse liver dihydroorotate dehydrogenase. Orotic acid is a necessary precursor leading eventually to UTP in cells. When levels of UTP are low, this starves the viral RNA polymerase of substrate for replication. This anti-cellular effect of decreasing intracellular UTP will result in inhibition of other unrelated viruses sensitive to this effect [14]. Although not the focus of this report, we have recently found D282 to be inhibitory to other RNA viruses in vitro, such as arena-viruses, flaviviruses and bunyaviruses (data not shown).
The effects of certain natural nucleosides to reverse antiviral activity was evident from the studies presented in Figure 4A. It was the observation that uridine treatment reversed antiviral activity that led to investigation of pyrimidine biosynthesis inhibition. The salvage pathway converts uridine to uridine monophosphate and eventually to UTP. Reversal of activity by cytidine is supported by the observation in Figure 6 that cytidine treatment partially restored UTP levels in cells treated with D282, thus allowing for some virus replication. Cytidine most likely is metabolized to uridine nucleotides (UTP in particular) by deamination pathways. D282-treated cells undergo normalization when treated with exogenous uridine (that is, UTP at 100% of normal), whereas cells treated with cytidine had high levels of CTP. This indicates that cells are able to regulate UTP levels in the cell but not CTP levels to the same degree of control (if any). Curiously, CTP levels were greatly elevated in cells treated with 400 μM D282, whereas lower doses showed suppression of CTP levels. This suggests an additional mode of action of D282 at high concentration that causes the persistence of CTP in cells (some possibilities being the prevention of deamination or catabolism of the triphosphate form).
The inhibition of viral CPE required higher concentrations of D282 in cells treated with dihydroorotic acid compared to cells treated with D282 alone (Figure 3B), which did not appear to be statistically significant. Flooding the cells with excess dihydroorotic acid could allow some of it to be metabolized to orotic acid and eventually to UTP, thus allowing more virus replication to occur.
Antiviral activity was enhanced by adenosine treatment of cells also exposed to D282, and this occurred to a lesser extent with guanosine and xanthosine. These data would need to be validated by virus yield reduction to determine to what extent virus inhibition was enhanced. We did not study intracellular pool changes in cells treated with these inhibitors to know what effects might have occurred.
Several unrelated compounds have been discovered over the years that inhibit the pyrimidine biosynthetic pathway, and consequently found to have broad-spectrum antiviral activity. These include 6-azauridine [26,27], N10169 [27] and inhibitors of dihydroorotate dehydrogenase such as teriflunomide (formerly A77 1726, the active metabolite of leflunomide) [28–30], brequinar [31], compound A3 [14] and NITD-982 [31]. In our laboratory we found teriflunomide to be 50% inhibitory to influenza A/Victoria/3/75 (H3N2) virus in a CPE assay at 5 μM (data not shown), which is similar to the potency of D282. Not surprisingly, D282 was cell-inhibitory to actively dividing cells and will almost certainly be immunosuppressive, as are other inhibitors of dihydroorotate dehydrogenase [32–34].
To date, there have been few successes in treating viral diseases in vivo with inhibitors of pyrimidine biosynthesis. Leflunomide was recently reported to reduce respiratory syncytial virus (RSV) production in cotton rats by over 1,000-fold [28]. In mice, leflunomide reduced inflammation during RSV infection [28]. Leflunomide is also being investigated for treating BK virus infections in renal transplant recipients [32,35]. These are encouraging results that need to be investigated further. Swine infected with transmissible gastroenteritis virus may have received partial benefit from treatment with 6-azauridine [36]. N10169 was ineffective in treating mice infected with influenza B and Banzi viruses, and similarly devoid of activity against a vaccinia virus infection in hamsters [27]. Similarly, we showed here that D282 was ineffective in treating influenza infections in mice. NITD-982 did not protect mice infected with dengue virus [31]. The authors attributed the lack of activity in mice to exogenous uptake of pyrimidine from the diet (uridine would reverse antiviral activity, as was demonstrated in the in vitro studies of this report) or else to high plasma protein-binding activity of the compound. Treatment of other infections in vivo with pyrimidine biosynthesis inhibitors has been largely unexplored.
Using pyrimidine biosynthesis inhibitors for treatment of virus infections involves immunosuppressive effects as well [32–34]. There appears to be benefit to immunosuppressive treatment in RSV infections in rodents [28,37]. In the case of BK virus infections in humans, the patients require immunosuppression anyway to prevent organ transplant rejection, so using a compound such as leflunomide that has both immunosuppressive and antiviral properties may be desirable [32,35]. Investigators have studied whether reduction in the cytokine storm caused by influenza virus would benefit the host. There are some indications that this is the case with certain immunosuppressants used alone or combined with antiviral agents [38,39]. Immunosuppression during certain infections such as those caused by vaccinia results in exacerbation of disease [40,41]. To avoid this problem, Schnellrath and Damasco [42] suggested that brequinar, a pyrimidine pathway inhibitor, might be used topically against cutaneous vaccinia infections.
Reversal of immunosuppressive or antiviral activity by dietary pyrimidines (specifically uridine) could account for low efficacy of pyrimidine pathway inhibitors in mice [31]. Whether this is a greater problem in mice than in other animal species is not known. The majority of in vivo animal models of virus infection use mice. Ferrets may be used for influenza infection studies [43] and represent an alternative infection model. Other possible explanations for the lack of in vivo efficacy of D282 may be associated with uptake and metabolism of the compound, and its residence time in target tissues, which were not investigated in these studies. However, because of weight loss occurring in mice that were intranasally treated with D282 (Table 2), this argues for an in vivo biological effect (although unfavourable, since it represents toxicity).
There are a number of chemically unrelated compounds that affect pyrimidine biosynthesis by virtue of inhibition of dihydroorotate dehydrogenase [14,27–31]. D282 is yet another one of these inhibitors. The potential of this class of compounds for treatment of virus infections in vivo may be limited, but yet has only been minimally investigated. Although D282 was not active in vivo in the current study, this class of compounds may have potential for treatment of other unrelated virus infections in vivo and merits further investigation, particularly in using these types of compounds in combination with clinically approved antiviral drugs.
Footnotes
Acknowledgements
This work was funded by contract N01-AI-15435 and contract N01-AI-30063 (awarded to Southern Research Institute), both from the Virology Branch, National Institute of Allergy and Infectious Diseases, National Institutes of Health, USA. The contents of this article do not necessarily reflect the policy of the government and no official endorsement shall be inferred.
The authors declare no competing interests.