
Abstract
Background:
Host genes serving potential roles in virus replication may be exploited as novel antiviral targets. Methods: Small interfering RNA (siRNA)-mediated knockdown of host gene expression was used to validate candidate genes in screens against six unrelated viruses, most importantly influenza. A mouse model of influenza A virus infection was used to evaluate the efficacy of a candidate FDA-approved drug identified in the screening effort.
Results:
Several genes in the PI3K-AKT-mTOR pathway were found to support broad-spectrum viral replication in vitro by RNA interference. This led to the discovery that everolimus, an mTOR inhibitor, showed in vitro antiviral activity against cowpox, dengue type 2, influenza A, rhino- and respiratory syncytial viruses. In a lethal mouse infection model of influenza A (H1N1 and H5N1) virus infection, everolimus treatment (1 mg/ kg/day) significantly delayed death but could not prevent mortality. Fourteen days of treatment was more beneficial in delaying the time to death than treatment for seven days. Pathological findings in everolimus-treated mice showed reduced lung haemorrhage and lung weights in response to infection. Conclusions: These results provide proof of concept that cellular targets can be identified by gene knockout methods, and highlight the importance of the PI3K-AKT-mTOR pathway in supporting viral infections.
Introduction
Seasonal influenza is thought to affect approximately 20% of the world's population, causing an estimated 500,000 deaths worldwide each year [1]. Vaccines may provide excellent protection against diseases associated with slowly replicating or genetically stable viruses, such as smallpox or poliovirus. However, vaccine efficacy is diminished for prophylaxis of highly mutable and rapidly replicating viruses such as HIV or HCV, and is additionally reduced in vulnerable groups, for example, the elderly and immunoincompetent [2]. Delays in vaccine production with late seasonal antigenic shift, as with the recent H1N1 pandemic strain, may leave populations at risk prior to production [3,4].
Current anti-influenza drugs target viral proteins, and fall into 2 classes: M2 ion channel blockers (amantadine and rimantadine) and neuraminidase (NA) inhibitors (oseltamivir/Tamiflu® and zanamivir/Relenza®). Although both classes of drugs have been effective in controlling influenza, their use can be compromised due to occurrences of escape mutations or toxic side effects [5,6]. While M2 ion channel blockers can be effective against influenza A, influenza B encodes an alternative protein (NB), which is not inhibited by amantadine [6,7]. One of the influenza strains of the 2008–2009 season was resistant to the NA inhibitor oseltamivir, the leading stockpiled influenza therapy [8]. This resistance emerged spontaneously in Scandinavia, and has spread widely. Such resistance to oseltamivir typically is associated with two additional enabling mutations [9]. Limitations associated with vaccines and the rising incidence of antiviral resistance in circulating influenza strains highlight the need to develop additional antiviral therapies.
The purpose of the current study was to identify cellular pathways used in viral replication. We used gene-trap insertional mutagenesis [10,11] to discover novel cellular genes that can be targeted to block viral infection. Cells are infected with a replication-deficient Moloney murine leukaemia virus (MMLV) encoding a promoter-less selectable marker, neomycin aminotransferase (neo). Integration of the MMLV vector between host promoters and early attendant exons disrupts (traps) cellular genes, allowing expansion of mutagenized cells with subverted gene expression following neomycin selection. Resulting gene-trap libraries comprise a mixed-cell population library harbouring approximately 40,000 trapped events. We have previously employed gene-trap libraries to identify host genes whose disruption confers survival following lytic viral infection [12–14] or bacterial intoxication [15].
We found five members of the phosphatidylinositol 3′-kinase (PI3K)-AKT-mammalian target of rapamycin (PI3K-AKT-mTOR) pathway to be associated with lytic infections of both RNA and DNA viruses, including influenza A. These data are concordant with the known involvement of this pathway in replication of a diverse array of viruses [16,17]. It has been found that following virus entry, the influenza A NS1 protein can bind the PI3K regulatory subunit (p85β), allowing the catalytic domain (p110) to activate AKT by phosphorylation [18–23]. The downstream role(s) of AKT and mTOR in influenza replication have not been clearly defined, although AKT activation may serve to promote mTOR-dependent stimulation of viral protein translation [16] and/or inhibition of the BCL2-associated agonist of cell death to prevent premature apoptosis [24], prior to viral egress. In the present study, we extended these observations to in vivo infection with highly lethal strains of influenza in mice using the mTOR inhibitor everolimus.
Materials and methods
Compounds
Everolimus was obtained from Sigma (St Louis, MO, USA). It was prepared as a 100 mM stock in DMSO and diluted into cell culture medium for in vitro studies. Everolimus was similarly prepared, then diluted in saline to a final 1.8% DMSO concentration for experiments in mice. Ribavirin was acquired from the former ICN Pharmaceuticals (Costa Mesa, CA, USA), and prepared in saline for treatment of mice. Oseltamivir (as Tamiflu®) was obtained by prescription from a local pharmacy and used for research purposes only. Entire capsules of Tamiflu were added to water then diluted down to appropriate mg/kg/day doses. Calculations of mg/kg/day doses of oseltamivir were based upon the liberated form of the drug (oseltamivir carboxylate, 75 mg per capsule), not the prodrug form of the drug in the capsule (oseltamivir phosphate).
Viruses and cell lines
Cowpox virus (Brighton strain), influenza A/WS/33 (H1N1) virus, human rhinovirus type 16 (HRV16, 11757 strain), and respiratory syncytial virus (RSV, strain A2) were obtained from the American type culture collection (ATCC, Manassas, VA, USA). Herpes simplex virus type 2 (HSV-2, 186 strain) was kindly provided by David Knipe (Harvard University, Cambridge, MA, USA) and dengue fever virus type 2 (DFV-2, 16681 strain) was provided by Guey Perng (Emory University, Atlanta, GA, USA).
Influenza A/NWS/33 (H1N1) was originally obtained from Kenneth Cochran (University of Michigan, Ann Arbor, MI, USA) and was lethal to mice. Influenza A/ Solomon Islands/03/2006 (H1N1) virus was obtained from the Centers for Disease Control and Prevention (Atlanta, GA, USA). The virus became lethal to mice after seven serial passages in the lungs of infected animals. Influenza A/Duck/MN/1525/81 (H5N1) virus, a low pathogenic North American strain, was provided by Robert Webster (St Jude Children's Research Hospital, Memphis, TN, USA) and was passaged 3× in mice to enhance its virulence. Virus pools were pre-titrated in mice prior to performing these studies to determine approximate dose of virus lethal to 50% of inoculated mice (MLD50).
The following reagent was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: TZM-bl cells from John C Kappes, Xiaoyun Wu and Tranzyme Inc. (Durham, NC, USA). Madin-Darby canine kidney (MDCK), Vero E6, Hep3B and HepG2 cells were obtained from the ATCC (Manassas, VA, USA). Viral stocks of cowpox virus, HSV-2 and RSV were grown in Vero E6 cells. Influenza A/WS/33 (H1N1) stocks were produced in HepG2 cells, whereas the other influenza A viruses were amplified in MDCK cells. DFV-2 and HRV16 stocks were grown in Hep3B and TZM-bl cells, respectively. Cowpox, DFV-2, HSV-2 and influenza A viral stocks were produced by infecting the indicated cell lines, harvesting clarified supernatants following the appearance of approximately 75% cytopathic effects in the monolayers, and titering by standard plaque or end point dilution (for influenza) assays.
Gene-trap library construction
The U3neoSV1 retrovirus shuttle vector [25] was obtained from H Earl Ruley (Vanderbilt University, Nashville, TN, USA). RIE-1, Vero E6 and HepG2 cells served as suitable parental cell lines for the preparation of gene-trap libraries, as they are efficiently killed by infection with reovirus (RIE-1 cells), influenza A (HepG2 cells), or either HSV-2, RSV or poliovirus infection (Vero E6 cells). Gene-trap libraries were prepared from parental, virus-sensitive cells as described [12–14].
Generation of cells lines resistant to lytic viral infection from gene-trap libraries
Clonal reovirus-resistant RIE-1 library cell lines were prepared as described previously [13,14]. Vero E6 library cells were used to select cell lines resistant to HSV-2 and HepG2 library cells were used for influenza A studies. Briefly, gene-trap libraries, each harbouring approximately 104 gene entrapment events, were expanded to 80–90% confluency until approximately 103 daughter cells represented each clone. Vero E6 were infected with HSV-2 using a multiplicity of infection (MOI) of 0.005 and HepG2 cells were infected with influenza A using an MOI of 0.001. Infection proceeded at 37°C until >90% of cells were dead, at which point the medium was changed every 2-3 days to remove dead cells. Dead cells were removed by aspiration for 2–3 weeks until surviving clones were visually observed. Individual clones were detached with trypsin and isolated into separate wells of 24-well plates. Following expansion, resistance confirmation was performed by replica plating clones into duplicate 24-well plates and re-infecting clonal cell lines with a 20-fold higher MOI than used in the initial viral selection for 2 h at 37°C before changing the medium. Clones that showed >70% survival following re-infection were selected for expansion to identify trapped genes.
U3neoSVl shuttle vector rescue and sequencing Genomic DNAs from clonal virus-resistant cell lines were extracted using a QIAamp DNA Blood Mini kit (Qiagen, Inc., Valencia, CA, USA). Shuttle vectors and genomic DNA flanking the U3neoSV1 integration site were recovered by restriction enzyme digests of genomic DNA, self-ligation, transformation into Escherichia coli, and sequencing the resultant carbenecillin-resistant plasmids to identify trapped genes, as described [12–14].
RNA interference and qRT-PCR studies
Candidate genes identified in gene-trap studies were tested by RNA interference against six different viruses. Vero cells were used for studies with cowpox virus, HSV-2 and RSV (strain A2). Hep3B, HepG2 and TZM-bl cells were used for respective studies involving DFV-2 (Strain 16681), influenza A/WS/33 (H1N1) and HRV16 (11757 strain).
Cells were seeded in six-well plates (105 cells/well) and transfected in duplicate wells with small interfering RNAs (siRNAs) targeting IRS1, AKT1, AKT2, mTOR, RAPTOR or FKBP8 (siRNAs from Dharmacon, Inc., Lafayette, CO, USA), along with relevant negative and positive control siRNAs. In all experiments, cells were transfected in quadruplicate with AllStars Negative control siRNA (Qiagen, Inc.) or a non-targeting siRNA control from Dharmacon, Inc. Negative controls were used to normalize quantitative reverse transcriptase PCR (qRT-PCR) results to 100%. The viral genes targeted by positive control siRNAs are as follows: D5R and D7R (cowpox), PrM (DFV-2), UL29 (HSV-2), PA (influenza A), 2C and VP4 (HRV16), and the P gene (RSV). Duplicate wells were seeded for uninfected and infected controls, lacking transfection reagents.
Transfections were performed using either Lipofectamine 2000 (Invitrogen) or HiPerfect (Qiagen), according to the manufacturer's suggested protocols. Lipofectamine 2000 was used for transfecting siRNAs into Vero cells, Hep3B cells and HepG2 cells, and HiPerfect was more effective with TZM-bl cells. Cells were transfected using a final concentration of 50 nM siRNA in the culture supernatant.
Cells were infected at 48 h post-transfection, using MOIs of 1 (cowpox and HSV-2), 0.1 (influenza A), 0.01 (HRV16 and RSV) or 0.001 (DFV-2). Cells were infected for 1 h at either 37°C (cowpox, DFV-2, HSV-2, influenza A or RSV) or 33°C (HRV16). Following the 1-h incubation period, cells were washed with PBS and fresh medium was added. At 3 days post-inoculation, culture supernatants were clarified by centrifugation and 200 μl was lysed in preparation for RNA extraction, using an epMotion 5075 Workstation (Eppendorf, Hauppauge, NY, USA) and the PureLink 96 Total RNA Purification Kit (Invitrogen). Total RNA was reverse transcribed using random hexamers (Applied Biosystems) and qRT-PCR was performed using an Eppendorf Mastercycler RealPlex2 system, with TaqMan assays developed that detect viral DNAs. To generate standards curves, amplicons produced during real time PCR detection of each viral cDNA were cloned into the pCRII vector (Invitrogen). qRT-PCR was performed using freshly prepared standards, serially diluted over 8 logs of copy numbers.
In vitro antiviral studies with everolimus
To assess the effects of everolimus on viral replication and cellular toxicity, cells were treated overnight over a range of concentrations in 24-well plates. Cell lines used for the respective studies were: HepG2 (influenza A), Vero E6 (cowpox, HSV-2 and RSV), Hep3B (DFV-2) and TZM-bl (HRV16). Following overnight treatment with everolimus or 0.1% DMSO (solvent control), the medium was changed and cells were infected with the indicated viruses for 1 h, using MOIs indicated above in RNA interference studies. Subsequently, cells were washed with PBS and grown for 3 days in fresh medium containing the indicated everolimus concentrations. At 3 days post-inoculation, lysates from clarified supernatants were processed for quantitative real time PCR analysis to quantitate relative viral production levels, as described above. Viral production in culture supernatants from everolimus-treated cells was expressed relative to solvent controls (100%).
Animal experiment design
Female BALB/c mice (18–20 g) were obtained from Charles River Laboratories (Wilmington, MA, USA). They were anaesthetized by intraperitoneal injection of ketamine/xylazine (50/5 mg/kg) followed by infection intranasally with a 90-μl suspension of influenza virus in cell culture medium. The infection inoculations equated to three to four 50% mouse lethal challenge doses (MLD50). The compounds were administered once a day for 7 or 14 days starting 2 h before infection. From past experience it was known that ribavirin is effective at 75 mg/kg/day and oseltamivir at 10 mg/kg/day [26]. Everolimus was evaluated at a dose previously shown to inhibit both tumour proliferation and transplant rejection in mice, 1 mg/kg/day [27–29]. The compound was pre-determined in toxicity assays performed in uninfected mice to be safe for administration, and is near its maximum tolerated dose. Ten drug-treated infected mice and 20 placebo-treated controls were observed daily for death through 21 days. An additional 5 mice per group were maintained for determining lung infection parameters on day 6 of the infection. On that day, five mice from the placebo and treatment groups were sacrificed, the lungs were weighed and scored for lung haemorrhage on a scale of 0 (normal) to 4 (maximum plum colouration over entire lung) [30] and then frozen at −80°C. At a later date, the lungs were homogenized and titrated for the presence of virus by end point dilution method [31] in 96-well microplates [32]. Virus titres are reported as log10 50% cell culture infectious dose (CCID50)/g of tissue.
Statistical methods and analysis of animal model studies
Kaplan-Meier survival curves were generated and compared by the Mantel-Cox log-rank test followed by pairwise comparisons using the Gehan-Breslow-Wilcoxon test. The relative experimental significance was adjusted to a Bonferroni corrected significance threshold based on the number of treatment comparisons made. Mean day of death and mean lung haemorrhage score comparisons were analysed by the Kruskal-Wallis test combined with Dunn's multiple comparisons test. Mean lung weights and log10 lung virus titres were evaluated by ANOVA, assuming equal variance and normal distribution. Following ANOVA, individual treatment values were compared to placebo control by the Tukey-Kramer multiple comparisons test. Analyses were made between treated and placebo groups using Prism® software (GraphPad Software, San Diego, CA, USA).
Results
Implication of PI3K-AKT-mTOR pathway genes in viral replication by gene-trapping
Gene-trap insertional mutagenesis studies performed in our collaborative laboratories have facilitated the identification of over 1,000 candidate cellular genes mediating cell lysis following viral infection or bacterial intoxication. Five genes associated with the PI3K-AKT-mTOR pathway (Figure 1) were disrupted (trapped) in clonal cell lines surviving lytic viral infections with either influenza A/WS/33 (H1N1), HSV-2 or reovirus. Trapped genes included the insulin receptor substrate 1 (IRS1, influenza A/WS/33), AKT2 and mTOR (HSV-2), regulatory associated protein of mTOR (RAPTOR) and FK506 binding protein 8 (FKBP8; reovirus).
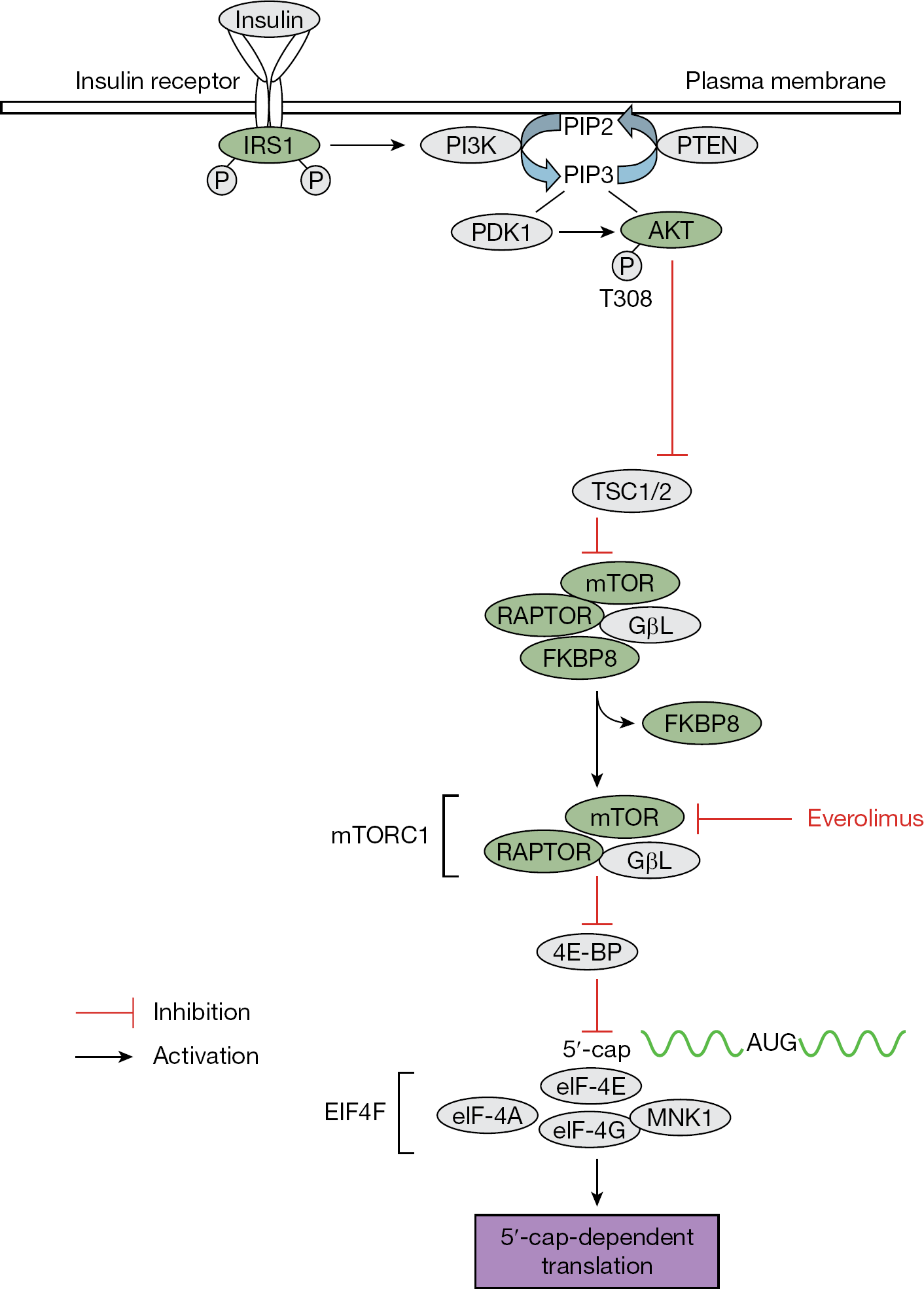
Trapped genes associated with the PI3K-AKT-mTOR pathway
In vitro RNA interference studies
As an initial screen to characterize the spectrum of utilization of PI3K-AKT-mTOR pathway genes, target gene expression was silenced with siRNAs prior to infection. Following a 48-h transfection period, cells were infected with cowpox, DFV-2, HRV16, HSV-2, influenza A/WS/33 (H1N1) or RSV. siRNA screening results indicated that each of the genes tested in the PI3K-AKT-mTOR pathway were required for efficient replication of 2–4 out of the 6 viruses tested, suggesting potential targets for broad-spectrum viral inhibition within this pathway (Table 1). Knockdown of mTOR expression resulted in resistance to cowpox and HRV16 replication; however, inconsistent results were found in mTOR siRNA knockdown experiments studying influenza A replication.
In vitro inhibition of viral replication with siRNAs targeting PI3K-AKT-mTOR pathway genesa
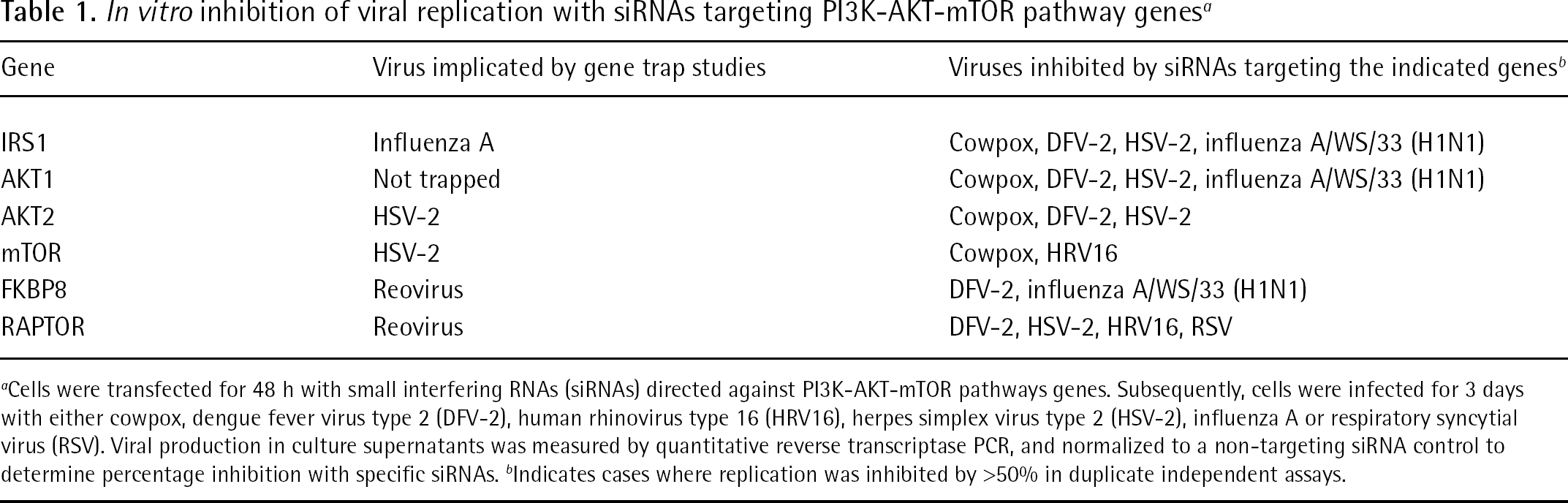
Cells were transfected for 48 h with small interfering RNAs (siRNAs) directed against PI3K-AKT-mTOR pathways genes. Subsequently, cells were infected for 3 days with either cowpox, dengue fever virus type 2 (DFV-2), human rhinovirus type 16 (HRV16), herpes simplex virus type 2 (HSV-2), influenza A or respiratory syncytial virus (RSV). Viral production in culture supernatants was measured by quantitative reverse transcriptase PCR, and normalized to a non-targeting siRNA control to determine percentage inhibition with specific siRNAs. b Indicates cases where replication was inhibited by >50% in duplicate independent assays.
In vitro everolimus antiviral testing
As siRNA knockdown of several genes within the PI3K-AKT-mTOR pathway resulted in limitation of several viruses' capacity to replicate, we evaluated the antiviral efficacy of everolimus. Everolimus is an FDA-approved hydroxyethyl derivative of rapamycin known to inhibit mTOR, which has been used to treat renal cell carcinoma [33] and to prevent allograft rejection following heart and kidney transplants [34,35]. It has also been associated with a reduced incidence of cytomegalovirus infection in transplant recipients [36,37]. As indicated in Figure 2, everolimus treatment showed antiviral efficacy against influenza A virus, cowpox virus, DFV-2, HRV16 and RSV. HSV-2 replication was not inhibited at any everolimus concentration tested. No observable cellular toxicity was observed microscopically in association with everolimus treatment at any concentration (up to 450 nM), compared to solvent controls.
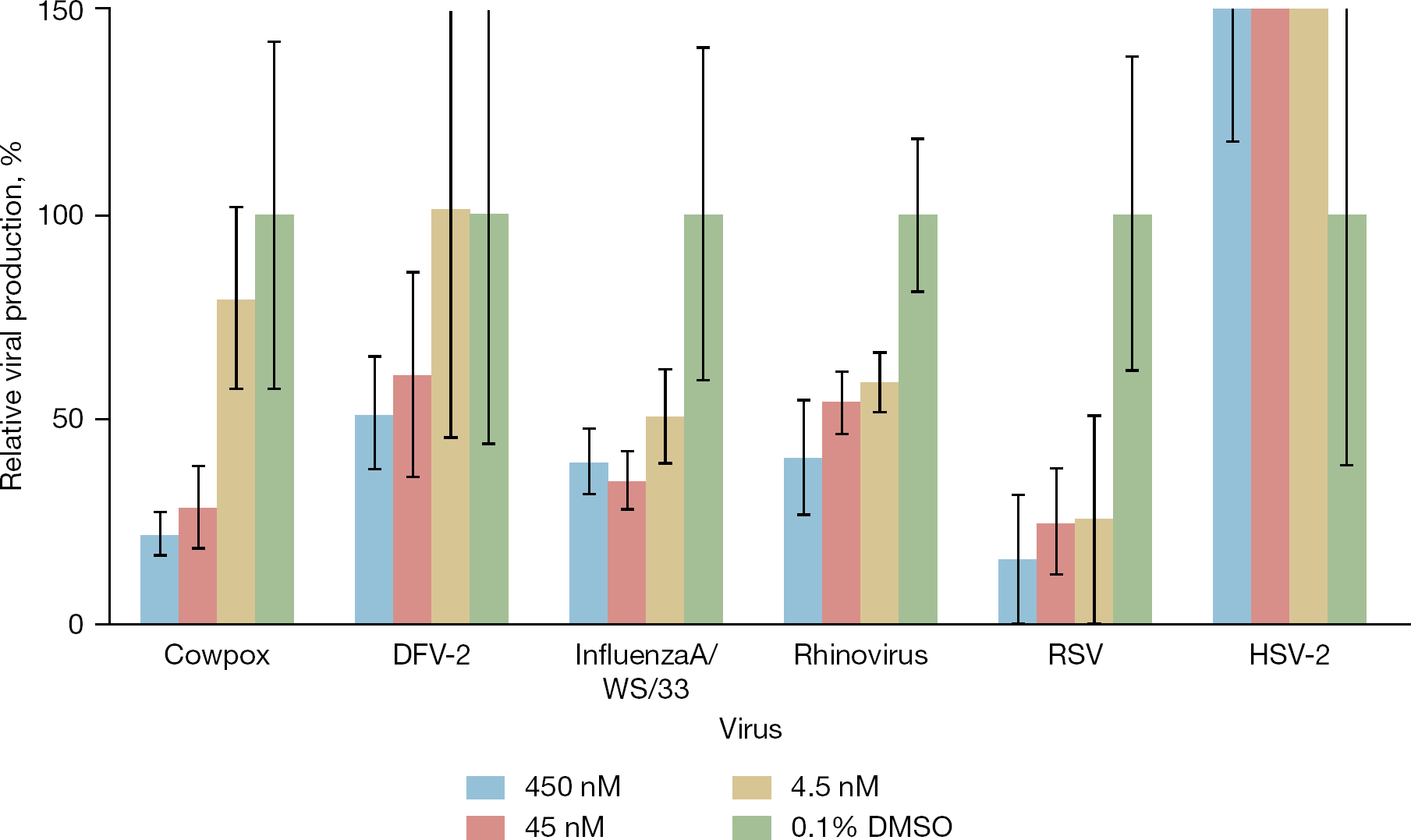
Viral inhibition with everolimus
Mouse infection studies
Treatment of mice was performed using everolimus and ribavirin or oseltamivir against influenza A H1N1 and H5N1 virus infections (Figure 3). Mice were administered everolimus intraperitoneally for either 7 or 14 days to determine which regimen might be more effective. Seven days of everolimus treatment delayed the time to death of mice infected with influenza A/ NWS/33 (H1N1) virus by 1.4 days, whereas 14 days of treatment resulted in a 2.8-day delay in death, with one mouse surviving (Figure 3A). Treatment with ribavirin protected all animals from death. Treatment of the influenza A/Duck/MN/1525/81 (H5N1) virus infection produced similar outcomes (Figure 3B). The 7- and 14-day treatments with everolimus delayed the time to death by 1.3 and 2.6 days, respectively, with one mouse surviving the infection in the 14-day treatment group. Ribavirin protected all mice from death. Results with the A/NWS/33 (H1N1) strain were confirmed using the influenza A/Solomon Islands/03/2006 (H1N1) virus, which was a prominent H1N1 strain in circulation prior to the emergence of the 2009 pandemic H1N1 virus. Everolimus treatment caused a 1.8-day delay in the time of death with one mouse surviving infection (Figure 3C), which was similar to that seen for infection with other strains of influenza A virus. Oseltamivir was used as a positive control and provided 90% protection against the infection.
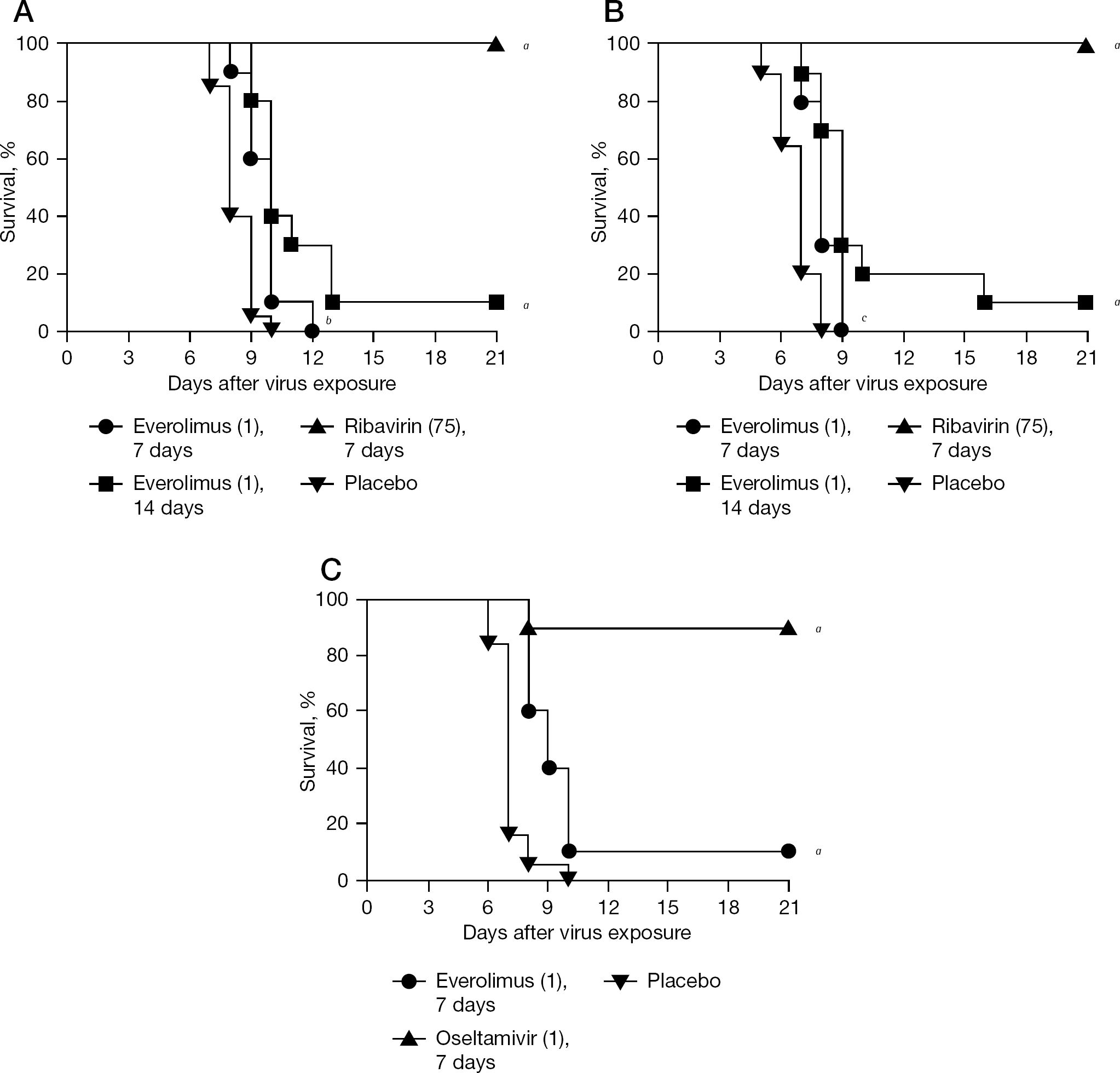
Survival curves for influenza A virus-infected mice treated with everolimus, ribavirin or oseltamivir
Lung samples were harvested from groups of mice during the influenza A/Duck/MN/1525/81 (H5N1) virus infection (Figure 3B), and were analysed to determine lung haemorrhaging, lung weights and lung virus titres (Figure 4). Lung haemorrhage scores and virus titres were significantly less in everolimus-treated mice than in placebo-treated mice on day 3, and lung weights were significantly reduced on day 6 (Figure 4). Lung virus titres in everolimus-treated animals exceeded titres in the placebo group on day 6. Treatment with ribavirin resulted in significant reductions in lung infection parameters on both days. Overall, the ability of ribavirin to reduce lung infection parameters exceeded that of everolimus, which translated into a survival benefit (Figure 3B).
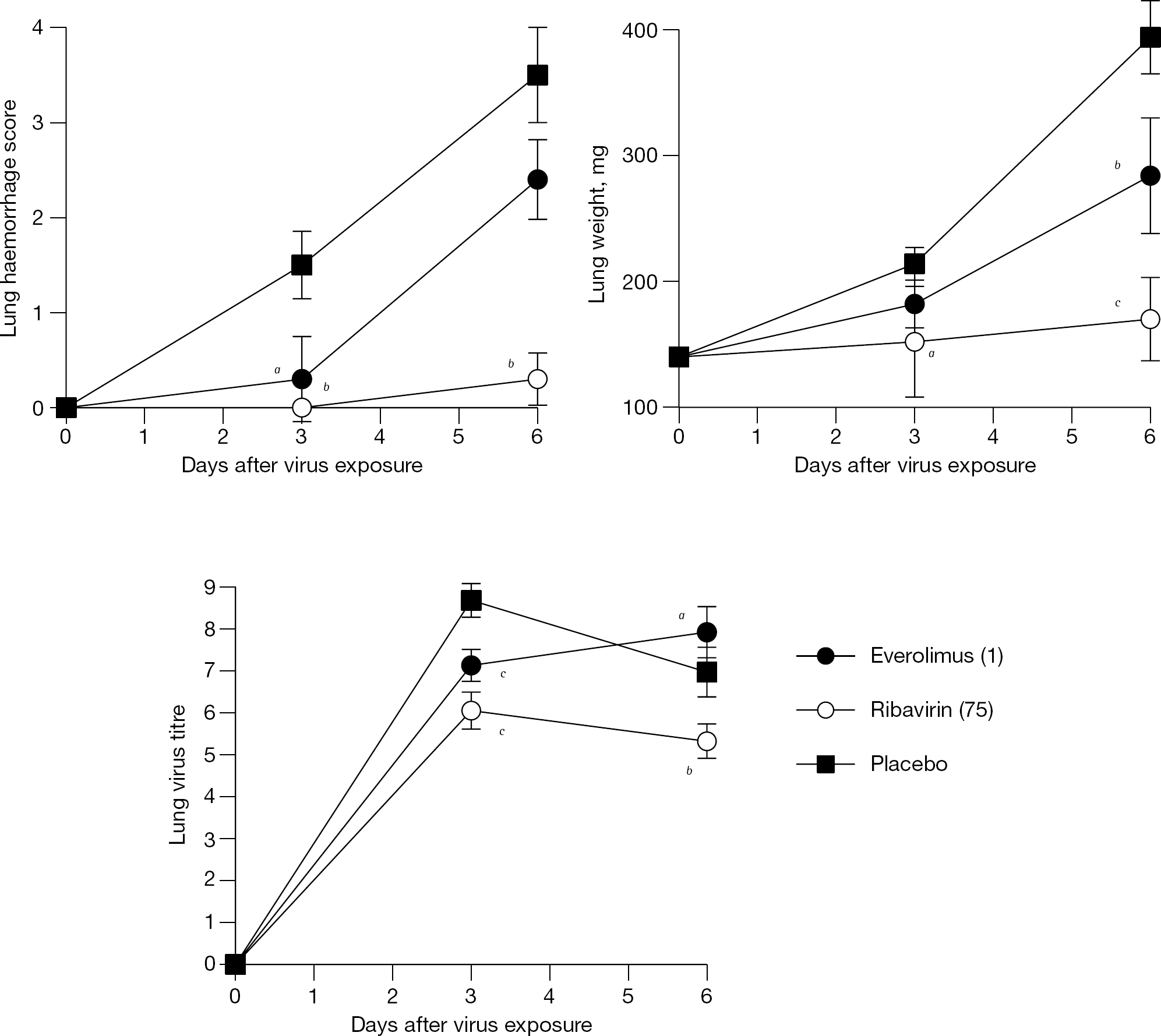
Mean lung infection parameters measured during an influenza A/Duek/MN/1525/81 (H5N1) virus infection in mice
Discussion
Signalling through the PI3K-AKT-mTOR pathway is known to augment replication of several viruses, and can be initiated through ligand binding/activation of receptor tyrosine kinases such as the insulin receptor. Upon receptor activation, the insulin receptor substrate (IRS1) binds and activates PI3K (Figure 1). Active PI3K converts PIP2 to PIP3 which then recruits both phosphoinositide-dependent protein kinase 1 (PDK1) and AKT1 or AKT2 to the membrane. PDK1 can then activate AKT1 or AKT2 by phosphorylation [38] to reverse mTOR inhibition by TSC1/2. Following FKBP8 dissociation, the active mTORC1 blocks eIF4F inhibition by 4E-BP, thereby promoting 5′ cap-dependent translation (reviewed in [16,17]).
We found that members of the PI3K-AKT-mTOR pathway were disrupted in gene-trapped cell clones surviving lytic infection with several different families of virus. RNAi was used to validate that knockdown of the identified genes conferred a phenotype of resistance to virus infection, with six unrelated RNA and DNA viruses. siRNA targeting of two trapped genes (IRS1 and AKT2) were active when tested against the original trapping virus, confirming the gene-trapping events (Table 1). Influenza A virus infection was inhibited by siRNA knockdown of IRS1, FKBP8 and AKT1 (but not AKT2). These data are consistent with IRS1 serving as an upstream activator of the PI3K-AKT-mTOR pathway (Figure 1) and are in agreement with data presented here and in the literature implicating the pathway in viral infections. FKBP8 was originally described as an endogenous mTORC1 inhibitor [39], in which case silencing FKBP8 expression would be predicted to enhance pathway activation, making it difficult to rationalize how FKBP8 silencing inhibits influenza. However, recent studies have challenged the findings that FKBP8 blocks mTORC1 function [40,41]. In separate studies, it was shown that FKBP8 binds the HCV NS5A protein, and that disrupting the FKBP8-NS5A interaction inhibits HCV replication and promotes apoptosis [42–44].
We have observed that while siRNA screening may be useful as a preliminary test to highlight potential antiviral targets, toxicities inherent to the transfection process may generate relatively large experimental errors, making independent confirmation with known target antagonists desirable. Although mTOR siRNA transfection did not inhibit influenza replication, collectively, the silencing data (Table 1) suggested that the PI3K-AKT-mTOR pathway is broadly utilized during replication of diverse viruses, providing rationale for in vitro testing of everolimus against the panel of six viruses. Investigation of potential antiviral activity of everolimus was also based upon the premise that cellular pathways used by viruses may be effective targets for intervention, and that repurposed drugs (such as everolimus) may be used to inhibit important cellular targets. Everolimus showed in vitro activity against multiple viruses including influenza A, prompting us to evaluate everolimus in vivo.
Treatment of influenza-infected mice with everolimus resulted in significant delays in the mean days to death compared to placebo-treated animals against infections with three different influenza A virus strains. In fact, all three infections showed a very similar response to treatment. However, treatment with everolimus was not able to provide complete protection from death. Mice treated with everolimus showed significant reductions in lung haemorrhaging and lung viral titres at day 3 post-infection with A/ Duck/MN/1525/81 (H5N1), but not day 6 post-infection. Everolimus also significantly reduced lung weights at day 6 post-infection. In contrast, ribavirin significantly reduced lung haemorrhaging, lung weights and lung viral titres at both time points.
Results from gene-trap studies and siRNA validation screens indicated that the PI3K-AKT-mTOR pathway is utilized during the replication of several unrelated RNA and DNA viruses, highlighting the potential for targeting alternative pathway members in controlling viral infection. However, the weak antiviral activity of everolimus suggests that mTOR may not be the ideal target within this pathway. To date, no drugs targeting cellular proteins have been developed to control influenza virus infection in humans, and much additional work remains to identify viable host targets and associated antagonists for anti-influenza chemotherapy. As insight is gained into critical host factors mediating the viral-host standoff, development of safe and effective anti-influenza drugs directed against durable host targets may help to advance this currently unmet need.
Footnotes
Acknowledgements
This work was supported, in part, by Public Health Service Small Business Innovation Research (SBIR) grant AI084705 (awarded to Zirus, Inc. and The University of Texas Medical Branch at Galveston) from Division of AIDS, National Institute of Allergy and Infectious Diseases (Bethesda, MD, USA). DHR was supported by gifts from Maggie Chassman, Buisson Foundation and the Public Health Service. We thank Heather Greenstone and Amy Krafft for supporting this study, in part, by contract N01-AI-30063 (awarded to Southern Research Institute, Birmingham, AL, USA) from the Virology Branch, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA. The animal experiments were conducted in accordance with the approval of the Institutional Animal Care and Use Committee of Utah State University in the AAALAC-accredited Laboratory Animal Research Center following the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The views expressed in the article do not necessarily represent the views of the Veterans Administration.
Selection of mammalian genes conferring resistance to lytic viruses was performed by multiple investigators at the CDC (MWS and NJM), the University of Georgia (TWH and JLM) and Vanderbilt University (JS and DHR) before initiation of Zirus, Inc. TWH, NJM and JLM were employees at Zirus, Inc., for in vitro viral replication studies which highlighted PI3K-AKT-mTOR pathway genes among hundreds of candidate genes tested, most of which were not critical for infection. Mouse studies were performed at the University of Utah by DFS, who does not have equity or employment by Zirus, Inc. DHR is a scientific founder of Zirus, Inc., and performed genomic analysis of insertion sites of the gene-trap vector blinded to the virus used for selection. DHR's laboratory is reviewed by an external auditor at Vanderbilt University, and the reports are available from David Raiford, Associate Dean for Faculty Affairs, Vanderbilt Medical School. Thus, none of the authors declare competing interests with respect to gene identification or validation.