Considerable attention has been focused on the development of phosphonate-containing drugs for application in many therapeutic areas. However, phosphonate diacids are deprotonated at physiological pH and thus phosphonate-containing drugs are not ideal for oral administration, an extremely desirable requisite for the treatment of chronic diseases. To overcome this limitation several prodrug structures of biologically active phosphonate analogues have been developed. The rationale behind the design of such agents is to achieve temporary blockade of the free phosphonic functional group until their systemic absorption and delivery, allowing the release of the active drug only once at the target. In this paper, an overview of acyclic and cyclic nucleoside phosphonate prodrugs, designed as antiviral agents, is presented.
Introduction to acyclic and cyclic nucleoside phosphonate prodrugs
Nucleoside analogues are synthetic compounds that are structurally similar to natural nucleosides, the building blocks of RNA and DNA. Once inside the cell, nucleoside analogues undergo (often three sequential) phosphorylation steps by viral and cellular kinases to generate, most often, nucleoside 5-triphosphates. These can act as competitive inhibitors of viral and cellular DNA or RNA polymerases or alternatively can be incorporated into growing DNA or RNA strands, causing chain termination [1]. Unfortunately, many nucleoside analogues are not phosphorylated effectively in vivo, in particular, the first phosphorylation step is often an inefficient and rate-limiting step in the conversion to the 5′-triphosphate. To circumvent this limitation, nucleotides with a phosphate group already attached to the nucleoside have been designed. However, these nucleotides become potential substrates for phos-phatases, leading to removal of the phosphate group. Replacement of the phosphate moiety with an isosteric and isoelectronic phosphonate group, results in a nucleoside phosphonate chemically and enzymatically much more stable than the corresponding phosphate. Different to the O-P linkage, the CH2-P-bond, due to its chemical nature, is not susceptible to phosphodiesterase and phosphatase hydrolysis. Enzymatically and chemically stable phosphonate analogues, which mimic the nucleoside monophosphates, can bypass the initial enzymatic phosphorylation and can potentially be more effective antiviral agents. Like a nucleoside monophosphate, a nucleoside phosphonate can be further phosphorylated by cellular nucleotide kinases. The nucleoside phosphonates are classified into major groups: acyclic nucleoside phosphonates (ANPs) and cyclic nucleoside phosphonates (CNPs) [2].
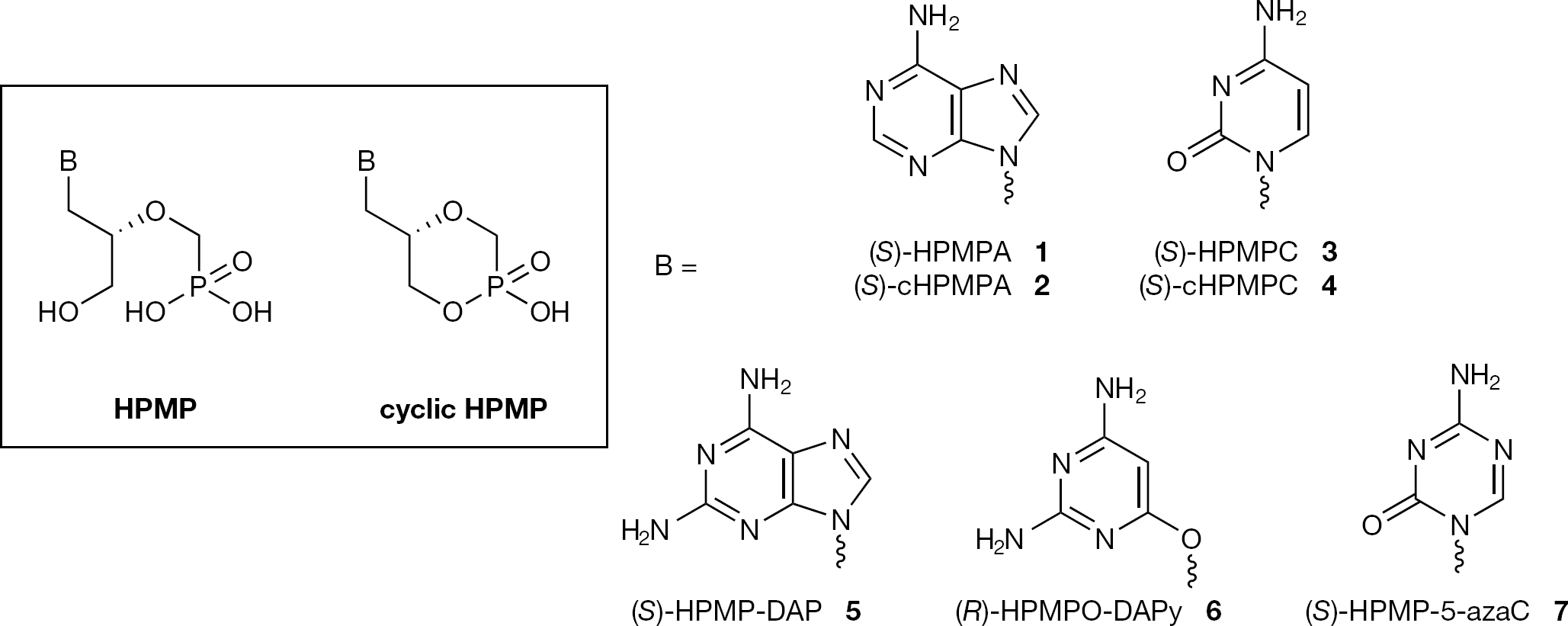
ANPs, originally developed by the Holý group in the 1980s, exhibit a broad spectrum of antiviral activities, particularly against DNA viruses and retroviruses [3,4]. The common structural attribute of ANPs is a nucleobase attached to an aliphatic side chain containing a phosphonomethyl residue. A methylene bridge between the phosphonate moiety and the rest of the molecule excludes the possibility of enzymatic dephosphorylation. The absence of a glycosidic bond in the structure of ANPs further increases their resistance to chemical and biological degradation. Flexibility in the acyclic chain is assumed to allow these compounds, once diphosphorylated, to adopt a conformation appropriate for interaction with active sites of different target enzymes involved in the biosynthesis of DNA (DNA polymerase for DNA viruses, reverse transcriptase for retroviruses). The diphosphorylated ANPs mimic nucleoside triphosphates and may therefore act as chain terminators of the viral DNA, inhibiting viral replication. After the description of the first member of ANPs, (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine (1; (S)-HPMPA) in 1986 [5], new generations of ANPs were prepared and evaluated for their antiviral activity [6]. The ANPs can be classified, according to their structure, in three different series: 3-hydroxy-2-(phosphonomethoxypropyl) (HPMP, Figure 1), 2-(phosphonomethoxyethyl) (PME) and 2-(phosphonomethoxypropyl) (PMP) series (Figure 2). The HPMP series, characterized by the presence of a hydroxymethyl chain, exhibit (generally as the (S)-enantiomer) an antiviral activity against a wide spectrum of DNA viruses encompassing, in particular, adeno-, pox-, polioma-, papilloma- and herpesvirus infections. Among this series, derivatives of adenine (1, (S)-HPMPA), cytosine (3, (S)-HPMPC, Cidofovir, Vistide®, used to treat human cytomegalovirus [HCMV] retinitis in AIDS), 2,6-diaminopurine (5, (S)-HPMP-DAP), 2,4-diamino-3-hydroxy pyrimidine (6, (R)-HPMPO-DAPy) and 5-azacytosine (7, (S)-HPMP-5-azaC) were found to be especially potent [3,7]. In particular, the DAP (2,6-diaminopurine) derivatives show an antiviral potency and activity spectrum comparable to that of their adenine counterparts [8]. Thus, (S)-HPMP-DAP (5) is equivalent to (S)-HPMPA (1), that is, with regard to its activity against poxviruses such as vaccinia virus (VV) [9] and orf virus [10]. Similarly to (S)-HPMPA (1) and (S)-HPMPC (3), (R)-HPMPO-DAPy (6) proved to be a potent and selective inhibitor of adenovirus replication in vitro [11] and exhibited selective and potent activity against orf virus in both human and ovine cell monolayers and organotypic ovine raft cultures [10]. In vivo, (R)-HPMPO-DAPy (6), similarly to (S)-HPMPC (3) was shown to lead to healing of cutaneous vaccinia lesions in athymic-nude mice (which corresponds to an experimental model infection for disseminated vaccinia in immunosuppressed patients inadvertently vaccinated with live smallpox vaccine) [9]. The antiviral activity of (S)-HPMP-5-azaC (7) was also shown to be comparable to that of the reference drug (S)-HPMPC (3) against herpes simplex viruses 1 and 2 (HSV-1, HSV-2) and VV, or two- to sevenfold more active against varicella zoster virus (VZV), HCMV, human herpes virus type-6 (HHV-6) and adenovirus type-2 (Ad2). For all these DNA viruses, (S)-HPMP-5-azaC (7) showed a 2-16-fold higher selectivity index (ratio of 50 % cytostatic concentration [CC50] to 50% effective concentration [EC50]) against DNA viruses in cell culture than (S)-HPMPC (3) [12]. In an effort to increase the cell permeability of HPMP-based nucleosides, investigators have also generated a cyclic version of this series, forming an internal phosphonate ester bond with the hydroxymethyl chain (cyclic HPMP; Figure 1). As a representative example, cyclic (S)-HPMPC (4) has been shown to have advantages when compared to (S)-HPMPC (3), such as reduced nephrotoxicity due to diminished uptake in the renal proximal tubular cells [13]. Researchers have identified a cellular cCMP phosphodiesterase that can convert (S)-cHPMPC (4) to (S)-HPMPC (3)in vivo [14].
Structures of acyclic nucleoside phosphonate HPMP and cyclic HPMP series
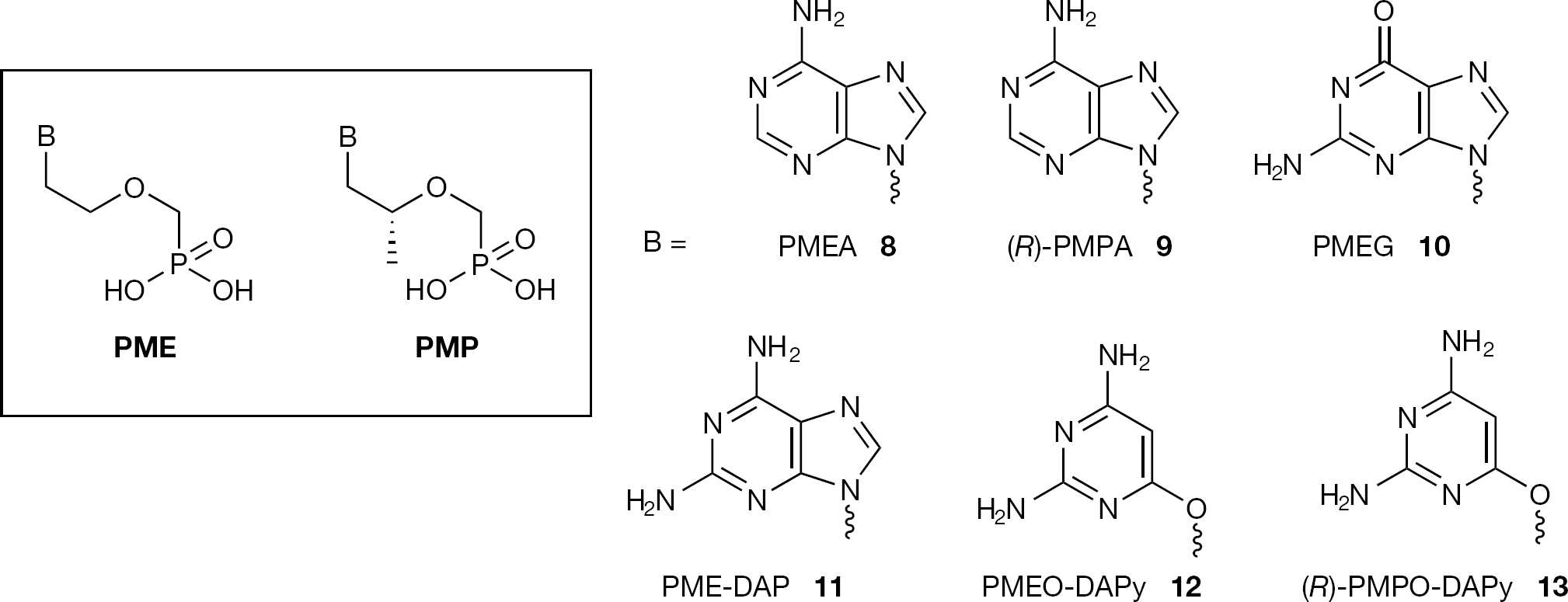
Structures of acyclic nucleoside phosphonate PME and PMP series
The PME and PMP series of ANPs, which lack, respectively, the hydroxymethyl and hydroxyl groups in their acyclic side chain, are based on PMEA (8, adefovir) and (R)-PMPA (9, tenofovir) structures (Figure 2). The PME structures, unlike the HPMP and PMP series, do not contain a chiral carbon centre and thus do not exist as (R) and (S) enantiomers. The PMP series exerts its antiviral activity mostly as the (R)-enantiomer. These nucle-otides and their analogues have shown potent activity against retro- and hepadnaviruses. PME-DAP (11) was found to be more active as an antiretrovirus agent than PMEA (8), but also more toxic, so that its therapeutic index, based on its in vivo activity against murine sarcoma virus (MSV), was equivalent to that of PMEA (8) [15]. It was reported with even higher antiretrovirus potency than (R)-PMPA (9) and has proved active against (both wild-type and lamivudine resistant) HBV with a potency comparable to that of (R)-PMPA (9) [16,17]. PMEG (10) was shown to be a potent inhibitor of DNA polymerase [18]. In animal models PMEG (10) has an antiproliferative effect. However its use for the treatment of human papilloma virus infections, which cause proliferative lesions, is limited by its toxicity [19]. ‘O-linked’ ANPs such as 2,4-diamino-6-[2-(phosphonomethoxy)ethoxy]pyrimidine (12, PMEO-DAPy) and 2,4-diamino-6-(R)-[2-(phosphonomethoxy) propoxy]-pyrimidine (13, (R)-PMPO-DAPy) were found to inhibit the replication of herpes viruses (HSV-1, HSV-2, VZV) as well as retroviruses such as HIV-1 and HIV-2 [20]. In particular, the antiretroviral activity of PMEO-DAPy (12) and (R)-PMPO-DAPy (13) appeared interesting, as this was comparable to that of PMEA (8) and (R)-PMPA (9), and was also observed in vivo [21].
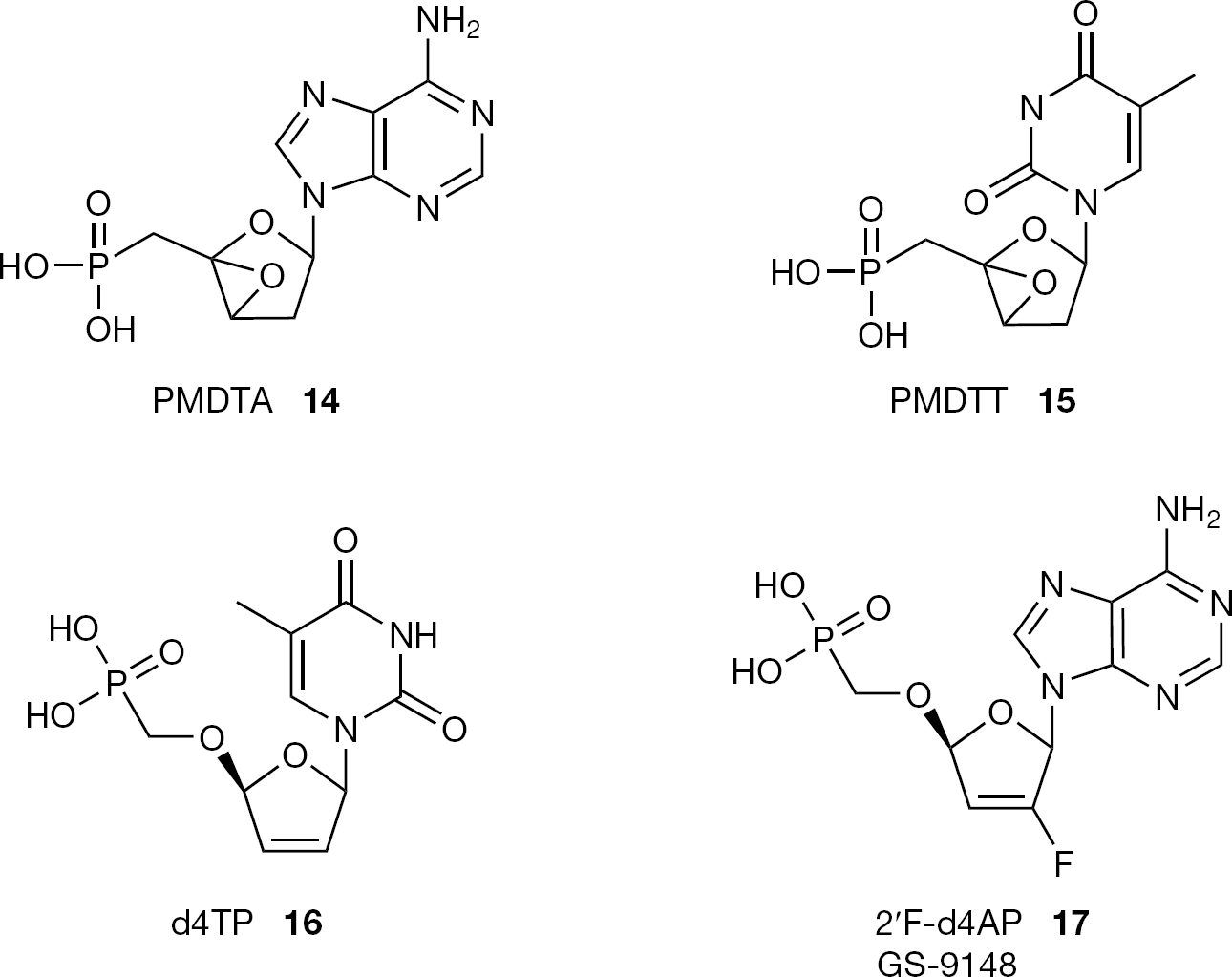
CNPs are real nucleoside analogues as they contain a nucleobase and a sugar moiety. Compared to the large number of the ANPs described in the literature, only a few examples of CNPs endowed with antiviral activity have been reported (Figure 3) [2]. The lack of antiviral activity of CNPs is generally explained by their poor substrate properties for cellular and viral kinases; whereas, the potent antiviral activity of ANPs is ascribed to their intracellular phosphorylation to their diphos-phates and to a refractory incorporation of the modified nucleosides in nucleic acids. To try to overcome the drawback of CNPs, compounds where the phosphono-alkoxy group of the nucleoside phosphonates is bound at the 3′-position were designed. Interesting examples of this class of drugs are the (L)-2-deoxythreosyl phosphonate nucleosides PMDTA (14) and PMDTT (15), reported as selective anti-HIV agents [22]. The absence of a hydroxymethyl substituent in the 4′-position of the nucleoside can probably avoid OH hindrance during enzymatic phosphorylation allowing the formation of the diphosphate species.
Structures of cyclic nucleoside phosphonates PMDTA (14), PMDTT (15), d4TP (16) and 2′F-d4AP (17, GS-9148)
In an effort to identify new nucleoside inhibitors of reverse trascriptase, phosphonomethoxy analogues of cyclic pyrimidine nucleotide were synthesized and compared for their antiviral activity [23]. Among them, only d4TP (16;Figure 3) showed an antiviral activity below 200 μM. This approach has been recently extended to purine analogues leading to the discovery of the [5-(6-amino-purin-9-yl)-4-fluoro-2,5-dihydro-furan-2-yloxymethyl] phosphonic acid (17, GS-9148; Figure 3), a ribose-modified HIV-1 nucleotide reverse transcriptase inhibitor (NRTI) selected from a series of nucleoside phosphonate analogues for its low mito-chondrial toxicity, minimal cytotoxicity in renal proximal tubule cells and other cell types, for its synergy in combination with other antiretroviral drugs and for its unique resistance profile against multiple resistant HIV-1 strains [24].
Natural nucleosides are hydrophilic molecules and do not rapidly penetrate cell membranes by non-facilitated diffusion. Instead, they permeate the cell by carrier-mediated endocytosis, which is an active or facilitated transport mechanism that requires energy and a specific receptor or protein on the cell surface. Acyclic and cyclic nucleoside phosphonate analogues contain a phosphonic acid group completely ionized at physiological pH, resulting in a molecule largely impermeable to mucosal and cellular membranes. However, carrier-mediated transport often requires very close structural resemblance to natural products. Due to their low oral bioavailability, ANP and CNP analogues are of limited value in the treatment of chronic diseases, for which the oral therapies are highly desired.
Prodrugs are used, in general, to bypass physicochemical, pharmaceutical, pharmacokinetic and pharmaco-dynamic barriers to drug formulation and delivery, such as poor aqueous solubility, chemical instability, insufficient oral absorption, rapid presystemic metabolism and inadequate tissue penetration. In an attempt to overcome the limited oral bioavailability of ANPs and CNPs ionizable phosphonate can be masked by derivat-ization, thus generating a prodrug with increased lipophilicity, capable of altering cell and tissue distribution/ elimination patterns of the parent drug [25].
Passive transcellular absorption is the most general route for absorption of lipophilic (pro)drugs through the intestinal mucosal membranes. After the (pro)drug molecule is dissolved in the aqueous media, absorption occurs by diffusion through the lipid bilayer and because it is driven by a concentration gradient, energy is not required. Absorption is thus dependent on the lipophilicity, intrinsic aqueous solubility and molecular weight of the drug molecule. Once the (pro)drug has reached the blood circulation it will pass through the endothelium of the capillary and distribute into tissues. Thus, also the distribution is dependent on several structural and physicochemical properties, including the chemical and enzymatic stabilities, the molecular size, logP, hydrogen-bonding, charge state and finally on its ability to be a substrate for influx and efflux transporters and to bind plasma proteins. The liver is the major organ for drug metabolism, which also occurs in the gastrointestinal (GI) tract and to some extent in lungs, skin and kidneys. First-pass metabolism consisting of Phase I oxidation reactions catalyzed by cytochrome P450-enzymes and Phase II conjugation reactions catalyzed by several enzymes occurs in the liver. Excretion, elimination or clearance of a drug molecule from the body, starts immediately after the drug has entered the circulation and one organ is mainly responsible for it, the kidney. Renal clearance occurs via filtration of the blood in the glomerulus of the kidneys and the further reabsorption of the compounds from the kidney tubule back to the systemic circulation with the molecular weight and lipophilicity of the drug molecule playing a key role in these events.
In order to achieve oral bioavailability and intracellular delivery an ideal oral prodrug must survive the GI tract, be absorbed across intestinal mucosa and be delivered into the systemic circulation following its active/passive transport, and then be distributed into cells where it has to be converted to the parent drug, releasing a non-toxic promoiety, which, in turn, has to be rapidly excreted from the body. Thus, stability is one of the major factors influencing the design of prodrugs. The bioactivation mechanism for most prodrug structures is enzymatic or at least requires enzymes to initialize the bioactivation process, which can then further continue chemically.
Several prodrug approaches have been utilized to overcome the limitation of phosphonate-containing drugs. General reviews about prodrug strategies elaborated for phosphate-, phosphonate- and phosphinate-containing compounds [26–29] and more specific reviews directed toward delivery of nucleosides and nucleotides have been published [25,30–32]. The aim of this review is to survey, specifically, the status and the recent progress in the design and development of acyclic and cyclic nucleoside phosphonate prodrugs, focusing on the most promising strategies currently under development in the antiviral area.
Bis-(POM) and bis-(POC) ester prodrugs
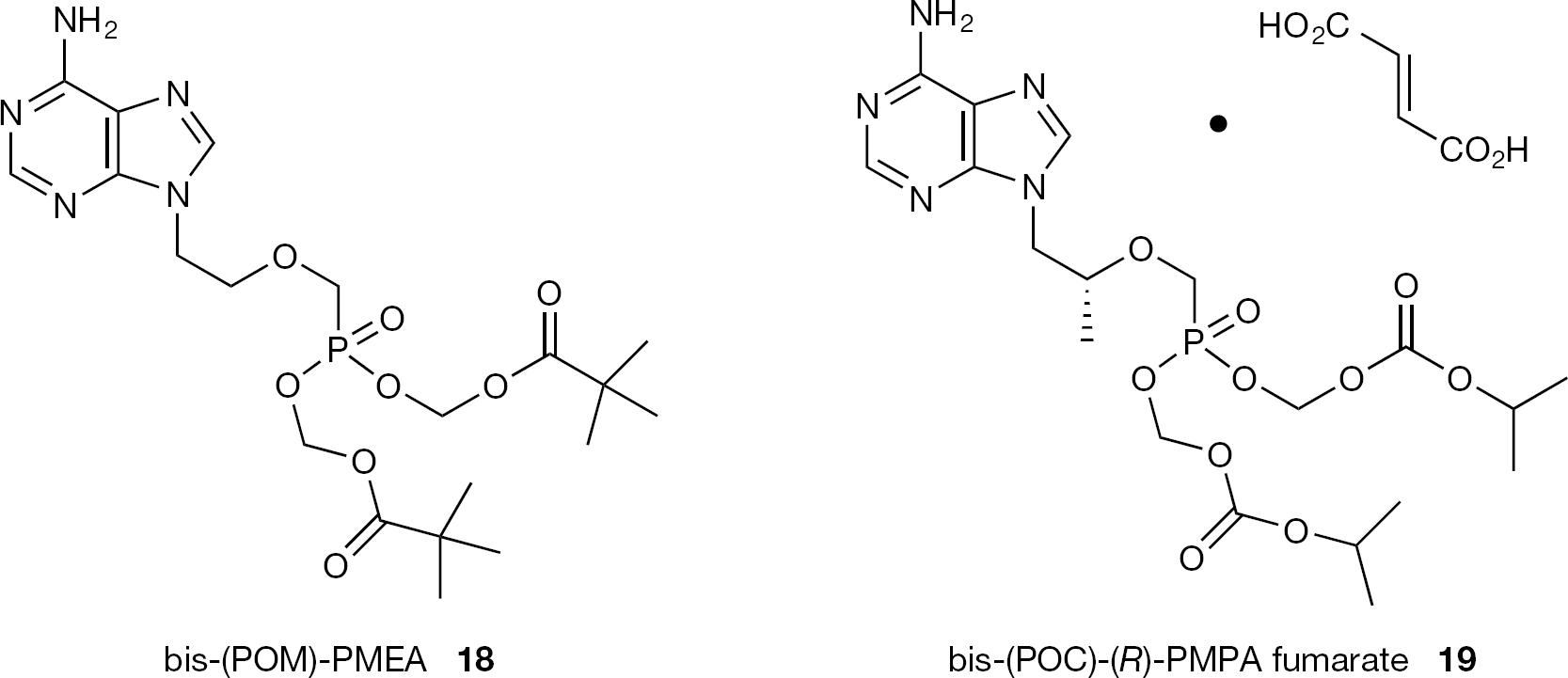
The development of acyloxy ester and alkoxycarbonyl ester prodrugs of ANPs has been successful. Among these ester prodrugs, two compounds are currently marketed for antiviral therapy: the bis (pivaloyloxymethyl) ester of adefovir (18, bis-(POM)-PMEA; Hepsera®), approved in 2002 for the treatment of HBV infections and the diisopropyloxycarbonyloxymethyl ester of tenofovir fumarate (19, bis-(POC)-(R)-PMPA fumarate; Viread®), licensed in 2001 for the treatment of HIV infections and approved to treat chronic HBV infections since 2008 (Figure 4).
Structures of bis-(POM)-PMEA (18) and bis-(POC)-(/?)-PMPA fumarate (19)
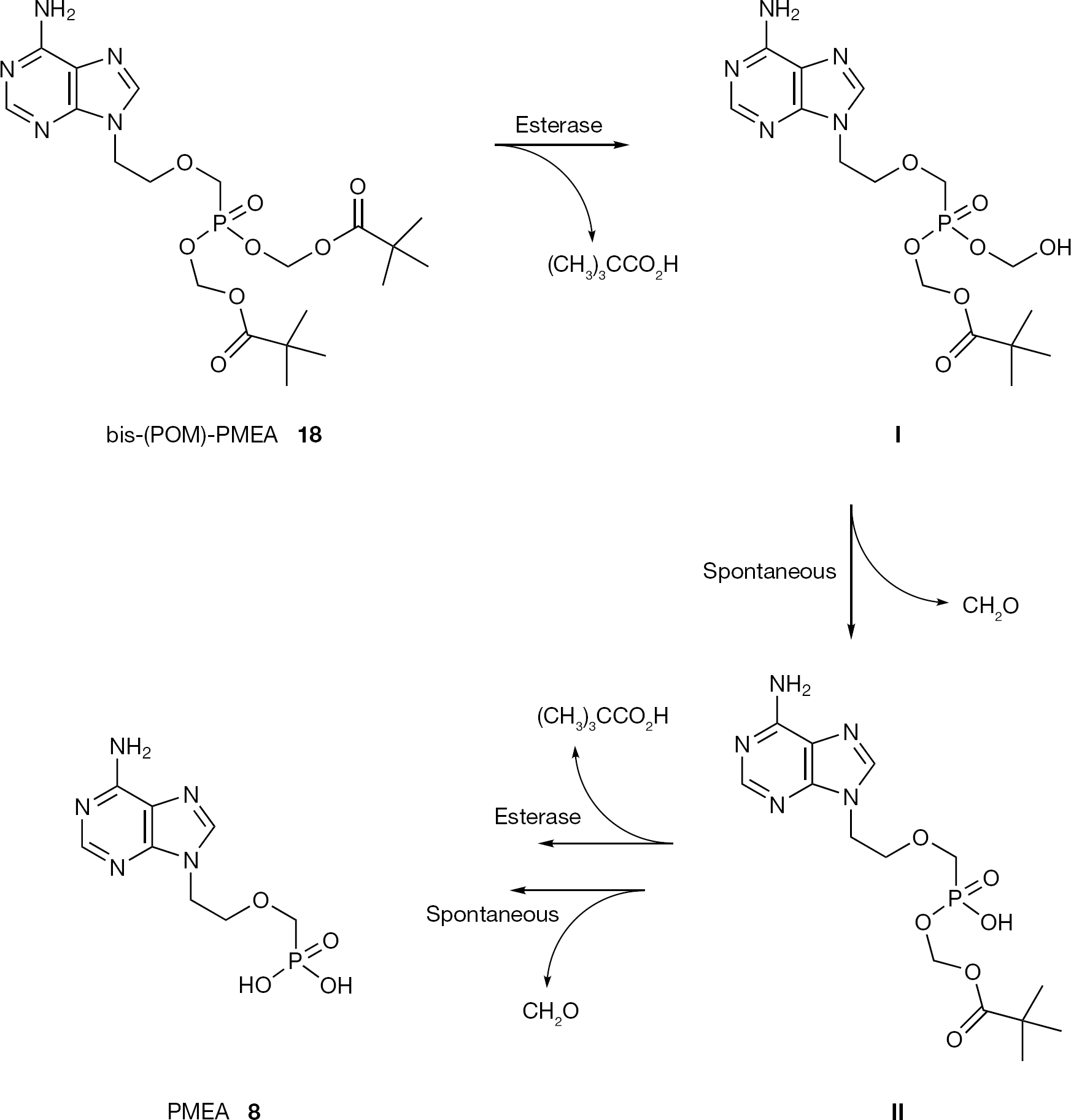
In general, bis-POM derivatives are reported to exhibit a 9- to 13-fold greater in vitro antiretroviral activity than their corresponding unmodified compounds and to show remarkable increased bioavailability in vivo. Bis-(POM)-PMEA (18), selected among a series of acyloxy methyl ester prodrugs, was first considered as a possible candidate for the treatment of HIV infection due to its ability to reduce the viral load in plasma [33]. However, the toxicity due to its decomposition products (formaldehyde and pivalic acid) generated some concern (Figure 5) [34–36].
Metabolic pathway for bis-(POM)-PMEA (18)
The pivalic acid was demonstrated to be responsible for altering carnitine homeostasis [37]. Additionally, it has been shown that bis-(POM)-phosphotriesters were chemically unstable and highly susceptible to serummediated hydrolysis, factors that limit their potential utility for intracellular drug delivery [38]. For these reasons, bis-(POM)-PMEA (18) was considered too toxic to permit long-term use at the dosage required in order to inhibit HIV and it was instead approved by the FDA only for the treatment of HBV. The pivaloyloxymethyl prodrugs of (R)-PMPA and (R)-PMP-DAP were also evaluated but due to toxicity concerns they were not selected for clinical trials.
The diisopropyloxycarbonyloxymethyl ester (bis-(POC)) is a modification of the bis-(POM)-ester, offering the advantage of not generating pivalic acid during its bioconversion. The bis-(POC) motif was successfully applied to (R)-PMPA (8) leading to bis-(POC)-(R)-PMPA, which was selected for clinical trials and subsequently formally approved as its fumarate salt (19) for the treatment of HIV. Bis-(POC)-(R)-PMPA fumarate (19) is also currently commercially available in combination with emtricitabine (Truvada®), and emtricitabine and efavirenz (Atripla®).
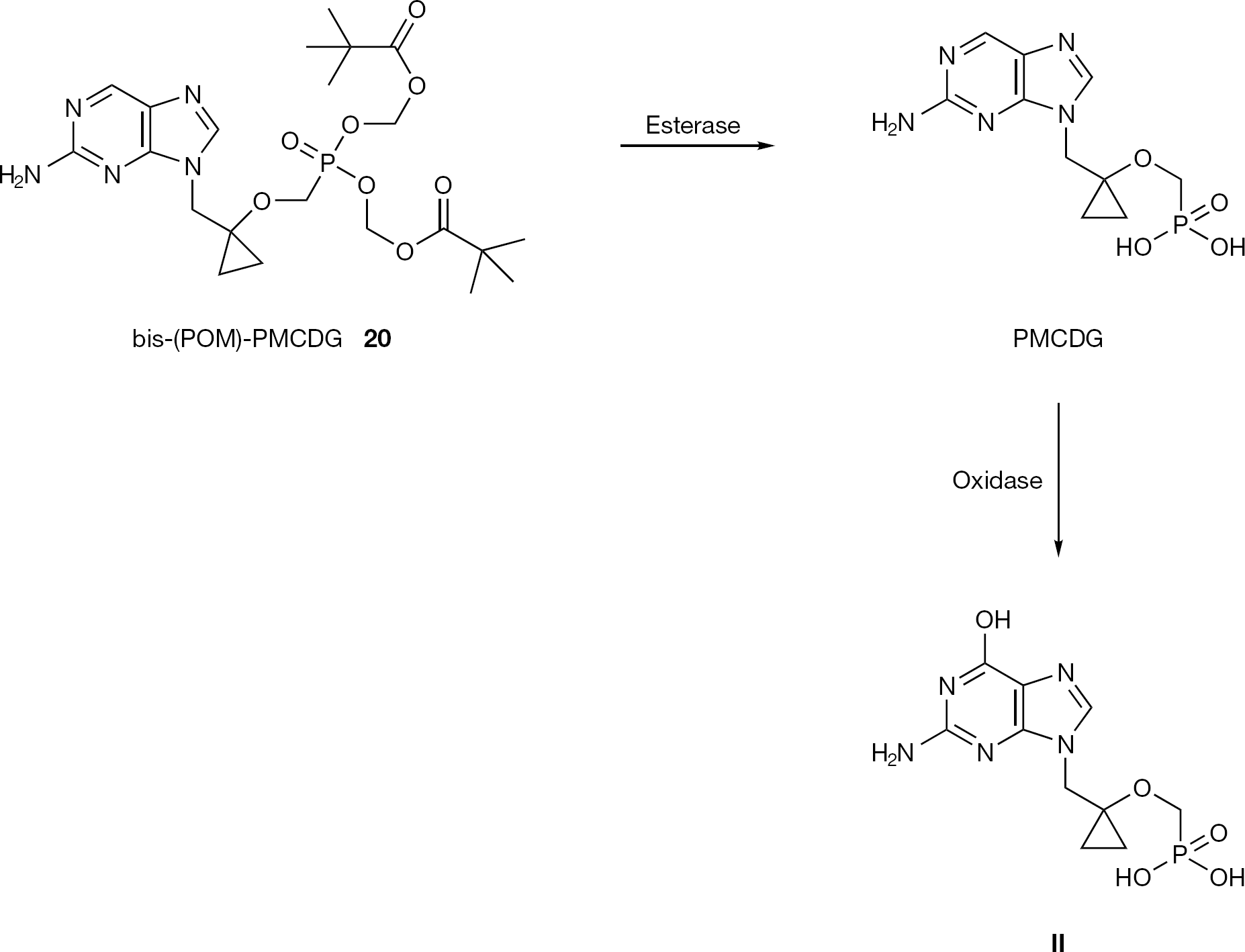
Subsequently, PMCDG dipivoxil (20, bis-(POM)-PMCDG; Figure 6) emerged as a possible candidate as an oral prodrug for HBV treatment and it is currently in Phase II clinical trials for evaluation of efficacy in humans [39,40]. Bis-(POM)-PMCDG is rapidly converted to the parent drug (PMCDG) in the liver and intestine, probably by esterases. The parent drug is further metabolized to a nucleotide analogue of guanosine monophosphate (II), by an oxidase such as aldehyde or xanthine oxidase (Figure 6). After phosphorylation to di- and triphosphate forms, the molecule inhibits viral replication following incorporation into viral DNA.
Structure of bis-(POM)-PMCDG (20) and its metabolic pathway
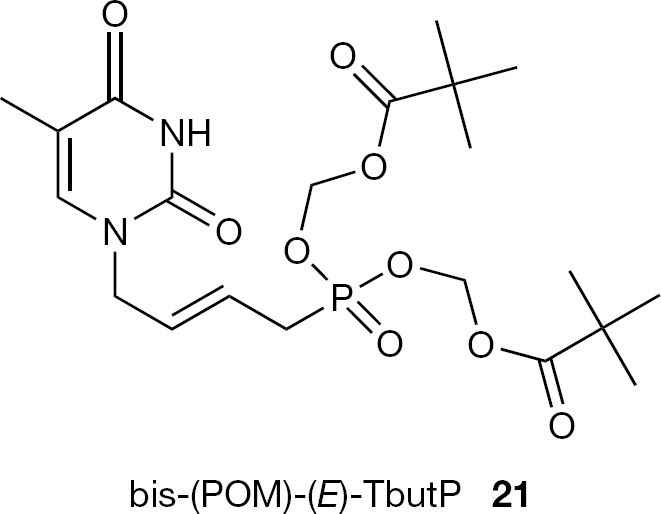
More recently, bis-POM esters of (S)-HPM-DAP and its cyclic analogue were reported by Krečmerová et al. [41]. Although these compounds proved to be less active than the corresponding alkoxyalkyl ester prodrugs (see Alkoxyalkyl ester prodrugs), they appeared to be less cytotoxic and cytostatic. Finally, bis-POM esters of novel ANPs have been described. Among them, bis-(POM)-(E)-TbutP (21) emerged as a potent antiviral agent against several herpes viruses, (HSV-1, HSV-2, VZV), representing a new potential antiviral lead for further optimization (Figure 7) [42].
Structure of bis-(POM)-(E)-TbutP (21)
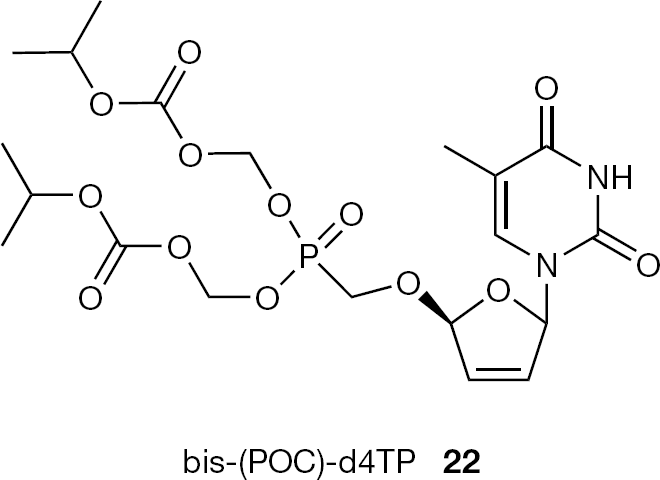
The antiviral potency of CNPs can also be improved considerably by the application of bis-(POC) prodrug technology. As a representative example, bis-(POC)-d4TP (22) has been reported to improve 29-fold the HIV antiretroviral activity of the corresponding nucleoside (Figure 8) [23].
Structure of bis-(POC)-d4TP (22)
Alkoxyalkyl ester prodrugs
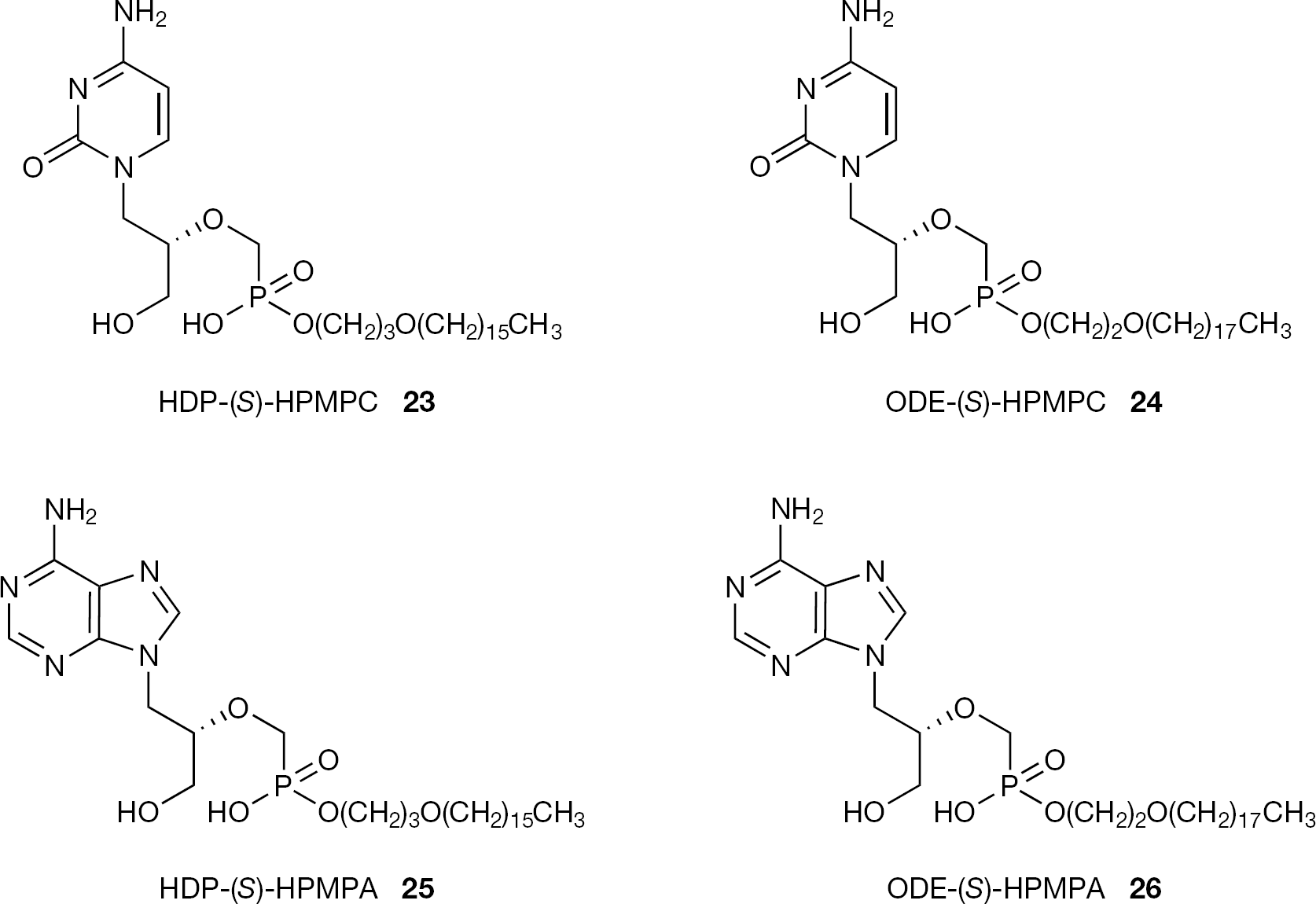
In order to enhance cellular drug uptake of ANPs, the Hostetler group and collaborators have developed a series of very promising ether lipid conjugates of ANPs, including (S)-HPMPC (3), (S)-HPMPA (1), PMEA (8), (R)-PMPA (9) [43]. Two alkoxyalkyl esters of (S)-HPMPC, hexadecyloxypropyl-(S)-HPMPC (23, HDP-(S)-HPMPC) and octadecyloxyethyl-(S)-HPMPC (24, ODE-(S)-HPMPC) have been shown to be more active than (S)-HPMPC (3) against HCMV and other herpes viruses, exhibiting 2.5- to 4-fold increases in antiviral activity depending on the in vitro antiviral assay used (Figure 9) [44]. These alkoxyesters were also found to be 25- to 910-times more active than (S)-HPMPC (3) against variola virus (smallpox) with EC50 values ranging from 0.04 to 0.1 μM for HPD-(S)-HPMPC (23) and from 0.01 to 0.03 μM for ODE-(S)-HPMPC (24) [45,46]. Similar results were also reported with monkeypox and cowpox viruses in vitro. When tested in vitro against VV, HPD-(S)-HPMPC (23) showed an increase in antiviral activity of 58-fold when compared to the parent drug, whereas ODE-(S)-HPMPC (24) was even more active with a 231-fold increase (with respect to (S)-HPMPC; 3) and with an EC50 value of 0.2 μM. The authors state that such an increase in antiviral activity of 23 and 24 when compared to (S)-HPMPC (3) is due to the the increased cellular uptake and conversion to (S)-HPMPC diphosphosphate.
Structures of HDP-(S)-HPMPC (23), ODE-(S)-HPMPC (24), HDP-(S)-HMPA (25) and ODE-(S)-HMPA (26)
In vivo, these compounds proved as effective as parental (S)-HPMPC (3) in the treatment of HCMV infection in a variety of murine models [47]. They also proved to be effective when tested in animal models of orthopox diseases such as cowpox, vaccinia and ectromelia virus infections [48,49]. When given orally to mice infected with ectromelia virus by small particle aerosol, HDP-(S)-HPMPC (23) and ODE-(S)-HPMPC (24) were almost fully protective by oral doses of 5 mg/ kg and 10 mg/kg, administered 4 h after infection and sustained daily for five days. A dose of 10 and 12 mg/ kg of HDP-(S)-HPMPC (23) and of ODE-(S)-HPMPC (24), respectively, given orally for five days prior to or a few days after nasal infection with cowpox, were reported to be able to significantly reduce the mortality. HDP-(S)-HPMPC (23) with the name CMX001 is currently under development by Chimerix Inc. as an oral drug for the treatment of HCMV and smallpox infection. Recently, CMX001 has completed Phase I clinical trials in healthy volunteers and Phase II trials are now in progress in HCMV stem cell transplant patients and BK virus infection in kidney transplant patients. However, CMX001 recently failed in an in vivo efficacy trial (monkeypox model) likely due to metabolic differences between rodents and monkeys [50].
Besides (S)-HPMPC derivatives, alkoxyalkyl derivatives of another ANP, (S)-HPMPA (1) have been described (Figure 9). Esterification to HDP-(S)-HPMPA (25) or ODE-(S)-HPMPA (26) resulted in large increases in antipoxvirus activity raging from 160 to 270-fold versus (S)-HPMPA (1) [51,52]. Similar results have been observed with CMV infection [53]. Among these derivatives, ODE-(S)-HPMPA (26) was the most active compound in adenovirus-infected cells with EC50 values of 0.04–0.16 μM compared with 0.19–1.1 μM of HDP-(S)-HMPA (25). When compared to the alkoxyalkyl of (S)-HPMPC (3), the corresponding alkoxyalkyl ester of (S)-HPMPA (1) displayed similar activities against human and murine CMV.
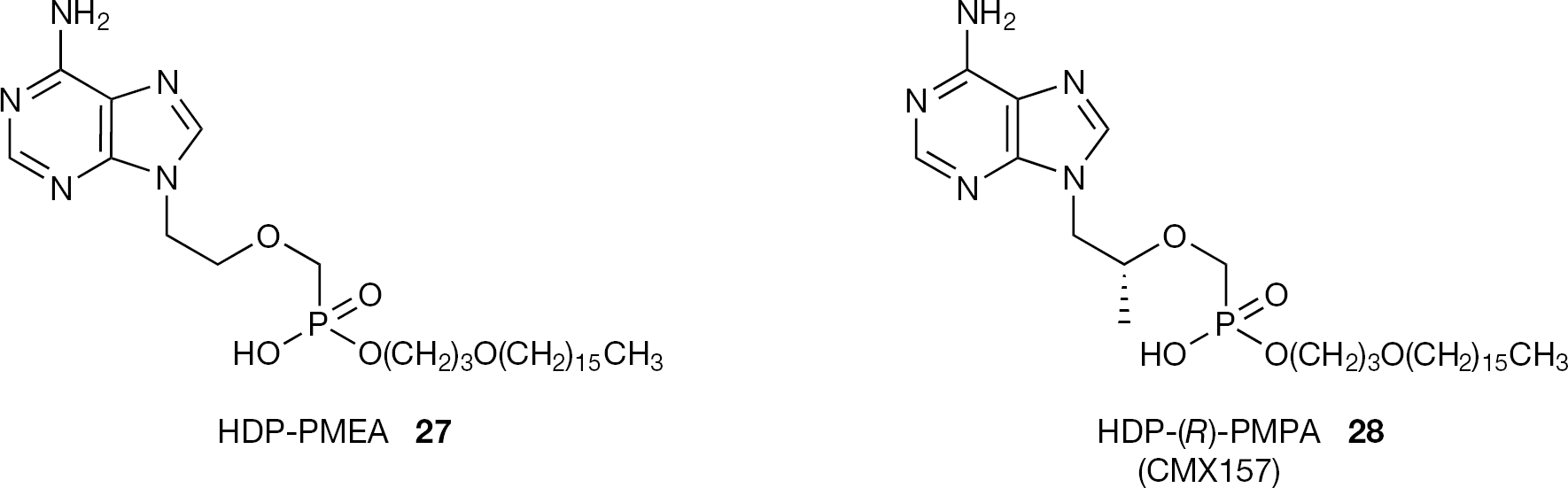
Both (S)-HPMPC (3) and (S)-HPMPA (1) have been reported to inhibit replication of all double-stranded DNA viruses [3,8] and the increased antiviral activity observed after alkoxyalkyl esterification does not appear to be specific for a particular type of virus [43]. Although (S)-HPMPA (1) was reported to be inactive against HIV, HDP-(S)-HPMPA (25) and ODE-(S)- HPMPA (26) are described as highly active in vitro (with low nanomolar EC50 values), indicating that the alkoxy alkylester strategy can eventually widen the range of antiviral activity. The same strategy has been applied to PMEA (8) and (R)-PMPA (9;Figure 10). HDP-PMEA (27) and HDP-(R)-PMPA (28) have been found to be highly active against HIV-1. HDP-(R)-PMPA (28) under the name of CMX157, is currently in clinical development by Chimerix Inc. as an oral drug for treatment of HIV infection. Recently it was announced that the first results in human Phase I clinical trials demonstrate a favourable safety, tolerability and drug distribution profile for CMX157.
Structures of HDP-PMEA (27) and HDP-(R)-PMPA (28)
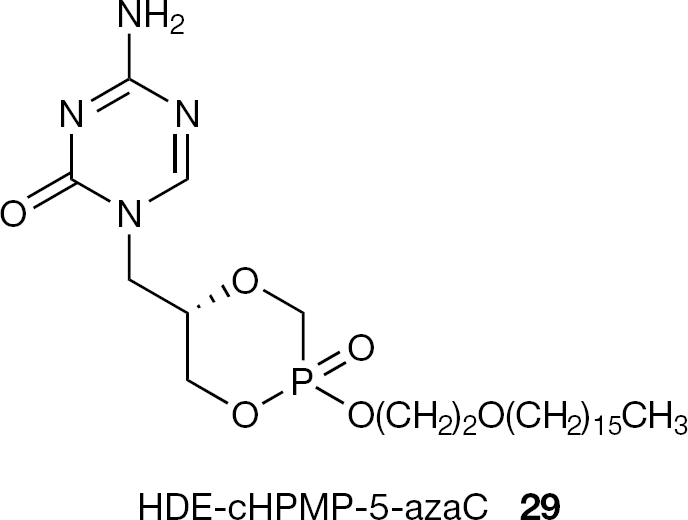
Other ANPs have been subjected to esterification with alkoxyalkyl groups. In particular, the antiviral activity of the cyclic version of (S)-HPMPC-5azaC (7) was reported to be further enhanced by introduction of alkoxyalkyl groups, the most active being the hexadecyloxyethyl (HDE) ester derivative (29) with EC50 values in the range of 0.003–0.008 μg for HSV (Figure 11) [54].
Structure of HDE-(S)-cHPMP-azaC (29)
Recently, Krečmerová et al. [41] selected (S)-HPMP-DAP (5) and its cyclic form for further evaluation, synthesizing a series of different prodrugs and then assessing their in vitro antiviral activity. From these studies the HDP and the POM mono ester of (S)-HPMP-DAP (5) and its cyclic analogue, emerged as the most active prodrugs against VV, HSV, VZV and HCMV.
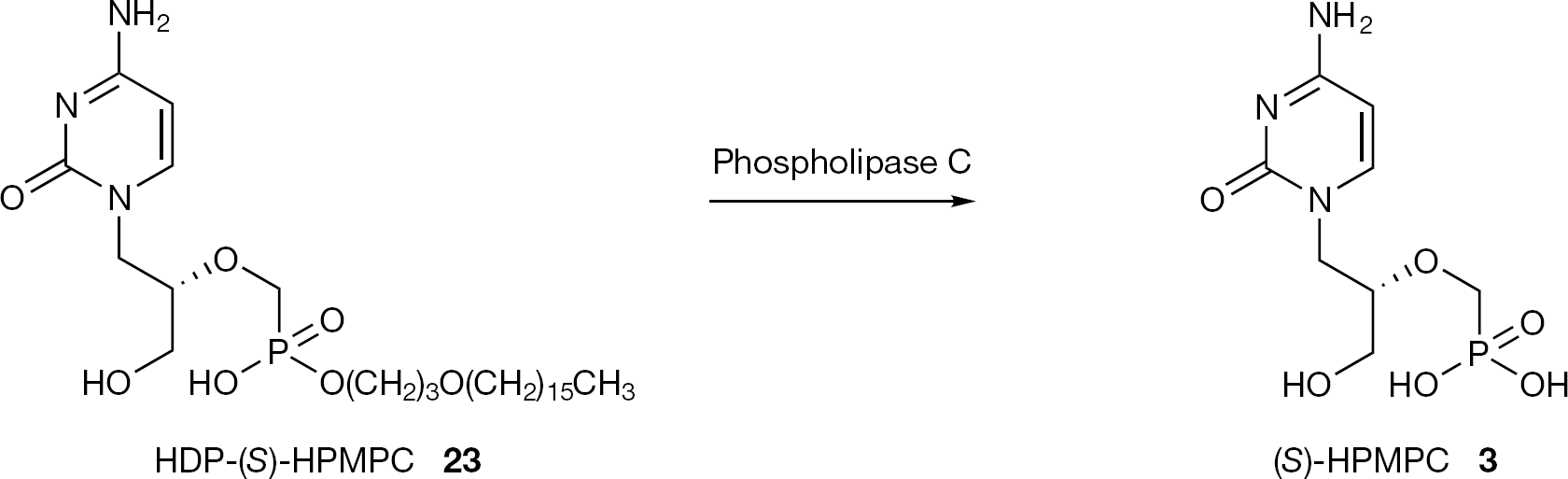
The esterification of ANPs with HDP and ODE chains was designed to increase oral bioavailability, based on the resemblance to lysophosphatidylcholine. As a representative example for this class of prodrugs, the metabolic pathway of HDP-(S)-HPMPC (23) is reported in Figure 12. Phospholipase C, which is reported to be the only enzyme responsible for the metabolic cleavage, is common in mammalian tissues. Phospholipase C is not present in plasma or pancreatic secretions, providing stability for 23 and other compounds of this type during oral absorption and transport in plasma to tissues.
Metabolic pathway for HDP-(S)-HPMPC (23)
Hexaethyleneglycol unit, hydroxylated decyl- and decyloxyethyl- chains were recently used by Krečmerová et al. [55] to mask either PMEA (8) or (S)-HPMPC (3). However, the antiviral activities of these prodrugs were, in general, lower or similar to the corresponding parent drugs. Considering these results, the authors conclude that these prodrugs are taken up less efficiently or are not suitable substrates for the phospholipase C.
Despite the notable success of the alkoxyalkyl prodrugs developed by Hostetler there are still challenges around this approach because of the potentially poor solubility in aqueous solutions, arising from a lipid moiety [49,56].
Aryl and phenyl ester prodrugs
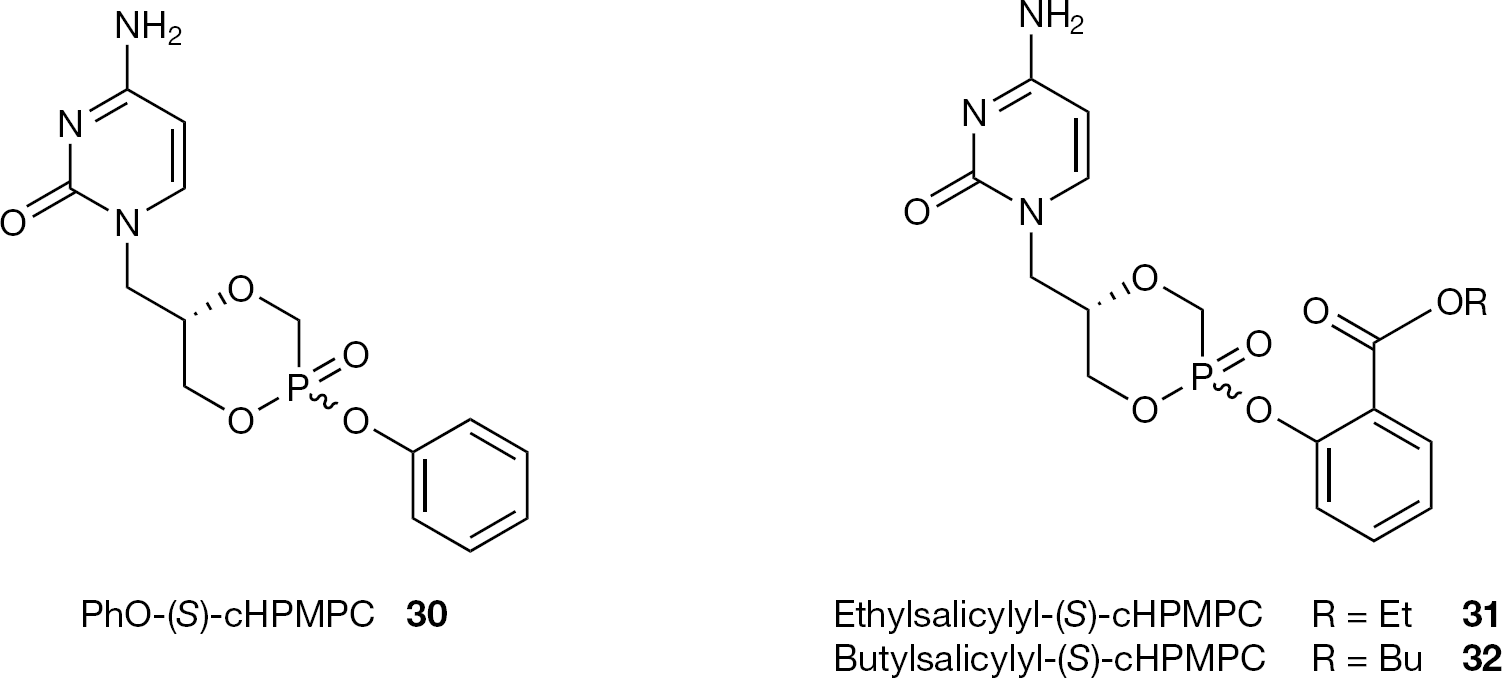
Representative examples of these prodrugs are the phenyl (30) and the salicylate ester prodrugs (31 and 32) of (S)-cHPMPC (4) reported by Oliyai et al. [57,58] (Figure 13).
Structures of PhO-(S)-cHPMPC (30), ethylsalicylyl- (31) and butylsalicylyl-(S)-cHPMPC (32)
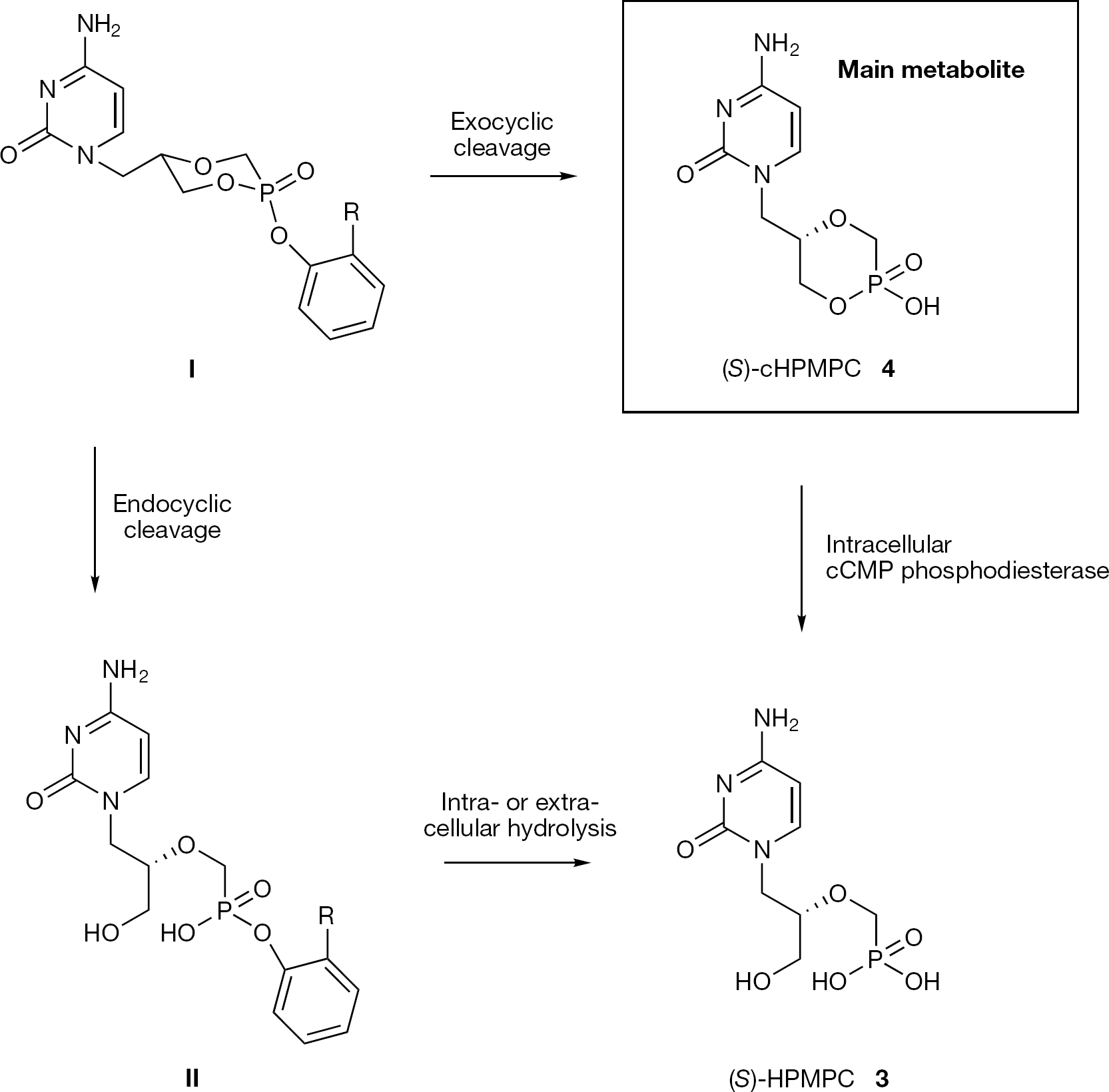
Esterification of (S)-cHPMPC (4) via a phosphoester bond, either with a phenol or a salicylic ester produces a new chiral centre at the phosphorus, leading to two different diastereoisomers of the resulting prodrug (SRp and SSp). The carboxylate function on the salicylate prodrugs provides an additional site for chemical modification to tune the lipophilicity, solubility and biological reactivity of the prodrug. These compounds were synthesized in a stereospecific manner and investigated for their physicochemical and pharmacokinetic properties. The authors demonstrated that the chemical stability was dependent on the phosphorus stereochemistry (with the axial isomer more stable than the equatorial) and not greatly upon the nature of the salicylate ester, which, on the contrary, plays a key role for the enzymatic stability. The axial isomer of salicylate prodrugs (I), selected for in vivo oral bioavailability evaluation, produces (S)-cHPMPC (4) as the major metabolite, together with (S)-HPMPC (3) and the mono ester of (S)-HPMPC (II), whereas, the corresponding equatorial isomer generates only (S)-cHPMPC (Figure 14). The results of these studies proved that salicylate ester prodrugs can successfully deliver (S)-cHPMPC (4) to the systemic circulation with an oral bioavailability of 4, ranging from 18.5 (for 31) to 46.3% (for 32). This prodrug approach presents the advantage to allow oral delivery of (S)-cHPMPC (4), while minimizing (S)-HPMPC (3) related toxicity.
Metabolic pathway for the axial isomer of aryl (S)-cHPMPC prodrugs
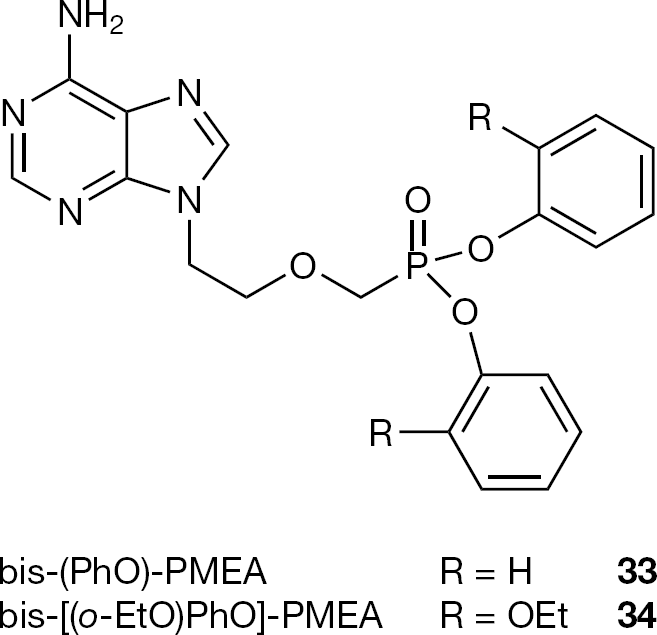
A number of esters of PMEA (8) have been synthesized and evaluated for their oral bioavailability [59]. Among them, the diphenyl ester (33) was identified as the preferred prodrug because it is well absorbed and efficiently converted to the parent compound with an oral biovailability of 50% (Figure 15). However, further studies revealed that the diphenyl ester (33), even if moderately absorbed, is poorly converted to PMEA (8) due to the oxidation of the ethyl side chain by P450 enzymes to generate the inactive metabolite 2-adenylacetic acid. The same metabolite was observed from the activation of bis-(oethoxy)phenyl ester (34) making these two aryl esters unsuitable for further evaluation as prodrugs [60].
Structures of bis-(PhO)- (33) and bis-[(o-EtO)PhO]-PMEA (34)
Cyclosaligenyl ester prodrugs (CycloSal)
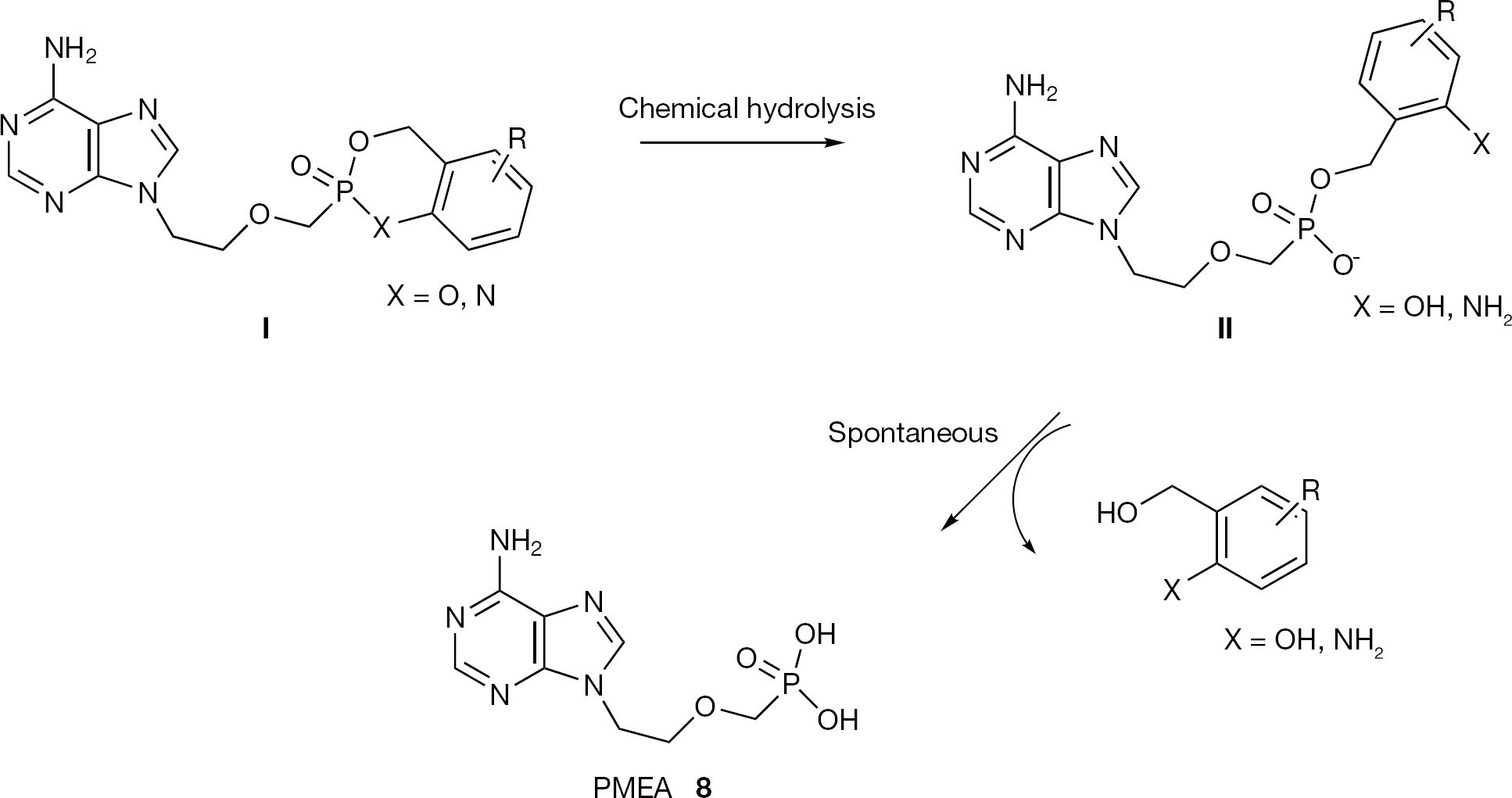
The cyclosaligenyl prodrug strategy was elaborated by Meier et al. [61], first to deliver monophosphates and only later was it applied to ANPs. In particular, the same group reported the synthesis, the stability and the biological evaluation of cycloSal-PMEA prodrugs. The structure of the cycloSal PMEA prodrugs present a salicyl alcohol as the only masking unit for both the two hydroxyl moieties of the phosphonic acid of PMEA (8;Figure 16). Due to an unexpected low hydrolytic stability of these cycloSal-PMEA prodrugs, the authors had to investigate the more stable cycloAmb-PMEA derivatives, in which the salicyl alcohol has been replaced with a 2-amino benzyl alcohol (Figure 16). From this study it was proven that the mechanism of activation of cycloSal phosphonate is identical to that of the cycloSal-phosphate with the PMEA (8) as the only product of hydrolysis (Figure 16).
General structures of cycloSal- (X=O) and cycloAmb-PMEA (X=N) prodrugs and their hydrolysis mechanism
Although considerably chemically and enzymatically more stable than cycloSal-PMEA prodrugs, cycloAmb-PMEA derivatives were, unexpectedly, reported to exhibit antiviral activity two- to threefold lower compared to the PMEA (8) and 10-fold less active than the cycloSal-PMEA prodrugs. The reduced antiviral activity was attributed by the authors [61] to the slow release of PMEA (8) from the cycloAmb-PMEA, the rate-limiting step being the cleavage of the intermediate benzyl phosphonate ester II. To achieve higher antiviral activity, different cycloAmb-PMEA derivatives with new substitution patterns were reported [62]. However these new prodrugs failed to accelerate the decay of the hydrolysis of the intermediate II, presenting only a slightly improved antiviral activity when compared to the previous cyclo-Amb prodrugs.
S-Acylthioethyl ester prodrugs (SATE)
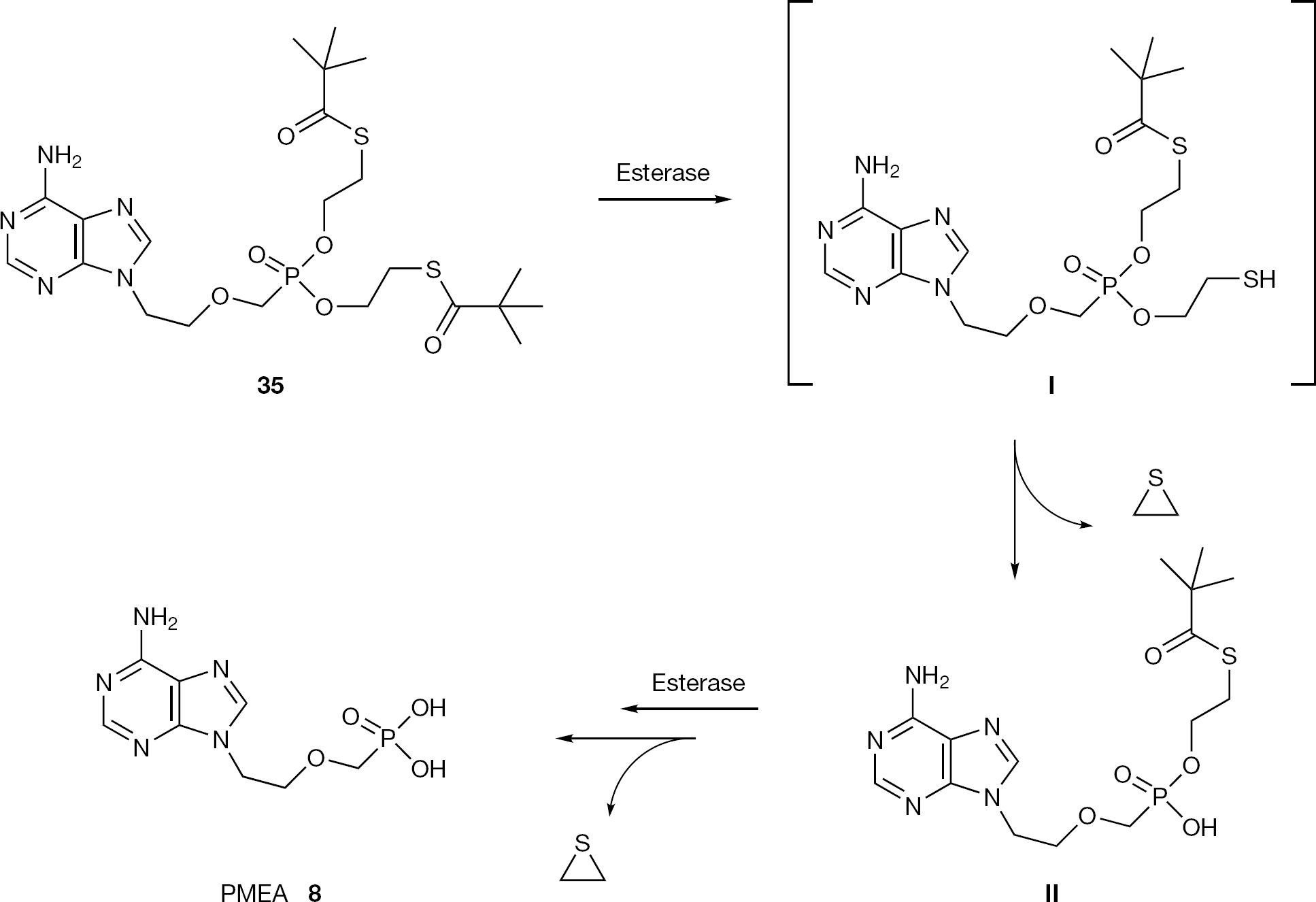
On the basis of the favourable results obtained with the S-acylthioethyl (SATE) prodrug for delivering monophosphate drugs, the SATE approach was applied successfully to ANPs. In particular, a series of bis-(SATE)-PMEA derivatives were reported. These compounds proved enzymatically more stable than the bis-(POM)-PMEA (18) [63]. Among them, the bis-(t-bu-SATE)-PMEA (35) emerged as the most promising compound, combining an antiviral potency against HIV similar to bis-(POM)-PMEA with a markedly greater chemical and enzymatic stability (Figure 17). The metabolic pathway for bis-(SATE) phosphonate prodrug 35, assumed to be identical to the one reported for the phosphate prodrug, is depicted in Figure 17 [64]. The prodrug, once inside the cell, preferentially forms an unstable 2-thioethyl intermediate (I) by the action of a carboxyesterase and then the 2-thioethyl moiety collapses to episulfide, releasing the mono SATE nucleotide (II). Exactly the same sequence of events leads to the release of the parent drug PMEA (8) from II. Other reports of SATE prodrugs of novel ANPs appeared recently indicating that this class of prodrugs may serve to study the in vitro delivery of phosphonate drugs [65]. The only concern for this class of prodrugs is a potential toxicity, which may limit their further development. In contrast, the SATE approach applied to CNPs did not give significant results. To eliminate the potential cell penetration issue associated with the ani-onic phosphonate moiety, several bis-SATE derivatives of adenosine phosphonate analogues were prepared [65]. Compared to the parent drugs, characterized by weak anti-HIV activity (EC50=100 μM), the anti-HIV activity of the prodrugs (EC50=40 μM) is moderately improved, but its cytotoxicity is increased from >100 μM to 50 μM.
Structure of bis-(t-bu-SATE)-PMEA prodrug (35) and its metabolic pathway
Aryl phosphonamidates and phosphonodiamidate prodrugs
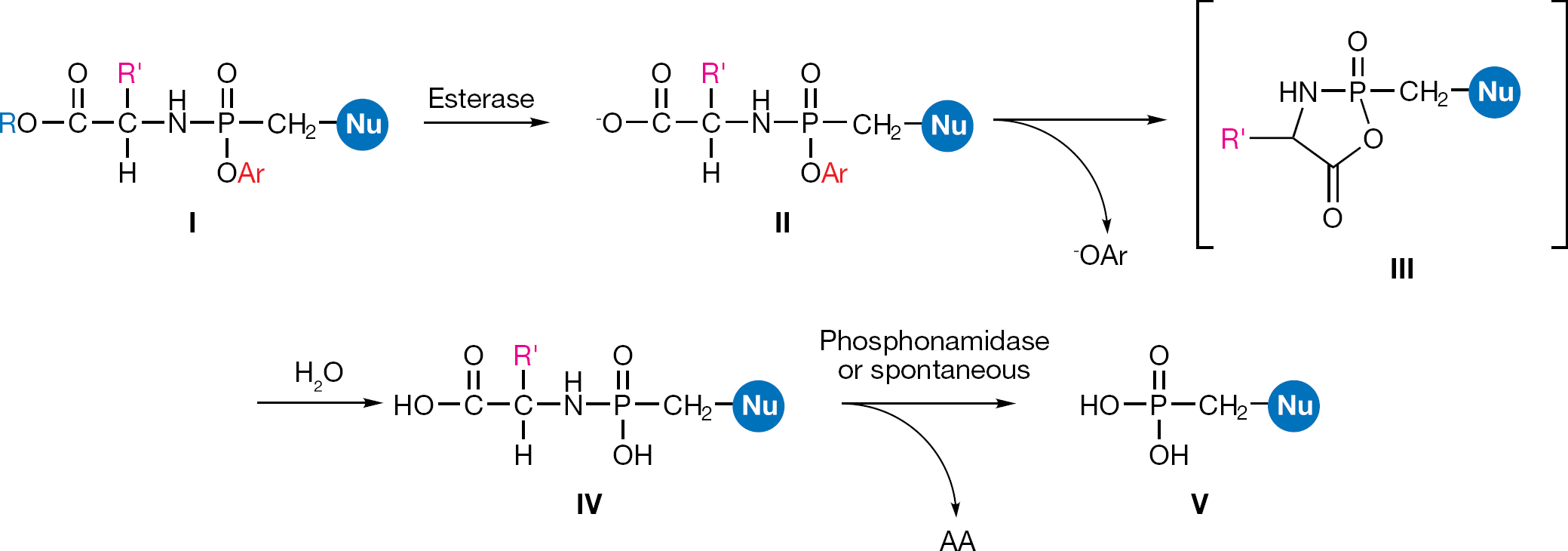
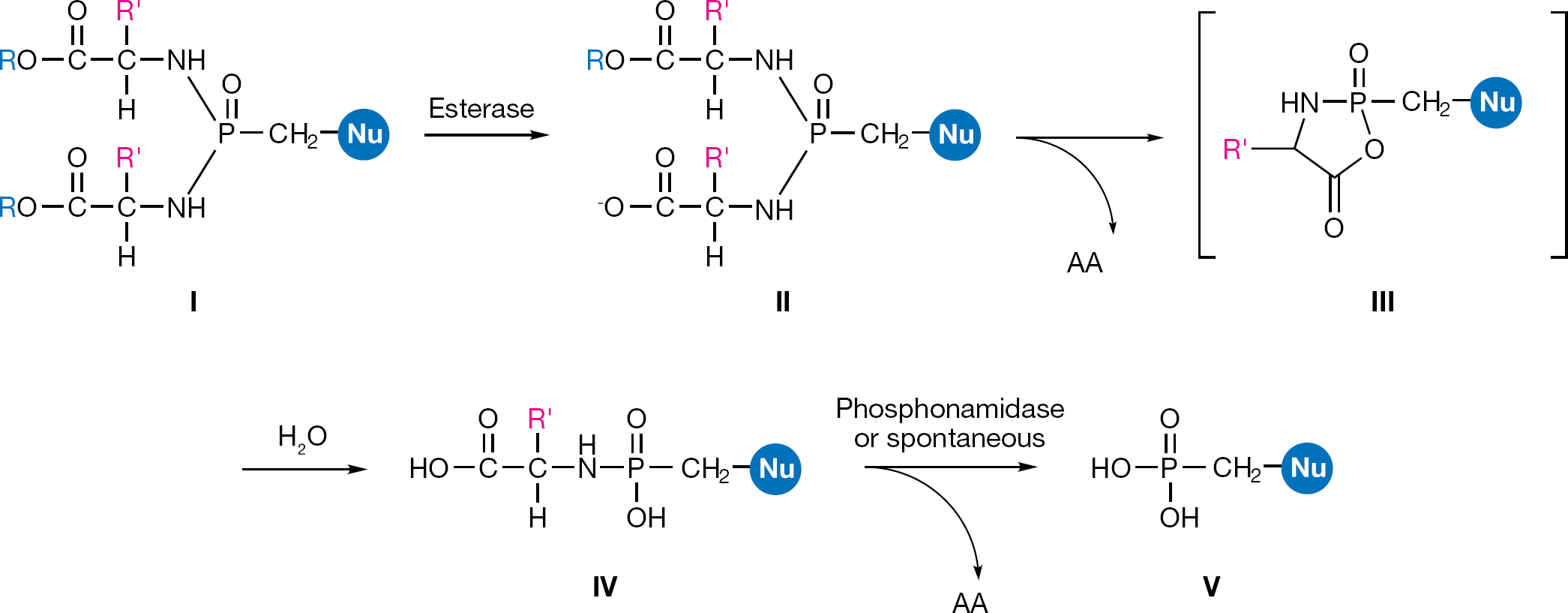
The ProTide (pronucleotide) approach, developed by McGuigan et al. [66] was succesfully applied to nucleoside phosphates and then investigated in application to nucleoside phosphonates. The ProTide of a nucleoside phosphonate is a phosphonamidate prodrug consisting of an amino acid ester promoiety linked via a P-N bond to a nucleoside aryl phosphonate. Such a prodrug should be able to facilitate passive diffusion through the cell membrane and when it is cleaved, it should deliver the nucleoside phosphonate inside the cell releasing the two masking groups. The metabolic activation of the phosphonamidates is generally assumed to follow the same two enzymatic steps involved in the activation of the phosphoramidates (Figure 18). The putative mechanism for the activation of the phosphonamidates then involves an initial carboxylic esterase or carboxy-peptidase-mediated hydrolysis of the carboxylic ester of the amino acid leading to intermediate II. The ester cleavage is followed by an internal nucleophilic attack of the acid residue on the phosphorus centre, displacing the aryloxy group and giving the transient formation of the putative five-membered cyclic intermediate III. This cyclic mixed anhydride is rapidly hydrolyzed to the corresponding aminoacyl phosphonamidate ester IV. The ester is then believed to undergo P-N cleavage, mediated by an enzyme endowed with a phosphonamidase activity or may result from simple hydrolysis in more acidic subcellular compartment, to eventually release the parent drug V.
General structure of aryl phosphonamidate prodrugs (I) and their metabolic pathway
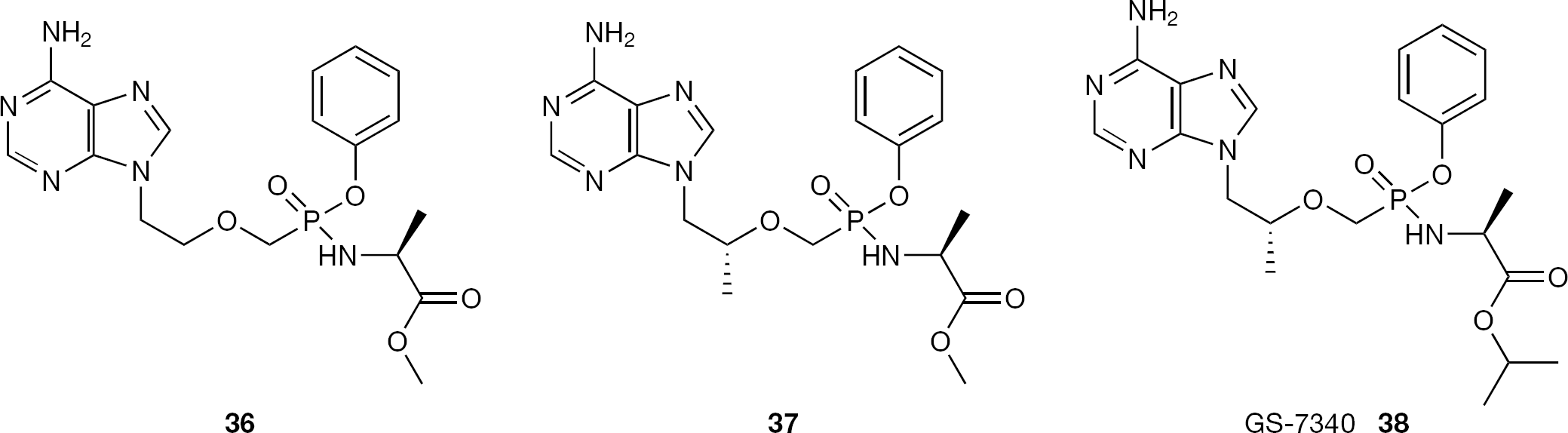
The ProTide technology was successfully applied by Ballatore et al. [66] to PMEA (8) and (S)-PMPA (9). In these studies, similar SARs were found for the phosphonamidates of PMEA and (S)-PMPA as earlier noted for nucleoside phosphoramidate analogues, with the (L)-alanine derivatives showing greatly enhanced antiviral potency against HIV compared with the parent nucleotide analogue (50-fold increase for phenyloxy methyl-(L)- alaninyl phosphonamidate PMEA prodrug (36) versus PMEA (8) and 50–100-fold increase for phenyloxy methyl-(L)-alaninyl phosphonamidate (R)-PMPA prodrug (37) versus (R)-PMPA (9;Figure 19)). Mirroring this work, Gilead Sciences reported an extensive study on the application of the ProTide approach to (R)-PMPA (9) investigating the mechanism of hydrolysis and the metabolism of this class of prodrugs [67]. As results of these studies, the phenyloxy isopropyl-(L)-alaninyl phosphonamidate prodrug of (R)-PMPA (38, GS-7340) emerged as a lead compound with improved biological properties (Figure 19). In addition, cathepsin A was found to be the primary enzyme that activates 38 in human lymphatic tissues [68].
Structures of PMEA phosphonoamidate prodrug 36 and (R)-PMPA phosphonamidate prodrugs 37 and GS-7340 (38)
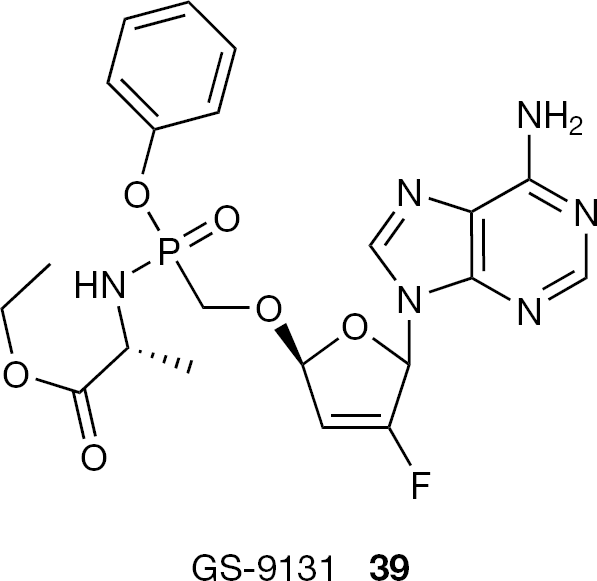
A series of aryl phosphonamidate prodrugs of GS-9148 (17), were also designed by Gilead to effectively deliver 17 and its active diphosphorylated metabolite into target cells [69]. The phenyloxy ethyl (L)-alaninyl phosphonamidate prodrug (39, GS-9131), improved the in vitro antiviral activity of 17 by approximately 50-fold against HIV, demonstrating the most favourable esterase (cathepsin) substrate properties in addition to good in vitro intestinal and hepatic stabilities [24,70] (Figure 20). Following oral dosing (3 mg/kg) of 39 in beagle dogs, high levels of GS-9148 diphosphate were observed inside the cells with a mean oral bioavailability of 26%. All these favourable properties lead to the selection of GS-9131 (39) as a clinical candidate.
Structure of phosphonoamidate derivative GS-9131 (39)
Phosphonodiamidate analogues having two identical amino acids as masking groups of the phosphonate moiety through a P-N bond were designed. They offer two distinct advantages compared to phosphonamidate analogues: due to their symmetric structure no phosphorus chirality arises and exclusively non-toxic promoieties are released. The activation mechanism is presumed to be similar to the one previously shown for the cognate phosphonoamidates. As first steps the hydrolysis of one amino acid ester lead through spontaneous cyclization to intermediate III, structurally identical to the one derived from the activation of an aryl phosphonamidate (see intermediate III in Figure 18). Then exactly the same pathway follows (Figure 21).
General structure of phosphonodiamidate prodrugs and their metabolic pathway
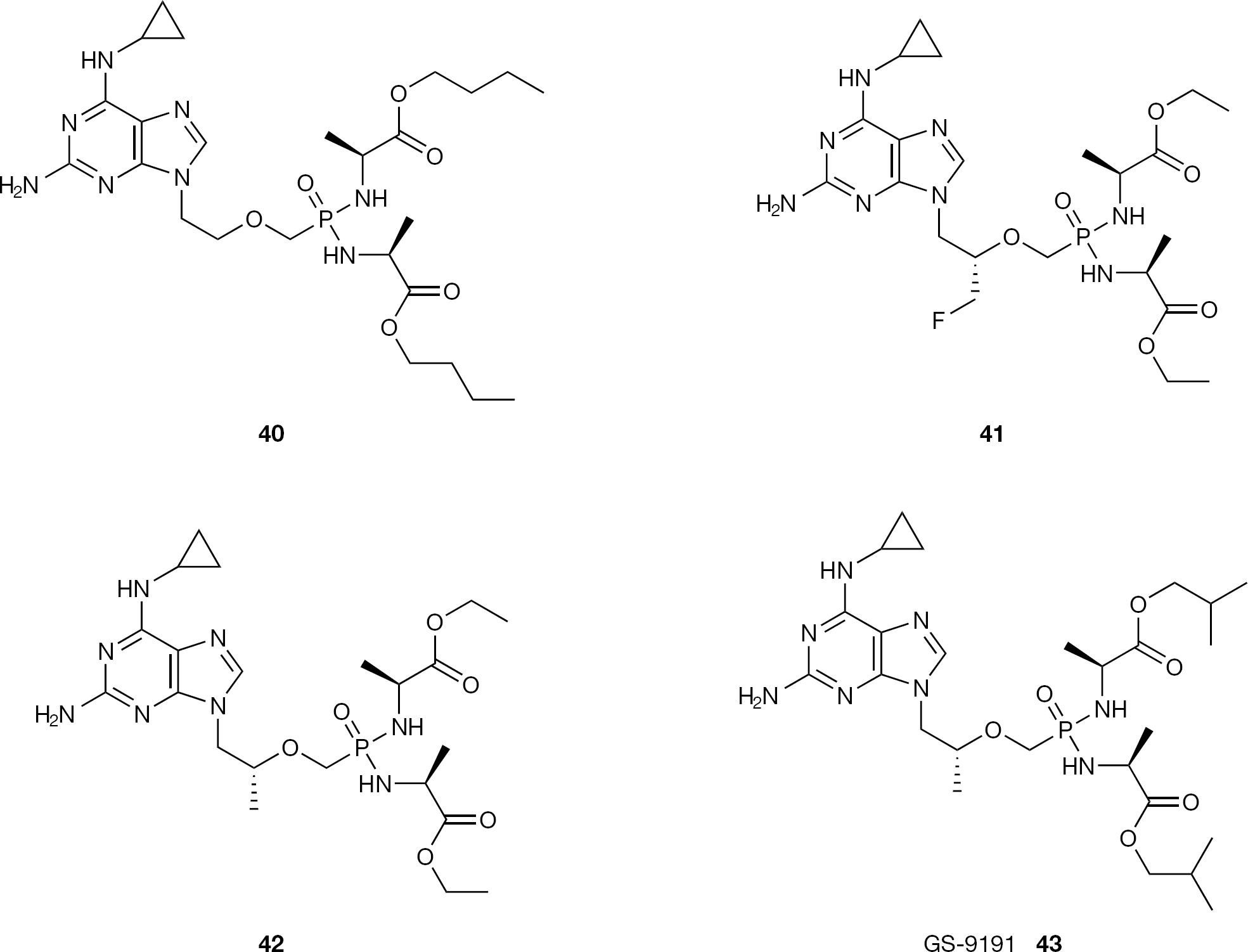
Several ANP phosphonodiamidates containing alkyl (L)-alanine were reported to exhibit potent antiviral activities, being bis(butyl-(L)-alaninyl)- (40) and bis(isobutyl-(L)-phenylalaninyl)phosphonodiamidate-PME-N6-(cyclopropyl)DAP (43, GS-9191) the most active of the series respectively against pox and papilloma viruses (HPV; Figure 22) [71,72]. After prodrug cleavage, the cPrPMEDAP is intracellularly deami-nated by N6-methyl-AMP amino hydrolase to yield the potent antiproliferative and antiviral agent PMEG (10). GS9191 is currently in Phase I clinical trials as a topical prodrug for the treatment of HPV lesion.
Structures of phosphonodiamidate prodrugs 40–43
Recently, the development of a novel and efficient one-pot synthesis of phosphonodiamidates directly from the phosphonic acid diesters, significantly improved the preparation of this class of prodrugs [73]. In this report, the methodology has been applied to the synthesis of 41 and 42, which exhibit anti-HIV activity with submicromolar EC50 values and no detectable cytotoxicity up to 100 μM, the highest concentration tested (Figure 22).
Diamidate prodrugs of GS-9148 (17) have been reported. Despite their potent in vitro anti-HIV activity these diamidates were quite ineffective in delivering 17 into the cells, producing, after intravenous administration to dogs, only approximately threefold higher intracellular levels of GS-9148 compared to that from the same dose of GS-9148 by itself [69].
Peptidomimetic prodrugs
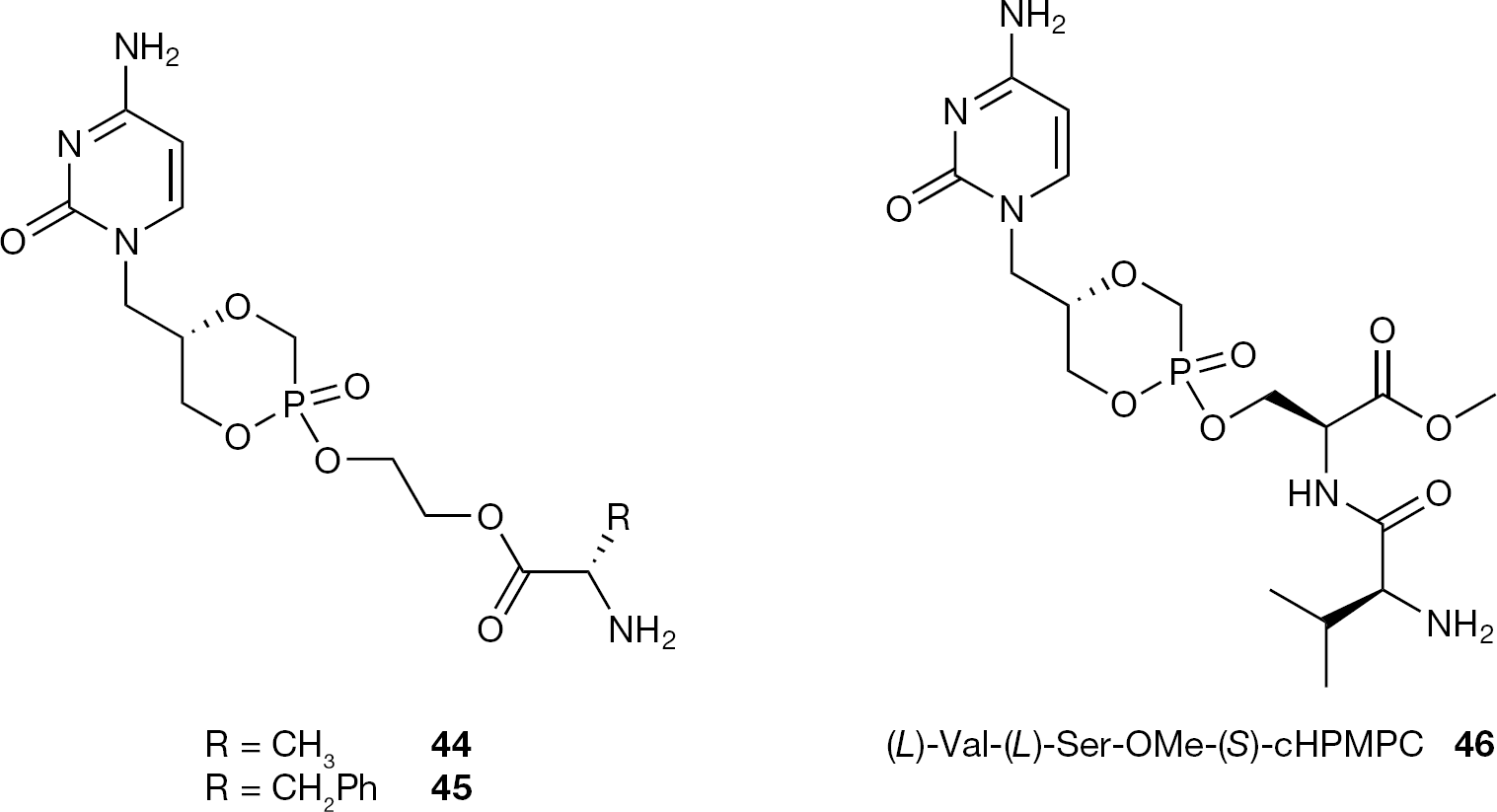
Very significantly enhanced oral bioavailability of valacyclovir resulting from esterification of the hydroxyl group of acyclovir with the carboxylic group of an (L)-valine, has encouraged the use of amino acids or dipeptides as promoieties. In this context, to increase the oral biovailability of acyclic nucleoside phosphonate McKenna and coworkers [74] have proposed an approach in which a non-toxic promoiety such as a dipeptide or an amino acid is conjugated to ANPs ((S)-HPMPC (3) or (S)-HPMPA (1)) by esterification of the phosphonic acid group with an alcoholic amino acid side chain [74]. The other phosphonic OH group was either left free or masked by intramolecular esterification affording (S)-cHPMPC or (S)-cHPMPA derivatives or by esterification with an ethyl group. In their initial investigation the synthesis and biological evaluation of several phosphono dipeptide ester prodrugs of (S)-cHPMPC with a dipeptide attached via the hydroxyl group of an (L)-serine as well as single amino acid ((L)-valine or (L)-phenyalanine) prodrugs coupled by an ethylene glycol (EG) linkage were reported (Figure 23) [75–77]. All the dipeptide and the EG-amino acid conjugates 44 and 45 showed, in general, an in vitro antiviral activity (HCMV) similar to the parent drug. Whereas 44 and 45 did not exhibit increased bioavailability compared to the parent compound after direct injection into the gastrointestinal tract of rats, interestingly, (L)-Val-(L)-Ser-OMe-(S)-cHPMPC (46) displayed an eightfold increase in oral bioavailability relative to (S)-cHPMPC (4) in in vivo murine model transport studies.
Structures of ethylene glycol aminoacid (44 and 45) and serine dipeptide (S)-cHPMPC prodrugs (46)
Since valacyclovir and valganciclovir are actively transported by the human peptide-specific intestinal transporter (hPEPT1) which is highly expressed in the gastrointestinal tract, Peterson et al. [78] investigated the possibility that hPEPT1 was involved in the transport of (L)-Val-(L)-Ser-OMe (S)-cHPMPC conjugate (46) observed in in vivo murine model transport studies. Under the conditions studied, the authors showed that the Val-Ser-OMe dipeptide (S)-cHPMPC stereoisomer conjugates as well as different serine mono amino acid (S)-cHPMPC and (S)-cHPMPA conjugates are recognized, but not transported by hPEPT1. According to the authors, this is probably due to the steric and/or polar structural specifics of the linked (S)-cHPMPC drug cargo, suggesting that an alternative transport mechanism operates for these prodrugs.
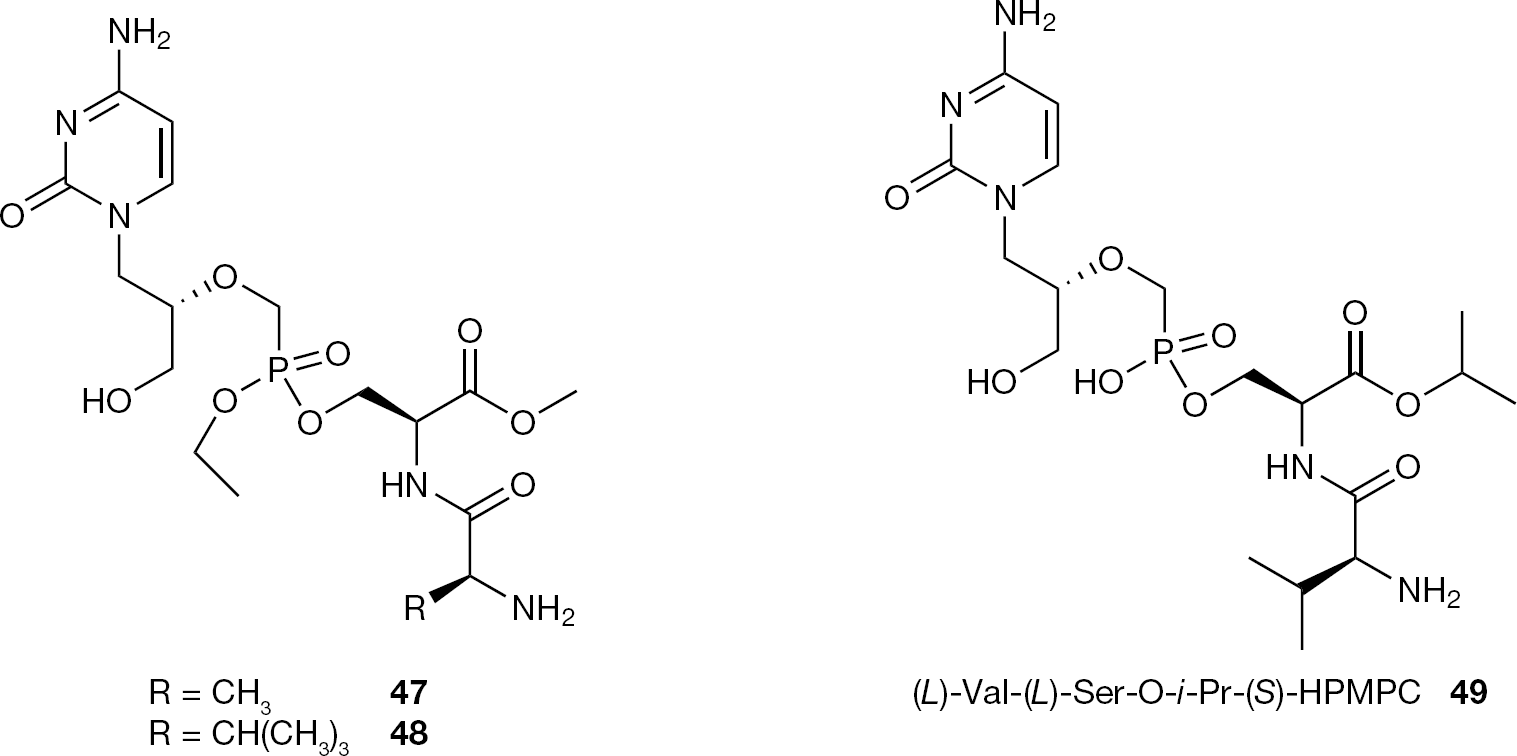
Despite an enhanced bioavailability, the dipetide (S)-cHPMPC conjugates having the second phosphonic OH masked with an ethyl group (47 and 48), proved to be not suitable as prodrugs due to the fact that the ethyl group was not cleaved during in vivo experiments and that the P-OEt monoester (S)-HPMPC metabolite did not exhibit significant antiviral activity in an in vitro vaccinia plaque reduction assay (Figure 24) [79].
Structures of serine dipeptide (S)-HPMPC prodrugs (47–49)
Intriguingly, among the serine dipeptide (S)-HPMPC conjugates series, despite the presence of an ionizable P-OH group, (L)-Val-(L)-Ser-O-i-Pr (S)-HPMPC (49;Figure 24) displayed the greatest oral biovailability with a 15-fold increase in total cidofovir species in the plasma (related to 3 and 4) after oral administration. This enhanced oral bioavailability was tentatively justified by the authors as the results of a higher chemical and enzymatic stability compared to its cyclic analogue [79].
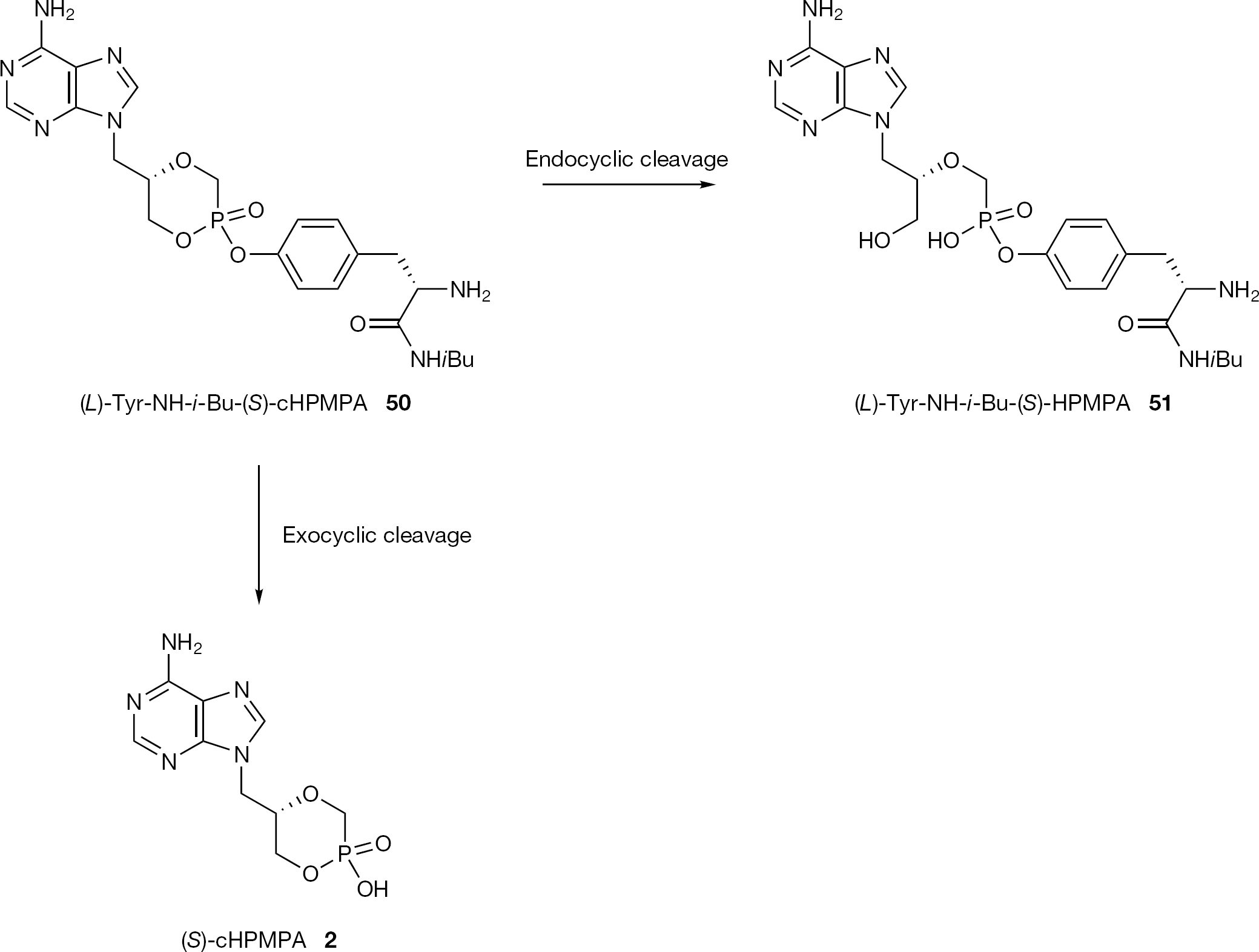
More recently, the same authors have reported cyclic (S)-HPMPC and (S)-HPMPA amino acid or dipeptide prodrugs in which the phosphonic acid has been masked by esterification with a tyrosine hydroxyl group [80]. In this report, the authors also provided a convenient method for the partial conversion of the prodrug into the more stable Rp diastereomer by a transesterification reaction from the corresponding Sp diastereomer. Along with this new series of peptidomimetic prodrugs, (L)-Tyr-NH-i-Bu (S)-cHPMPA (50, Figure 25) was converted in rat or mouse plasma solely to two active metabolites: (S)-cHPMPA 2 and the acyclic form of the respective prodrug (51) which was shown to possess in vitro antiviral activity as well (Figure 25). In addition, 50 had significantly enhanced oral bioavailability versus parent drug in a mouse model (39% versus <5%). Krečmerovα et al. [41] also successfully applied the peptidomimetic approach to cyclic (S)-HPMP-DAP (5). These results suggested that these peptidomimetic prodrugs are attractive candidates for further in vivo evaluation.
Structure of (L)-Tyr-NH-i-Bu-(S)-cHPMPA (50) and its metabolic pathway
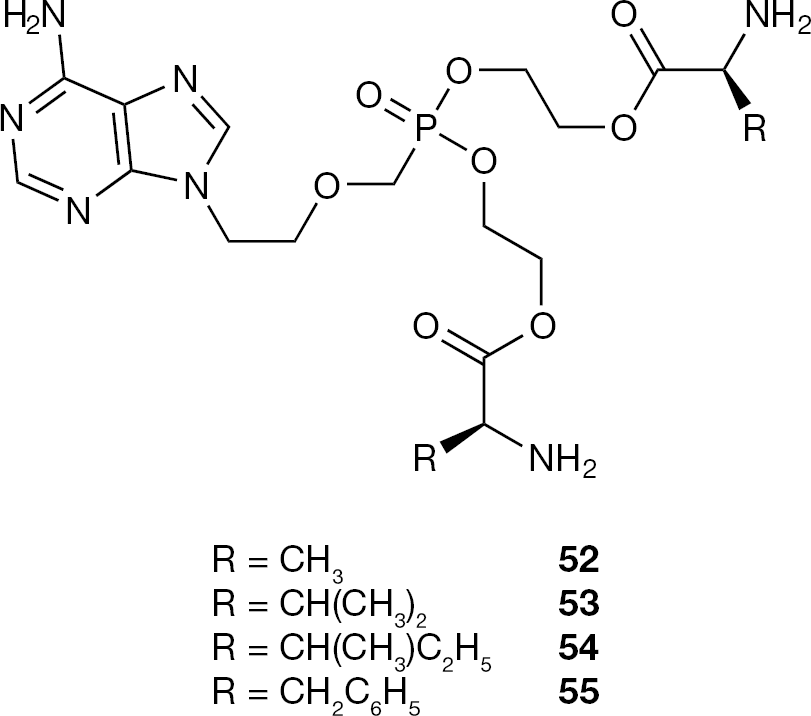
Independently, a different group described a series of bis-(L)-amino acid ester prodrugs of PMEA (8) as potent anti-HBV agents with reduced toxicity [81]. Several of these compounds demonstrated more potent anti-HBV activity and higher selective index (SI) than adefovir dipivoxil (18), which was used in this study as a positive control (Figure 26).
Structures of amino acid and peptide PMEA prodrugs 52–55
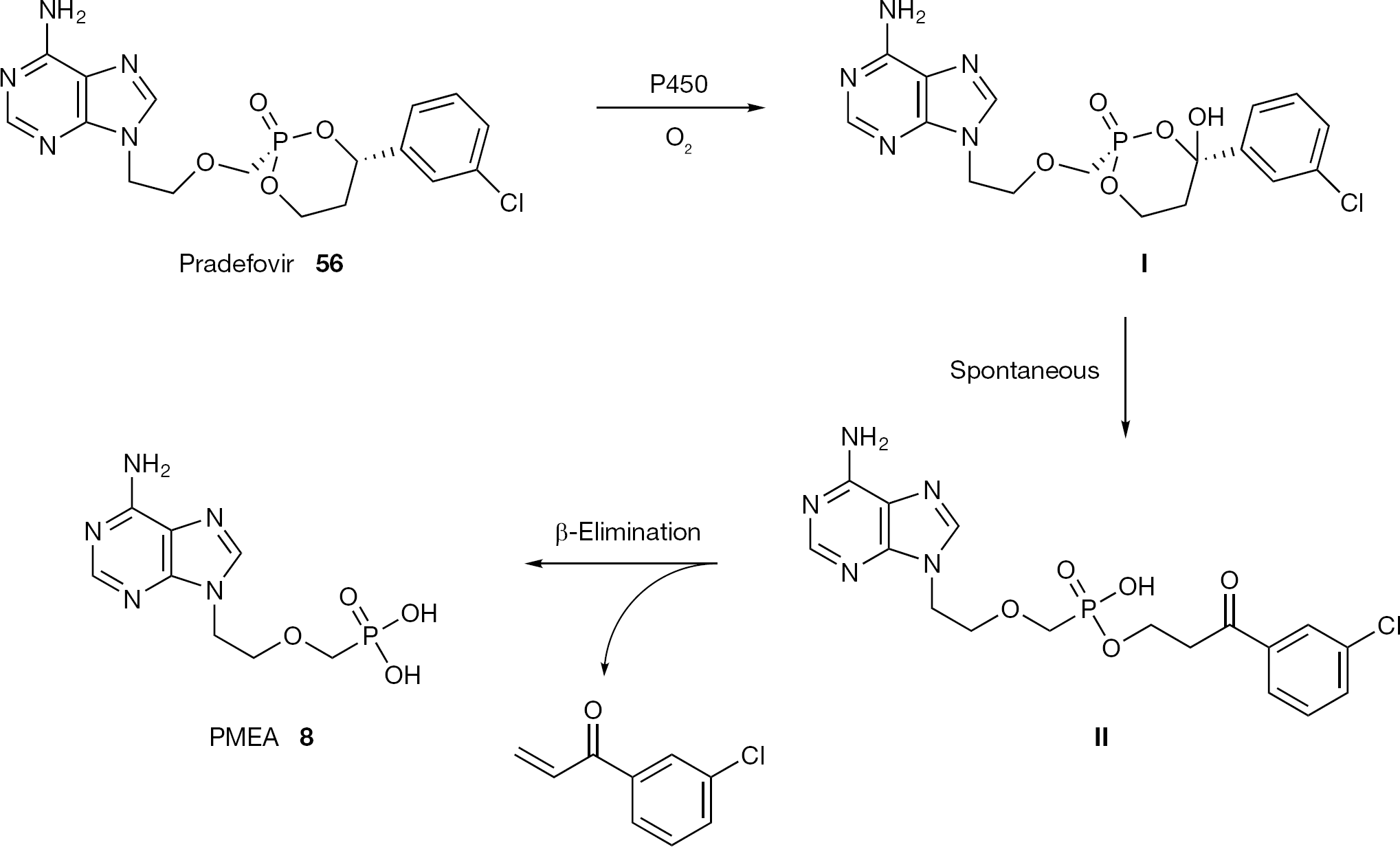
The cyclic 1-aryl-1,3-propanyl ester is a type of prodrug moiety recently reported to target phosphate and phosphonate-containing drug to the liver [82]. This prodrug class, called HepDirect prodrugs, is selectively activated by a liver specific cytochrome P450 isozyme CYP3A4 (Figure 27). In an effort to discover new drugs for treating diseases such as HBV, cyclic 1-aryl-1,3-propanyl ester prodrugs of PMEA (8), were developed. The lead prodrug pradefovir (56) identified among several Hept Direct PMEA prodrugs exhibited a high rate of activation in hepatocytes together with good oral biovailability in rat and dog species, which was further increased by using the mesylate salt to boost water solubility [83] (Figure 27). Evaluation of its individual isomers led to the selection of single prodrug isomer (4S, Rp) as the lead compound. Phase II studies of pradefovir in hepatitis B patients were completed in USA by Ligand Pharmaceuticals. The trials, however, were put on hold based on increased tumour incidence in animal studies [84]. Since January 2011 pradefovir is under clinical evaluation in hepatitis B patients by Chiva Pharmaceuticals in China.
Structure of pradefovir (56) and its metabolic pathway in hepatocytes
Discussion
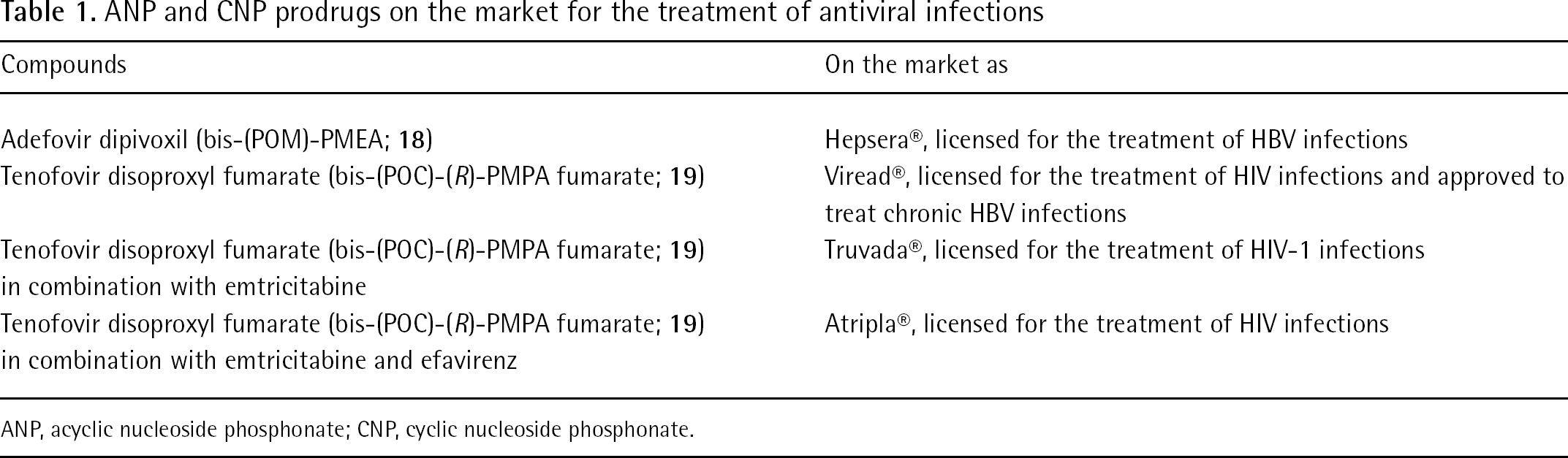
As a result of extensive efforts in the development of ANPs and CNPs prodrugs, two prodrugs of adefovir and tenofovir (Hepsera® and Viread®, respectively) and their two formulations (Truvada® and Atripla®), in combination with other drugs, are on the market (Table 1).
ANP and CNP prodrugs on the market for the treatment of antiviral infections
Compounds
On the market as
Adefovir dipivoxil (bis-(POM)-PMEA; 18)
Hepsera®, licensed for the treatment of HBV infections
However, further research is needed in this area. In fact, there is still some concern about adefovir dipivoxil (18) and tenofovir disopropoxil (19) due to their potential toxicity during long-term treatment. The same concern can be extended to the clinical candidate PMCDG dipivoxil (20). In contrast to the PME and PMP series, there is not a commercially available prodrug so far available for HPMP-derivatives; cidofovir (3), approved with the brand name of Vistide® for the treatment of cytomegalovirus retinitis in AIDS patients is applied as an intravenous infusion of the free acid. Cidofovir is also accepted as an effective therapy for smallpox infection, caused by variola virus, a member of the orthopoxvirus genus. Although this disease was eradicated after an intensive programme of vaccination in 1980, there is increasing concern that variola virus might be used as a bioterrorist weapon because of its ease of dissemination, contagiousness and high mortality rate. Lack of an oral form of cidofovir significantly limits its usefulness under the disruptive conditions of a large-scale biowarfare attack.
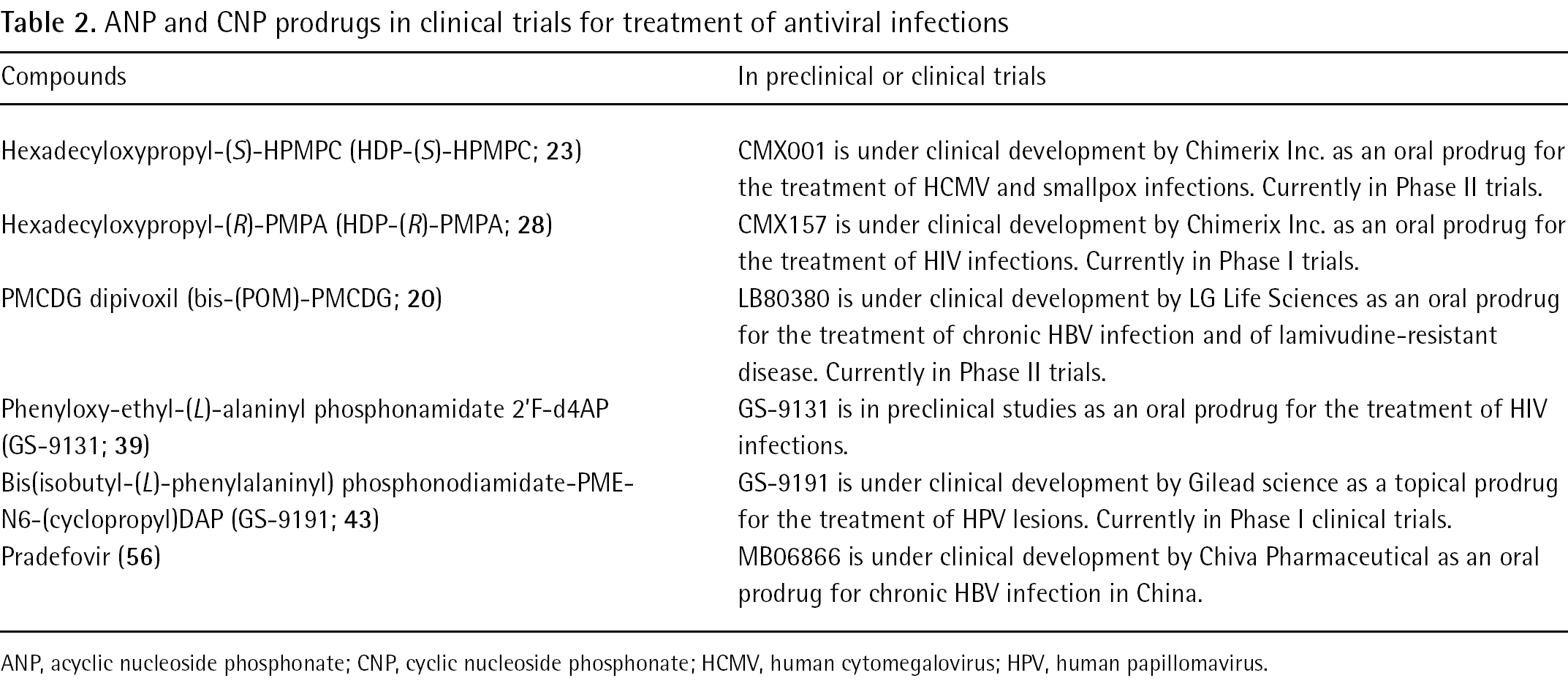
A considerable number of ANP and CNP prodrugs have been evaluated but only a few of them passed the preclinical studies and are now in clinical trials (Table 2). This likely arises from the complexity in designing prodrugs appropriate for clinical use in terms of stability, metabolism, toxicology and side effects. Some of them appear promising, such as the long chain alkyl ester prodrugs developed by Hostetler and applied to different ANPs or the phosphonamidate of 2′-F-d4AP. However, they may suffer from potential drawbacks such as lack of solubility for the long chain prodrugs or possible toxicity due to the releasing of the phenol in the case of the phosphonoamidate prodrugs for which additional diastereoi-somer issues need also to be taken in account.
ANP and CNP prodrugs in clinical trials for treatment of antiviral infections
Compounds
In preclinical or clinical trials
Hexadecyloxypropyl-(S)-HPMPC (HDP-(S)-HPMPC; 23)
CMX001 is under clinical development by Chimerix Inc. as an oral prodrug for the treatment of HCMV and smallpox infections. Currently in Phase II trials.
Hexadecyloxypropyl-(R)-PMPA (HDP-(R)-PMPA; 28)
CMX157 is under clinical development by Chimerix Inc. as an oral prodrug for the treatment of HIV infections. Currently in Phase I trials.
PMCDG dipivoxil (bis-(POM)-PMCDG; 20)
LB80380 is under clinical development by LG Life Sciences as an oral prodrug for the treatment of chronic HBV infection and of lamivudine-resistant disease. Currently in Phase II trials.
GS-9191 is under clinical development by Gilead science as a topical prodrug for the treatment of HPV lesions. Currently in Phase I clinical trials. MB06866 is under clinical development by Chiva Pharmaceutical as an oral prodrug for chronic HBV infection in China.
ANP, acyclic nucleoside phosphonate; CNP, cyclic nucleoside phosphonate; HCMV, human cytomegalovirus; HPV, human papillomavirus.
In these scenarios, other prodrug strategies like the phosphono-diamidate (one already in clinical trial) and the peptidomimetic conjugate prodrugs are thus of extreme importance and may offer valuable alternatives to the other prodrugs. Hep-direct prodrugs are the obvious option when targeting the liver is required.
In summary, a variety of ANP and CNP prodrugs with diverse chemical structures have been presented in this review. Although extensive efforts have been made in developing these prodrugs for the management of antiviral infections there are still exciting future prospects for either novel ANPs and CNPs and/or their prodrugs.
Footnotes
The authors declare no competing interests.
References
1.
De ClercqENeytsJ. Antiviral agents acting as DNA or RNA chain terminators. Handb Exp Pharmacol2009; 189:53–84.
2.
De ClercqE. The clinical potential of the acyclic (and cyclic) nucleoside phosphonates. The magic of the phosphonate bond. Biochem Pharmacol2011; 82:99–109.
3.
De ClercqEHolýA. Acyclic nucleoside phosphonates: A key class of antiviral drugs. Nat Rev Drug Discov2005; 4:928–940.
4.
HolýA. Phosphonomethoxyalkyl analogs of nucleotides. Curr Pharm Des2003; 9:2567–2592.
5.
De ClercqEHolýARosenbergI. A novel selective broad-spectrum anti-DNA virus agent. Nature1986; 323:464–467.
6.
De ClercqEAndreiGBalzariniJ. Antiviral potential of a new generation of acyclic nucleoside phosphonates, the 6-[2-(phosphonomethoxy)alkoxy]-2,4-diaminopyrimidines. Nucleosides Nucleotides Nucleic Acids2005; 24:331–341.
7.
De ClercqE. In search of a selective antiviral chemotherapy. Clin Microbiol Rev1997; 10:674–693.
8.
De ClercqE. The acyclic nucleoside phosphonates from inception to clinical use: Historical perspective. Antiviral Res2007; 75:1–13.
9.
De ClercqENeytsJ. Therapeutic potential of nucleoside/ nucleotide analogues against poxvirus infections. Rev Med Virol2004; 14:289–300.
10.
Dal PozzoFAndreiGHolýA. Activities of acyclic nucleoside phosphonates against orf virus in human and ovine cell monolayers and organotypic ovine raft cultures. Antimicrob Agents Chemother2005; 49:4843–4852.
11.
NaesensLLenaertsLAndreiG. Antiadenovirus activities of several classes of nucleoside and nucleotide analogues. Antimicrob Agents Chemother2005; 49:1010–1016.
12.
KrečmerováMHolýAPskalaA. Antiviral activity of triazine analogues of 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]cytosine (Cidofovir) and related compounds. J Med Chem2007; 50:1069–1077.
13.
CundyKCBALynchGShawJP. Pharmacokinetics, bioavailability, metabolism, and tissue distribution of cidofovir (HPMPC) and cyclic HPMPC in rats. Drug Metab Dispos1996; 24:745–752.
14.
MendelDBChilarTMoonKChenMS. Conversion of 1-[((S)-2-hydroxy-2-oxo-1,4,2-dioxaphosphorinan-5-yl) methyl] cytosine to cidofovir by an intracellular cyclic CMP phosphodiesterase. Antimicrob Agents Chemother1997; 41:641–646.
15.
NaesensLBalzariniJRosenbergI. 9-(2-Phosphonylmethoxyethyl)-2,6-diaminopurine (PMEDAP): A novel agent with anti-human immunodeficiency virus activity in vitro and potent anti-Moloney murine sarcoma virus activity in vivo. Eur J Clin Microbiol Infect Dis1989; 8:1043–1047.
16.
YingCDe ClercqENeytsJ. Lamivudine, adefovir and tenofovir exhibit long-lasting anti-hepatitis B virus activity in cell culture. J Viral Hepat2000; 7:79–83.
17.
YingCDeCENicholsonW. Inhibition of the replication of the DNA polymerase M550V mutation variant of human hepatitis B virus by adefovir, tenofovir, L-FMAU, DAPD, penciclovir and lobucavir. J Viral Hepat2000; 7:161–165.
18.
KramataPVotrubaIOtovaB. Different inhibitory potencies of acyclic phosphonomethoxyalkyl nucleotide analogs toward DNA polymerases α, δ, and ε. Mol Pharmacol1996; 49:1005–1011.
19.
KreiderJWBaloghKOlsonRO. Treatment of latent rabbit and human papillomavirus infections with 9-(2-phosphonylmethoxy)ethylguanine (PMEG). Antiviral Res1990; 14:51–58.
20.
HolýAVotrubaIMasojidkovaM. 6-[2-(Phosphonomethoxy)alkoxy]pyrimidines with antiviral activity. J Med Chem2002; 45:1918–1929.
21.
BalzariniJPannecouqueCDe ClercqE. Antiretrovirus activity of a novel class of acyclic pyrimidine nucleoside phosphonates. Antimicrob Agents Chemother2002; 46:2185–2193.
22.
WuTFroeyenMKempeneersV. Deoxythreosyl phosphonate nucleosides as selective anti-HIV agents. J Am Chem Soc2005; 127:5056–5065.
23.
MackmanRLZhangLPrasadV. Synthesis and anti-HIV activity of cyclic pyrimidine phosphonomethoxy nucleosides and their prodrugs: A comparison of phosphonates and corresponding nucleosides. Nucleosides Nucleotides Nucleic Acids2007; 26:573–577.
24.
CihlarTRayASBoojamraCG. Design and profiling of GS-9148, a novel nucleotide analog active against nucleoside-resistant variants of human immunodeficiency virus type 1, and its orally bioavailable phosphonoamidate prodrug, GS-9131. Antimicrob Agents Chemother2008; 52:655–665.
25.
LiFMaagHAlfredsonT. Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J Pharm Sci2008; 97:1109–1134.
26.
KriseJPStellaVJ. Prodrugs of phosphates, phosphonates, and phosphinates. Adv Drug Deliv Rev1996; 19:287–310.
27.
HeckerSJErionMD. Prodrugs of phosphates and phosphonates. J Med Chem2008; 51:2328–2345.
28.
HeG-XKriseJPOliyaiR. Prodrugs of phosphonates, phosphinates, and phosphates. Prodrug challenges and rewards part 1 and part 2. New York: Springer2007; pp. 223–264.
29.
SchultzC. Prodrugs of biologically active phosphate esters. Bioorg Med Chem2003; 11:885–898.
30.
ArizaME. Current prodrug strategies for the delivery of nucleotides into cells. Drug Des Rev Online2005; 2:373–387.
31.
PetersonLWMcKennaCE. Prodrug approaches to improving the oral absorption of antiviral nucleotide analogues. Expert Opin Drug Deliv2009; 6:405–420.
32.
WagnerCRIyerVVMcInteeEJ. Pronucleotides: Toward the in vivo delivery of antiviral and anticancer nucleotides. Med Res Rev2000; 20:417–451.
33.
StarrettJEJr.TortolaniDRRussellJ. Oral bioavailability determination, and in vitro evaluation of prodrugs of the antiviral agent 9-[2-(phosphonomethoxy) ethyl]adenine (PMEA). J Med Chem1994; 37:1857–1864.
34.
ZídekZKmoníčkováEHolýA. Cytotoxicity of pivoxil esters of antiviral acyclic nucleoside phosphonates: Adefovir dipivoxil versus adefovir. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub2005; 149:315–319.
35.
SmedleyJ. Is formaldehyde an important cause of allergic respiratory disease?Clin Exp Allergy1996; 26:247–249.
36.
YuPHZuoDM. Formaldehyde produced endogenously via deamination of methylamine. A potential risk factor for initiation of endothelial injury. Atherosclerosis1996; 120:189–197.
37.
BrassEP. Pivalate-generating prodrugs and carnitine homeostasis in man. Pharmacol Rev2002; 54:589–598.
38.
SrinivasRVRobbinsBLConnellyMC. Metabolism and in vitro antiretroviral activities of bis(pivaloyloxymethyl) prodrugs of acyclic nucleoside phosphonates. Antimicrob Agents Chemother1993; 37:2247–2250.
39.
ChoiJ-RChoD-GRohKY. A novel class of phosphonate nucleosides. 9-[(1-phosphonomethoxycyclopropyl)methyl]guanine as a potent and selective anti-HBV agent. J Med Chem2004; 47:2864–2869.
40.
YuenM-FKimJKimR. A randomized placebo-controlled, dose-finding study of oral LB80380 in HBeAg-positive patients with chronic hepatitis B. Antivir Ther2006; 11:977–983.
41.
KrečmerováMHolýAAndreiG. Synthesis of ester prodrugs of 9-(S)-[3-hydroxy-2-(phosphonomethoxy) propyl]-2,6-diaminopurine (HPMPDAP) as anti-poxvirus agents. J Med Chem2010; 53:6825–6837.
42.
TopalisDPradèreURoyV. Novel antiviral C5-substituted pyrimidine acyclic nucleoside phosphonates selected as human thymidylate kinase substrates. J Med Chem2011; 54:222–232.
43.
HostetlerKY. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: Current state of the art. Antiviral Res2009; 82:A84–A98.
44.
BeadleJRHartlineCAldernKA. Alkoxyalkyl esters of cidofovir and cyclic cidofovir exhibit multiple-log enhancement of antiviral activity against cytomegalovirus and herpesvirus replication in vitro. Antimicrob Agents Chemother2002; 46:2381–2386.
45.
HostetlerKY. Synthesis and early development of hexadecyloxypropyl-cidofovir: An oral antipoxvirus nucleoside phosphonate. Viruses2010; 2:2213–2225.
46.
KernERHartlineCHardenE. Enhanced inhibition of orthopoxvirus replication in vitro by alkoxyalkyl esters of cidofovir and cyclic cidofovir. Antimicrob Agents Chemother2002; 46:991–995.
47.
KernERCollinsDJWanWB. Oral treatment of murine cytomegalovirus infections with ether lipid esters of cidofovir. Antimicrob Agents Chemother2004; 48:3516–3522.
48.
QuenelleDCCollinsDWanW. Oral treatment of cowpox and vaccinia virus infections in mice with ether lipid esters of cidofovir. Antimicrob Agents Chemother2004; 48:404–412.
49.
BullerRMOwensGSchriewerJ. Efficacy of oral active ether lipid analogs of cidofovir in a lethal mousepox model. Virology2004; 318:474–481.
50.
TrostLCLampertBMRobertsonA. Interspecies comparison of the pharmacokinetics of CMX001, a lipid conjugated nucleotide analog with broad dsDNA antiviral activity. 23th ICAR Annual Meeting. 25–28 April 2010, San Francisco, CA, USA.
51.
QuenelleDCCollinsDJHerrodBP. Effect of oral treatment with hexadecyloxypropyl-[(S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine] [(S)-HPMPA] or octadecyloxyethyl-(S)-HPMPA on cowpox or vaccinia virus infections in mice. Antimicrob Agents Chemother2007; 51:3940–3947.
52.
BeadleJRWanWBCieslaSL. Synthesis and antiviral evaluation of alkoxyalkyl derivatives of 9-(S)-(3-hydroxy-2-phosphonomethoxypropyl)adenine against cytomegalovirus and orthopoxviruses. J Med Chem2006; 49:2010–2015.
53.
QuenelleDCCollinsDJPettwayLR. Effect of oral treatment with (S)-HPMPA, HDP-(S)-HPMPA or ODE-(S)-HPMPA on replication of murine cytomegalovirus (MCMV) or human cytomegalovirus (HCMV) in animal models. Antiviral Res2008; 79:133–135.
54.
KrečmerováMHolýAPohlR. Ester prodrugs of cyclic 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]-5-azacytosine: Synthesis and antiviral activity. J Med Chem2007; 50:5765–5772.
55.
TichýTAndreiGDracínskýM. New prodrugs of adefovir and cidofovir. Bioorg Med Chem2011; 19:3527–3539.
56.
LebeauIAndreiGDal PozzoF. Activities of alkoxyalkyl esters of cidofovir (CDV), cyclic CDV, and (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine against orthopoxviruses in cell monolayers and in organotypic cultures. Antimicrob Agents Chemother2006; 50:2525–2529.
57.
OliyaiRArimilliMNJonesRJ. Pharmacokinetics of salicylate ester prodrugs of cyclic HPMPC in dogs. Nucleosides Nucleotides Nucleic Acids2001; 20:1411–1414.
58.
OliyaiRShawJ-PSueoka-LennenCM. Aryl ester prodrugs of cyclic HPMPC. I: Physicochemical characterization and in vitro biological stability. Pharm Res1999; 16:1687–1693.
59.
SerafinowskaHTAshtonRJBaileyS. Synthesis and in vivo evaluation of prodrugs of 9-[2-(phosphonomethoxy) ethoxy]adenine. J Med Chem1995; 38:1372–1379.
60.
ShawJ-PLouieMSKrishnamurthyVV. Pharmakokinetics and metabolism of selected prodrugs of PMEA in rats. Drug Metab Dispos1997; 25:362–366.
61.
MeierCGörbigUMüllerC. cycloSal-PMEA and cycloAmb-PMEA: Potentially new phosphonate prodrugs based on the cycloSal-Pronucleotide approach. J Med Chem2005; 48:8079–8086.
62.
GörbigUBalzariniJMeierC. New cycloAMB-nucleoside phosphonate prodrugs. Nucleosides Nucleotides Nucleic Acids2007; 26:831–834.
63.
BenzariaSPelicanoHJohnsonR. Synthesis, in vitro antiviral evaluation, and stability studies of bis(S-acyl-2-thioethyl) ester derivatives of 9-[2-(phosphonomethoxy) ethyl]adenine (PMEA) as potential PMEA prodrugs with improved oral bioavailability. J Med Chem1996; 39:4958–4965.
64.
PérigaudCGosselinGLefebvreI. Rational design for cytosolic delivery of nucleoside monphosphates: ‘SATE’ and ‘DTE’ as enzyme-labile transient phosphate protecting groups. Bioorg Med Chem Lett1993; 3:2521–2526.
65.
LiHHongJH. Synthesis and anti-HIV evaluation of new acyclic phosphonate nucleotide analogues and their bis(SATE) derivatives. Nucleosides Nucleotides Nucleic Acids2010; 29:581–590.
66.
BallatoreCMcGuiganCDe ClercqE. Synthesis and evaluation of novel amidate prodrugs of PMEA and PMPA. Bioorg Med Chem Lett2001; 11:1053–1056.
67.
BirkusGKuttyNHeG-X. Activation of 9-[(R)-2-[[(S)-[[(S)-1-(Isopropoxycarbonyl)ethyl]amino] phenoxyphosphinyl]-methoxy]propyl]adenine (GS-7340) and other tenofovir phosphonoamidate prodrugs by human proteases. Mol Pharmacol2008; 74:92–100.
68.
BirkusGWangRLiuX. Cathepsin A is the major hydrolase catalyzing the intracellular hydrolysis of the antiretroviral nucleotide phosphonoamidate prodrugs GS-7340 and GS-9131. Antimicrob Agents Chemother2007; 51:543–550.
69.
MackmanRLRayASHuiHC. Discovery of GS-9131: Design, synthesis and optimization of amidate prodrugs of the novel nucleoside phosphonate HIV reverse transcriptase (RT) inhibitor GS-9148. Bioorg Med Chem2010; 18:3606–3617.
70.
RayASVelaJEBoojamraCG. Intracellular metabolism of the nucleotide prodrug GS-9131, a potent anti-human immunodeficiency virus agent. Antimicrob Agents Chemother2008; 52:648–654.
71.
KeithKAHitchcockMJLeeWA. Evaluation of nucleoside phosphonates and their analogs and prodrugs for inhibition of orthopoxvirus replication. Antimicrob Agents Chemother2003; 47:2193–2198.
72.
WolfgangGHIShibataRWangJ. GS-9191, a novel topical prodrug of the nucleotide analog PMEG (9-(2-phosphonylmethoxyethyl) guanine), with antiproliferative activity and possible utility in the treatment of HPV lesions. Antimicrob Agents Chemother2009; 53:2777–2784.
73.
JansaPBaszczyňskiODračícnskýM. A novel and efficient one-pot synthesis of symmetrical diamide (bis-amidate) prodrugs of acyclic nucleoside phosphonates and evaluation of their biological activities. Eur J Med Chem2011; 46:3748–3754.
74.
SerpiMKrylovISZakharovaVM. Synthesis of peptidomimetic conjugates of cyclic nucleoside phosphonates. Curr Protoc Nucleic Acid Chem2010; 43:15.4.1–15.4.13.
75.
McKennaCEKashemirovBAErikssonU. Cidofovir peptide conjugates as prodrugs. J Organomet Chem2005; 690:2673–2678.
76.
ErikssonUPetersonLWKashemirovBA. Serine peptide phosphoester prodrugs of cyclic cidofovir: Synthesis, transport, and antiviral activity. Mol Pharm2008; 5:598–609.
77.
ErikssonUHilfingerJMKimJ-S. Synthesis and biological activation of an ethylene glycol-linked amino acid conjugate of cyclic cidofovir. Bioorg Med Chem Lett2007; 17:583–586.
78.
PetersonLWSala-RabanalMKrylovIS. Serine side chain-linked peptidomimetic conjugates of cyclic HPMPC and HPMPA: Synthesis and interaction with hPEPT1. Mol Pharm2010; 7:2349–2361.
79.
PetersonLWKimJ-SKijekP. Synthesis, transport and antiviral activity of Ala-Ser and Val-Ser prodrugs of cidofovir. Bioorg Med Chem Lett2011; 21:4045–4049.
80.
ZakharovaVMSerpiMKrylovIS. Tyrosine-based 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]cytosine and -adenine ((S)-HPMPC and (S)-HPMPA) prodrugs: synthesis, stability, antiviral activity, and in vivo transport studies. J Med Chem2011; 54:5680–5693.
81.
FuXJiangSLiC. Design and synthesis of novel bis(L-amino acid) ester prodrugs of 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA) with improved anti-HBV activity. Bioorg Med Chem Lett2007; 17:465–470.
82.
ErionMDReddyKRBoyerSH. Design, synthesis, and characterization of a series of cytochrome P450 3A-activated prodrugs (HepDirect prodrugs) useful for targeting phosph(on)ate-based drugs to the liver. J Am Chem Soc2004; 126:5154–5163.
83.
ReddyKRMatelichMCUgarkarBG. Pradefovir: A prodrug that targets adefovir to the liver for the treatment of hepatitis B. J Med Chem2008; 51:666–676.
84.
TillmannHL. Pradefovir, a liver-targeted prodrug of adefovir against HBV infection. Curr Opin Investig Drugs2007; 8:682–690.