
Abstract
Hepatocellular carcinoma (HCC) is one of the most common cancers worldwide. The limited treatment options and poor prognosis of HCC patients underscore the importance of developing new therapeutic strategies. Infection with HBV and HCV are the major risk factors for developing HCC. While the precise molecular mechanisms that link HBV and HCV infections to the development and progression of HCC are not entirely understood, increasing evidence indicates that stimulation of angiogenesis by these viruses may contribute to HCC malignancy. In this review, we summarize the progress in understanding the role of HBV and HCV infection in liver and HCC angiogenesis, the mechanisms applied by these viruses to deregulate the angiogenic balance and the potential therapeutic options that come with this understanding.
Introduction
Liver cancer is one of the most common malignancies worldwide, ranking fifth in overall frequency (with over half a million new cases each year) and third in annual mortality [1]. Although a lot of research has been done on hepatocellular carcinoma (HCC), very little has translated from bench to bedside and the prognosis of patients with HCC remains dismal. Even now, the 5 year survival rate of these patients is only 7% in developed countries [2], barely up from the 4% it was in the 1974–1976 period [3].
Chronic infection with HBV and/or HCV is a major risk factor for the development of HCC. It has been estimated that these viruses play a crucial role in the development of well over 80% of all HCCs [4]. Depending on economical and geographical factors, 25 to 73% of HCC is associated with HCV and 12 to 60% with HBV [5]. HBV is the predominant viral factor inducing hepatitis in developing countries while HCV is most prevalent in industrialized countries (for example, Japan and USA). During the last 20 years, several developed countries have shown a sharp rise in deaths attributed to HCC. This rise is largely attributable to the increased number of people chronically infected with HCV due to intravenous drug use or transfusions with infected blood prior to 1992, when HCV screening was implemented [6]. Given this relatively recent peak of chronic hepatitis C patients and the slow progression of the disease, liver cancer deaths due to HCV infection are projected to double by 2022 [7].
Although poorly understood, there are significant similarities and differences in the way both viruses may contribute to liver carcinogenesis. HBV is a DNA virus which integrates in the host genome, and this integration itself can result in chromosomal instability and insertional mutagenesis [8,9] (see Figure 1 for more information on the HBV replication cycle). Viral integration also results in the expression of the viral HBx protein. This protein interferes strongly with cell signalling and transcription, thereby affecting cell cycle and cell growth (reviewed in [10]). Moreover, HBV patients will often develop chronic liver inflammation, resulting from immune responses against infected hepatocytes. This persistent inflammation will give rise to hepatocellular damage and fibrosis, eventually resulting in liver cirrhosis, a major risk factor for HCC [11]. HCV, by contrast, is a positive strand RNA virus which does not integrate into the host genome and which has an exclusively cytoplasmic life cycle (see Figure 2 for more information on the HCV life cycle). As is the case for HBV, chronic immunemediated inflammation is likely to be a driving factor in HCV-associated HCC development, as cell lysis and stimulation of mitosis lead to an accumulation of events needed for malignant transformation of the hepatocytes. However, it is unlikely that inflammation alone is sufficient to cause HCC and there is increasing evidence that several HCV proteins can interact with key regulators of the cell cycle (see HCV section). Furthermore, expression of these proteins may lead directly to HCC, even in the absence of an immune response, inflammation or cirrhosis, as transgenic mice expressing the entire HCV viral polyprotein showed a significantly increased risk of HCC despite exhibiting neither hepatic inflammation nor fibrosis or cirrhosis ([12] and reviewed in [13]).

HBV replication cycle and pro-angiogenic activity

HCV replication cycle and pro-angiogenic activity
As evidenced by the very poor 5 year survival rate, there are no good curative therapies available for HCC patients. Surgical resection is the current mainstay of curative treatment but is only performed on patients who present with a limited tumour and liver disease burden. Furthermore, resection is characterized by a very high (>70%) recurrence rate [14]. HCC is typically refractory to systemic chemotherapy and there is currently no standard treatment for patients with non-resectable HCC [15]. Hence, a better understanding of the molecular pathways of virus-induced HCC should be a prime research target as this understanding is crucial to the development of novel strategies to combat this aggressive cancer. As HCC is a highly vascularized solid tumour, characterized by active neovascularization, cellular factors regulating angiogenesis may present potential targets for these novel therapies. Recently, accumulating data have provided insights into the role of angiogenesis in HCC development and progression (reviewed in [16]), but the effect of HBV and HCV on angiogenesis is poorly characterized. In this review, we will summarize the molecular mechanisms by which HBV and HCV can influence angiogenesis in HCC and pre-HCC stages, and the potential implications for the design of therapeutic strategies.
Angiogenesis
Blood vessels are formed under physiological conditions during embryogenesis, wound healing and corpus luteum formation. Under these conditions angiogenesis is a tightly controlled process, regulated by pro- and anti-angiogenic molecules. However, unregulated angiogenesis may lead to several diseases including rheumatoid arthritis, psoriasis and macular degeneration. Moreover, angiogenesis is indispensable for solid tumour growth and metastasis [17–20].

Tumour-induced angiogenesis
Angiogenesis is a multistep process involving an extensive and complex interplay between cells, extracellular matrix components, soluble factors and cellular receptors (see Figure 3). During carcinogenesis, tumour hypoxia is the main driver of blood vessel formation, resulting in the expression of several proteins that stimulate the angiogenic process. Vascular endothelial growth factor (VEGF) is one of the most potent and best studied angiogenic factors. It activates endothelial cells by binding to specific tyrosine kinase receptors on the cell surface [21]. As a consequence, pericytes detach from the vessel wall and matrix degrading proteases (for example, matrix metalloproteinases [MMPs]) are produced and/or activated. Removal of the basement membrane and extracellular matrix by MMPs allows endothelial cells to migrate and proliferate into the perivascular space [22]. Finally, a new blood vessel is formed that connects with the existing vasculature and is stabilized by pericytes. These different steps are regulated by growth factors (for example, VEGF, angiopoietins [Ang], basic fibroblast growth factor [FGF2], platelet-derived growth factor [PDGF]), chemokines (for example, interleukin 8 [IL-8/CXCL8], stromal cell-derived factor-1 [SDF-1/CXCL12]), cytokines (for example, tumour necrosis factor-a [TNF-a], interleukin-6 [IL-6]), angiogenic enzymes (for example, cyclooxygenase-2 [COX-2], thymidine phosphorylase [TP]), proteases (for example, MMPs, plasminogen activator) and adhesion molecules (for example, inte-grins). While it goes beyond the scope of this paper to provide a comprehensive review on angiogenesis, we can refer the interested reader to several excellent reviews [17–20].
HBV
An estimated 350 to 400 million people worldwide are believed to be chronically infected with HBV, a partially double stranded DNA virus of the Hepadnaviridae. Although the relationship between HCC and persistent HBV infection has been well documented, a comprehensive understanding of the underlying mechanisms leading to HCC is difficult to establish as HBV-associated carcinogenesis can be seen as a multifactorial process that includes both direct and indirect mechanisms that might act synergistically [23]. An increasing number of data indicate that HBV-induced angiogenesis may contribute to HCC progression and malignancy (Figure 1). In vitro, conditioned medium of HBV-infected hepatocytes was shown to induce the formation of tube-like structures, resembling blood vessels, of human umbilical vein endothelial cells [24].
Of the different proteins coded in the HBV genome, the regulatory X protein, HBx has been the focus of much attention because it is strongly implicated in different steps of the hepatocarcinogenic process [25,26] and because the X gene is the most frequently integrated portion of HBV DNA found in hepatocyte chromosomes during the development of HCC [27]. HBx may also contribute to tumourigenesis in HCC through modulation of angiogenesis (Figure 1). After subcutaneous injection of mice with matrigel (a commercially available basement membrane matrix derived from an Engelbreth-Holm-Swarm Mouse Tumor [28]) containing either HBx-expressing hepatoma cells or control hepatoma cells, Lee et al. [29] observed significantly more blood vessels in the matrigel containing HBx transfectants, indicating that HBx alone could elicit an angiogenic response. Moreover, a significant enhancement of VEGF mRNA expression was observed in vivo in the liver of HBx transgenic mice [30]. Since VEGF transcription is regulated by hypoxia-inducible factor (HIF)-1α, which binds to hypoxia-response-elements in the 5′ and 3′ regions of the VEGF promoter, it was hypothesized that HBx-induced upregulation of VEGF could be mediated by HIF-1α.
HIF-1 is a transcription factor, which is composed of an oxygen-sensitive α subunit and a constitutively expressed β subunit. HIF-1 function is primarily regulated by HIF-1α protein stability. Under normoxic conditions, HIF-1α is ubiquitinated through interaction with the von Hippel-Lindau tumour suppressor protein (pVHL) and subsequently degraded by the pro-teasome. In contrast, under hypoxic conditions, such as those present in the tumour microenvironment, the HIF-1α subunit is stable and forms a heterodimer with the β subunit. HBx was found to stimulate angiogenesis directly through both the transcriptional activation and stabilization of HIF-1α [31,32]. Either treatment with a mitogen-activated protein kinase (MAPK) inhibitor PD98059 or coexpression of dominant-negative MAPK mutants could abolish HBx-induced HIF-1α transcription, suggesting that the MAPK signalling pathway plays a critical role in the HBx-induced transcriptional activation of HIF-1α [32]. On the other hand, coim-munoprecipitation studies showed that HBx could also directly interact with the HIF-1α protein [31] thereby inhibiting its interaction with pVHL, resulting in decreased ubiquitination and proteasomal degradation of HIF-1α. Both HBx-HIF-1α interaction and increased HIF-1α stability were mapped to the carboxy terminus of the HBx protein [33]. Upregulation of both VEGF and HIF-1α by HBV might play an important role in the clinical setting since patient studies have shown that VEGF plays a crucial role in angiogenesis of HCC [34] and that increased HIF-1α in HCC samples is positively correlated with vascular invasion [35]. Clinical investigations also revealed that the expression rate of VEGF and the mean microvessel density (MVD), a measure of tumour vascularization, were significantly higher in HBx-positive HCCs than in non-HBV related HCC tissues [36].
Besides VEGF, the HBx protein also promotes increased expression and secretion of angiopoietin-2 (Ang-2) in liver tissue, via the activation of MAPK [37]. Angiopoietins (Ang) are key regulators of vessel stabilization/activation by interacting with Tie2, a tyrosine kinase receptor, which is mainly found on endothelial cells [38]. Ang-1, which is constitutively expressed by many cell types, binds and activates Tie-2 resulting in vessel stabilization and survival [39]. Ang-2, which is upregulated during hypoxia, is an antagonist of Ang-1 that binds Tie-2 without activating the receptor, thereby causing vessel destabilization. The destabilized vessels are prone to regression in the absence of growth factors but are more sensitive to activation by angiogenic factors such as VEGF (reviewed in [40]). Supporting this, in vivo Ang-2 has been shown to stimulate angiogenesis in the presence of VEGF [41]. Thus, the balance between Ang-1 and Ang-2 determines the fate and angiogenic potential of a blood vessel [38]. Since the expression of VEGF and Ang-2 has been found to be positively correlated with microvessel density in HBV-associated HCC [42], it seems likely that HBx-induced stimulation of both angiogenic factors plays an important role in the progression of HBV-associated liver cancer.
HBx may also contribute to HCC angiogenesis by upregulation of several MMPs. MMPs contribute to neovascularization by degrading the basement membrane and other extracellular matrix components, allowing endothelial cells to migrate into the surrounding tissue [43], a critical step in angiogenesis. HBx expression has been shown to upregulate the MMP-2 [44–46], MMP-3 [47], MMP-9 [46,48,49] and MMP-14 [44–46] protein levels and activity. Finally, trans-fection of hepatoma cells with HBx resulted in the upregulation of COX-2 [50], an enzyme that catalyzes the production of prostaglandins with pro-inflammatory and pro-angiogenic activity, suggesting that HBx-induced expression of COX-2 may also play a role in HBV-related angiogenesis in HCC.
Due to the close in vivo proximity of the HBV-infected hepatocytes and endothelial cells in the liver, it seems likely that the presence of an increased amount of secreted angiogenic factors will significantly affect the angiogenic process in HCC.
HCV
Worldwide, more than 170 million people are chronically infected with HCV, a positive strand RNA virus belonging to the Flaviviridae. Each year, 4–5% of these chronic hepatitis C patients develop HCC.
A comparison of microvessel density in liver biopsies of matched patients with chronic hepatitis due to either HBV or HCV showed that angiogenesis was more frequent in HCV-positive patients compared with HBV-infected individuals [51,52]. In vitro, HCV-positive sera could stimulate migration and proliferation of human endothelial cells to a higher degree then HBV-positive sera [51]. Furthermore, a high level of CD34, an endothelial marker reflecting the degree of angiogenesis, in the liver was a risk factor for HCC in patients with HCV-associated chronic liver diseases [53]. Chronic hepatitis C patients were also shown to have significantly higher serum concentrations of the angiogenic proteins placenta growth factor (PlGF) and Ang-2 [54,55] and the number of newly formed blood vessels was directly related to fibrosis stage and inflammatory activity grade in liver biopsies from these patients [56]. Interestingly, high serum levels of VEGF, Ang-2 and soluble Tie2 (sTie2) in chronic hepatitis C patients could be significantly reduced by antiviral combination therapy [57]. Moreover, conditioned medium (CM) collected from HCV-infected Huh7 hepatoma cells proved to be more angiogenic in the chick chorioallantoic membrane (CAM) assay than the CM of uninfected Huh7 cells [58]. Despite this clear link between HCV and angiogenesis (Figure 2), and the importance of angiogenesis in HCV-mediated HCC, very little is known about the molecular mechanisms behind these events.
Nasimuzzaman et al. [58] showed that infection of Huh7 with HCV leads to the stabilization of HIF-1α under normoxic conditions, a finding which could be replicated in Huh7 cells expressing the HCV subgenomic replicon, indicating that HIF-1α stabilization is due to the expression of (a subset of) HCV non-structural genes. However, contrary to the stabilization of HIF-1α by HBV proteins, HIF-1α stabilization by HCV appears to be indirect. By using inhibitors of various cellular kinases, dominant negative mutants and antioxidantia it was shown that oxidative stress, signal transducer and activator of transcription 3 (STAT-3), nuclear factor kappa B (NF-κβ) and both the MAPK kinase (MEK) and phosphatidylin-ositol 3-kinases (PI3-kinases) contribute to HIF-1α stabilization. Given these results, the authors hypothesized that HCV-induced reactive oxygen species (ROS) [59] mimic the hypoxic condition needed for HIF-1α stabilization. HCV-induced ROS were proposed to activate the abovementioned signal transduction pathways which, in turn, stabilize HIF-1α
Although only a limited number of studies are available, HCV proteins, like HBV proteins, have been shown to induce the expression of certain other angiogenic factors (Figure 2). Ang-2 was shown to be upregulated by HCV infection [62]. MMP-2 was upregulated by the HCV core protein [63] or by the binding of the HCV envelope protein E2 to the CD81 receptor present on hepatic stellate cells [64] and MMP-9 expression could be enhanced by HCV core [65]. Finally, COX-2 was overexpressed in hepatocytes expressing either HCV core or NS5A protein [61,65]. Increased expression of COX-2 by the HCV subgenomic replicon was shown to be mediated by HCV-induced oxidative stress [66].
The in vivo importance of these findings is evidenced by clinical studies showing that patients with HCV chronic liver disease (seronegative for HBV) have an increased intrahepatic content of COX-2, MMP-2 and MMP-9 [65] which, along with VEGF and Ang-2, could play an important role in the stimulation of angiogenesis in HCV-associated HCC. Finally, it should be noted that the VEGF receptors VEGFR-1 and VEGFR-2 have been identified on HCC cells and that VEGF can induce the disruption of tight junctions between liver cells [67]. Thus, increased expression of VEGF, as seen in HCV-and HBV-infected livers, may also promote HCC spreading into normal liver parenchyma.
Hepatic inflammation due to virus infection
Although the previous sections mainly focus on the direct effects of hepatic viruses on angiogenesis in HCC, it should be noted that the chronic wound healing typically seen in virus-induced fibrinogenic chronic liver diseases may play a key role in the stimulation of angiogenesis in the inflamed liver and/or HCC tissue. Inflammation is characterized by the expression of a multitude of cytokines and growth factors, many of which posses a pro-angiogenic activity (for example, TNF-a, IL-1, IL-8) [68–70]. Furthermore, VEGF and Ang-2, angiogenic proteins upregulated by HBV and HCV, have also been shown to boost the inflammatory response [71,72]. Although very little data are available on this inflammation-angiogenesis link in relation to HBC/HCV infection, a positive correlation between angiogenesis and inflammatory grade has been observed in chronic hepatitis C patients [51,56].
Current status of anti-angiogenesis therapy for HCC
The concept of treating solid tumours by inhibiting tumour angiogenesis was already proposed by Judah Folkman in 1971 [73]. However, it took until 2004 for the first anti-angiogenic drug (that is, bevacizumab, a humanized antibody to VEGF) to be approved by the FDA for the treatment of metastatic colorectal carcinoma [74]. Currently, several other molecular therapeutics targeting angiogenesis are being used in cancer therapy and over 100 compounds have entered clinical trials [17]. As HCC, regardless of its aetiology, is a highly vascularized tumour, many angiogenesis-targeting drugs are being evaluated for the treatment of patients with advanced HCC (Table 1 and Figure 4) [75].
Anti-angiogenic agents in development for the treatment of HCC

FGFR, fibroblast growth factor receptor; HCC, hepatocellular carcinoma; PDGFR, platelet-derived growth factor receptor; TACE, transarterial chemo embolization; VEGFR, vascular endothelial growth factor receptor.

Structure of anti-angiogenic agents in development for the treatment of hepatocellular carcinoma
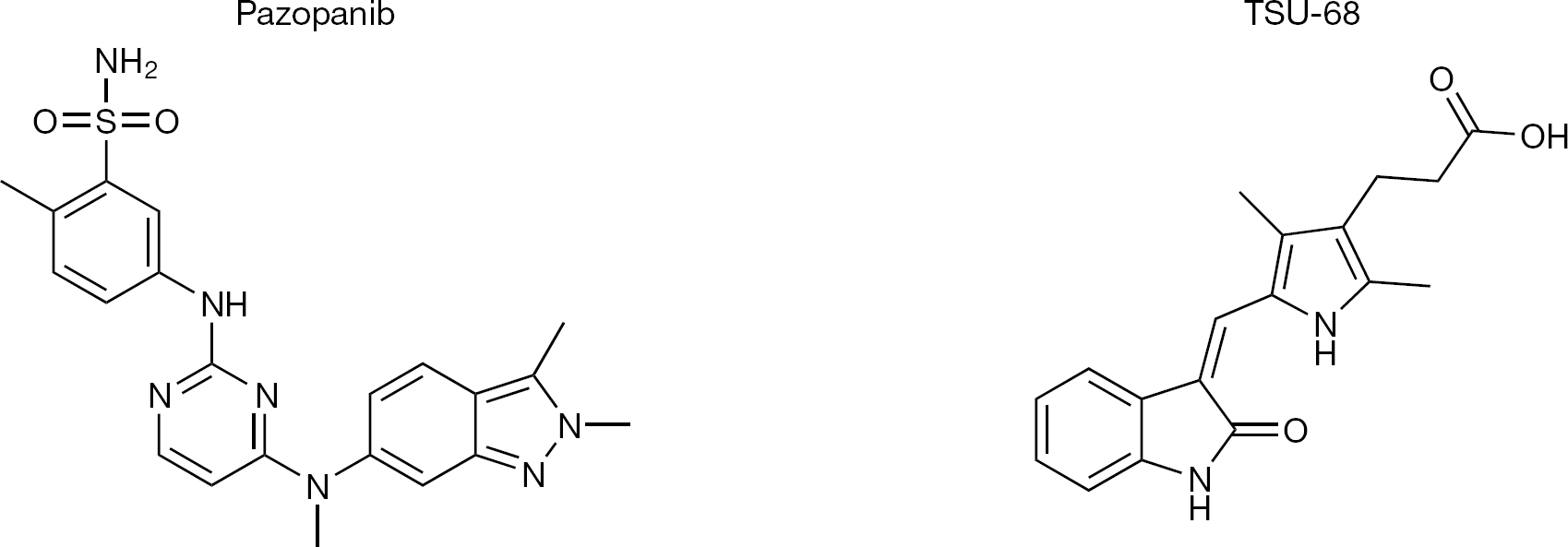
Sorafenib, a small molecule tyrosine kinase inhibitor blocking VEGF receptors among other targets, has been evaluated in two Phase III trials [76,77] and was found to increase the time to progression (TTP) by 1.4–2.7 months and the overall survival (OS) by 2–3 months. These benefits could be observed regardless of the cause or previous treatments of the HCC and have turned sorafenib into the standard first-line treatment option in patients with advanced HCC. The positive data obtained in the sorafenib trials open the door for other anti-angiogenic agents to be tested in clinical settings, either as a replacement, as a second-line option or in combination with sorafenib.
Bevacizumab has shown promising results in two Phase II trials when used as a single agent [78,79]. Both studies showed an increase in progression-free survival and a response rate higher than that seen in the sorafenib trial, suggesting that bevacizumab might be preferred over sorafenib for anti-angiogenic treatment of HCC. However, it should be noted that a clinical trial evaluating the use of a novel chemotherapeutic regimen (gemcitabin and oxaliplatin [GEMOX]) for the treatment of HCC showed very little benefit of adding bevacizumab to this regimen [80].
Sunitinib inhibits multiple kinases, including VEGFR-1 and −2 and theoretically has a stronger anti-angiogenic activity than sorafenib. Sunitinib showed promising results in Phase II trials, but recently presented data on a Phase III trial comparing the use of sunitinib and sorafenib for the treatment of advanced HCC showed sorafenib to be the superior drug as sunitinib did not result in an increase in OS [81]. Therefore it seems unlikely that sunitinib will replace the role of sorafenib as first-line HCC treatment.
Potential anti-angiogenesis treatments for virus-associated HCC
Although anti-VEGF (or VEGF[R]) treatment will likely remain an important part of anti-angiogenic therapy for HCC, a combination therapy tackling angiogenesis from multiple sides currently appears to be the more attractive option since VEGF pathway inhibitors have failed to produce enduring clinical responses in most patients, but instead resulted in transitory improvements followed by resistance [82]. Tumour resistance may be caused by circumvention of the angiogenic blockade by activation and/or upregulation of alternative pro-angiogenic pathways, including TP and FGF2 (reviewed in [82]). Based on data available on HBV/HCV and angiogenesis, some additional treatment options present themselves for virus-associated HCC.
COX-2, upregulated by both HBV and HCV, is highly expressed in HCC [83–85] and may thus be an attractive target. Moreover, high levels of COX-2 in tissue surrounding the liver tumour, as is the case in chronic HBV/ HCV infection, are associated with a decreased disease-free survival time in HCC patients [85]. In vitro, the growth of different HCC cell lines could be suppressed by the COX-2 inhibitor, NS-398. Moreover, a preliminary trial in 15 patients, all of whom had virus-induced HCC showed that addition of the COX-2 inhibitor celecoxib to their therapeutic, palliative regimen (fluorouracil and cyclophosphamide) significantly reduced their serum VEGF and FGF2 levels and resulted in an increased overall survival [86].
Another factor greatly influenced by both hepatic viruses is HIF-1α, which plays a central role in angiogenesis. HIF-1α may be an interesting therapeutic target, even more so given that the disease-free survival time of patients with high HIF-1 a expression is significantly shorter than that of patients with a low HIF-1a expression [87]. Development of HIF-1α inhibitors is ongoing, but only few specific inhibitors have currently been designed. Nonetheless, treatment of subcutaneous hepatoma tumours in BALB/c nude mice with HIF-1α antisense RNA in combination with doxorubicin was more effective in suppressing tumour growth and angiogenesis than doxorubicin alone [88].
Finally, Ang-2 is highly upregulated in HCV- and HBC-associated chronic liver infection, and with several anti-Ang-2 agents currently under development in clinical trials [89] and the preliminary evidence that VEGF and Ang-2 inhibitors can act in a complementary way to reduce tumour growth [90], it may be worthwhile to investigate the effect of Ang-2 inhibitors (alone or combined with VEGF antagonists) on HCC progression.
Conclusions
In this review, we highlighted data showing that hepatic viruses can stimulate the expression of angiogenic proteins, leading to increased blood vessel formation in fibrotic liver and HCC. While some of the reviewed data show a direct link of the virus and/or viral proteins with angiogenesis (for example, VEGF, Ang-2), it is important to note that several reports merely demonstrate an effect of HBV/HCV (viral proteins) on the levels or activity of cellular proteins with known angiogenic activity (for example, MMPs) without investigating the involvement of these factors in virus-induced angiogenesis. As such, it is not clear at this point which of these proteins actually drives angiogenesis in HCC. This further underlines the need for more research into the molecular mechanisms involved in the angiogenic activity of HBV and HCV, as an improvement of our understanding of HCC development and progression may ultimately lead to novel treatment strategies.
Footnotes
Acknowledgements
This work was funded by the ‘Geconcerteerde Onderzoeksacties’ (GOA) of the KU Leuven (GOA 10/014), the fonds voor Wetenschappelijk Onderzoek (FWO) Vlaanderen (G.0486.08) and the Centers of Excellence of the KU Leuven (PF/10/018). JP is a post-doctoral fellow of the FWO.
The authors declare no competing interests.