
Abstract
The development of over 20 antiretroviral drugs has led to efficient and successful suppression of HIV-1 replication. In addition to common viral targets, such as reverse transcriptase and protease, new targets have been recently exploited, including integrase, fusion and cellular CCR5. Hence, combination antiretroviral therapy is continually improved by the development of these new agents, especially for patients infected with drug-resistant HIV-1. In this review, we focused on fusion inhibitory peptides that have been developed since the first HIV-1 fusion inhibitor, enfuvirtide (T-20). T-20, approved for clinical use in 2003, is a polypeptide comprising 36 amino acids derived from the HIV-1 gp41 C-terminal heptad repeat and provides a novel treatment strategy for HIV-1 therapy. T-20 is able to suppress HIV-1 replication, including viruses resistant to reverse transcriptase or protease inhibitors. However, after prolonged T-20-containing treatment regimens, HIV-1 acquires resistance to T-20. Therefore, our laboratory and others have developed novel fusion inhibitors, termed next-generation fusion inhibitors, including electrostatically constrained, mutation introduced, and trimer-form peptides.
Introduction
Enfuvirtide (T-20) was the first fusion inhibitor and comprises a 36-amino-acid polypeptide derived from the C-terminal heptad repeat (C-HR) of the HIV-1 gp41. T-20 targets the HIV-1 gp41 envelope protein and inhibits fusion between the viral and cellular membranes [1,2] – an early, essential step in viral replication. T-20 was approved for clinical use in 2003 by the United States Food and Drug Administration (FDA) for HIV-1-infected adults and later, children with advanced HIV-1 infection [3,4]. The mechanism of this inhibition is believed to involve interference of gp41 folding during virus fusion after attachment [5]. Briefly, after binding of gp120 to cellular receptors, namely CD4, and subsequently chemokine receptors, such as CCR5 or CXCR4, gp41 is activated and exposed to initiate fusion of the virus and cellular lipid membranes (Figure 1A). The fusion peptide domain at the N-terminal region of gp41 comprises a conserved hydrophobic amino acid stretch, such as AVGIGALFLGFLGAA for HIV-1NL4–3 and HIV-1HXB2 strains, and penetrates into the target cell membrane. Two α-helical regions between the anchored fusion domain and transmembrane domain in gp41 interact with each other and lead to the formation of a six-helix bundle (Figure 1C). The formation of the six-helix bundle facilitates the fusion of the virus and cellular membranes. T-20 is believed to interfere with HIV-1 replication by inhibiting the formation of the six-helix bundle (Figure 1E).
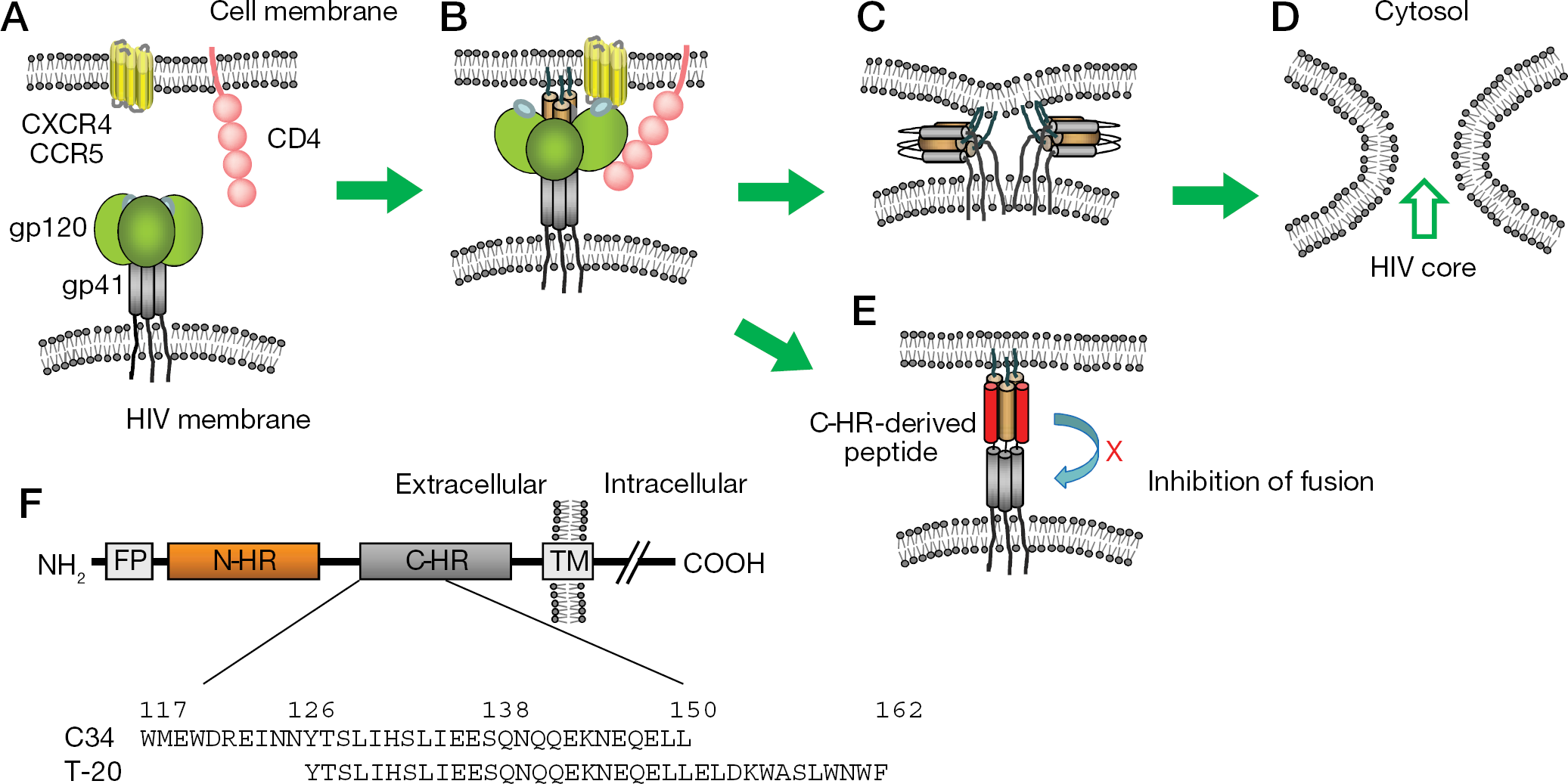
Entry steps of HIV-1 and schematic view of gp41
Reverse transcriptase inhibitors (RTIs) and protease inhibitors (PIs) are often used in combination for HIV-1 therapy, and resistance is a serious setback for efficient HIV-1 therapy. In the same class of inhibitors, apparent and non-apparent cross-resistance is frequently observed, where, even with distinct classes of inhibitors, multidrug resistance has been reported [6,7]. In addition to inhibitors targeting CCR5, such as maraviroc, and integrase, such as raltegravir, the T-20 fusion inhibitor has also been developed as a viable alternative for HIV-1 therapy. Because of its unique mode of action, T-20 suppresses replication of HIV-1 that are resistant to typical anti-HIV-1 drugs, such as RTIs and PIs. After prolonged therapy with T-20, several concerns have unfortunately emerged that include resistance and adverse effects. For the resistance to T-20 to develop, the genetic barrier for the emergence of resistant variants appears to be relatively low, where only one amino acid substitution, such as G36D, V38A and N43D, appears to be sufficient for resistance [8–17]. However, to develop a high level of resistance, at least two substitutions seem to be required [15]. Interestingly, the coding region for gp41 also contains a Rev-responsive element (RRE) – an RNA secondary structure that is important for unspliced RNA export from the nucleus, which is required for efficient viral protein synthesis and packaging of the genomic RNA [18–20]. Therefore, mutations in the gp41 gene also alter the RRE structure, and compensatory mutations that stabilize the RRE structure impaired by the primary mutations are required for most of the primary mutations, leading to moderate genetic restrictions [15,21]. Adverse effects of T-20 include inflammation at the site of injection [22,23]. Moreover, the injection process required for administration carries inherent risks, such as needle sticks and contamination. Therefore, the development of fusion inhibitors with few adverse effects, that nevertheless are effective even for T-20-resistant HIV-1 variants, are required. In this review, we describe new fusion inhibitory peptides and discuss resistance to these molecules in order to design next-generation inhibitors.
Electrostatically constrained peptides
Principle of glutamate-lysine electrostatically constrained peptides
C34 is a C-HR-derived peptide (Figure 2) that overlaps approximately two-thirds of the T-20 sequence and has been demonstrated to inhibit HIV-1 fusion more efficiently than T-20 in vitro [21,24,25]. Previously, our laboratory (Akira Otaka, Shinya Oishi and Nobutaka Fujii, Kyoto University Graduate School of pharmaceutical Science, Kyoto, Japan) remodelled C34 by introducing substitutions with glutamate (E) and lysine (K) at only the solvent-accessible sites of the helical bundle to maintain residues at the interactive site that are crucial for the interaction with the N-terminal heptad repeat (N-HR) [26,27]. In an α-helical heptad repeat, residues separated by three amino acid residues (i versus i+4) are located on the solvent side of the helix, closely positioned in space (Figure 3). Hence, we introduced consecutive EK motifs separated by three residues (E at positions i, i+1 and K at positions i+4, i+5) in the solvent accessible site of C34, resulting in a repeat of the following type: Z-EE-ZZ-KK (Z indicates the original amino acid in HIV-1, which are located in the interactive site). These modifications provided highly soluble and active derivatives [27]. A C34 derivative, SC34EK, which has two complete and three incomplete Z-EE-ZZ-KK motifs (Figure 2), showed enhanced anti-HIV-1 activity compared with the parental C34 and T-20 peptides (approximately 3- and-30-fold, respectively). Another C34 derivative, SC34, which contains a KE motif to maintain the original sequence of C34, also showed comparable activity to SC34EK. Circular dichroism (CD) analysis revealed that the α-helicity of SC34EK and the thermal stability of the N36/SC34EK complex were both enhanced [26,27]. Interestingly, the anti-HIV-1 activity of SC35EK, with five complete Z-EE-ZZ-KK motifs, was comparable to that of SC34EK [27], indicating that five complete repeats of the Z-EE-ZZ-KK motif are not required to enhance anti-HIV-1 activity. It is consistent that the 29-amino-acid SC29EK, which contains four complete Z-EE-ZZ-KK motifs also showed similar activity (see Applications of short peptides). Note that the peptides with introduced EK motifs inhibit the replication of T-20-resistant HIV-1 [27,28]. A similar activity profile was obtained for T-20EK, consisting of T-20 with the introduction of an EK motif [29]. We have previously demonstrated that the electrostatic interactions by the EK motif stabilized a-helical structure and enhanced specific binding affinity, even for the mutated target peptides, provide high selectivity and activity [26].

Amino acid sequences of fusion inhibition peptides and their activities
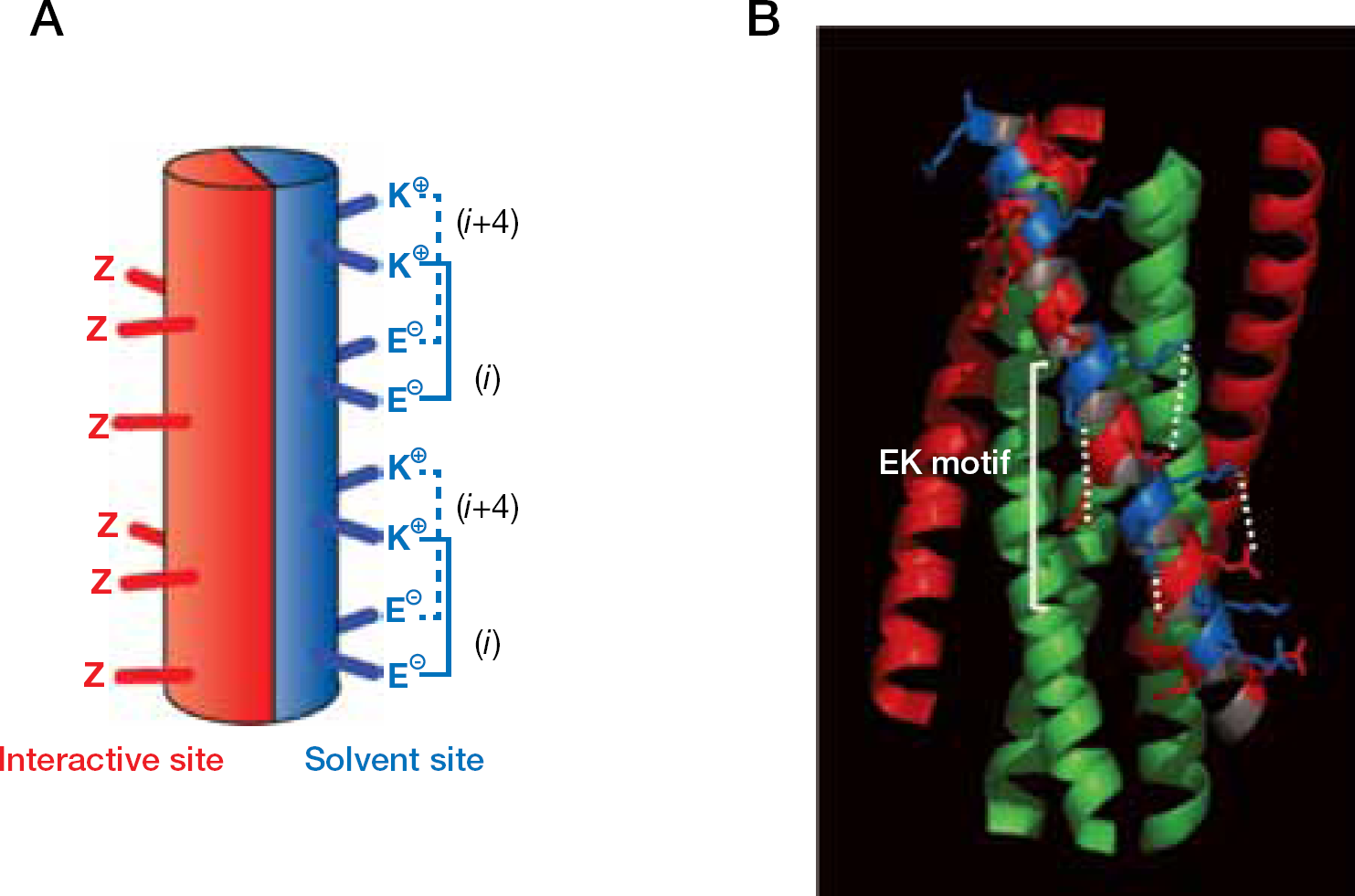
Crystal structure of SC34EK with an N-terminal heptad repeat region-derived peptide, N36
Applications of short peptides
To address how many complete Z-EE-ZZ-KK motifs are required to maintain the potent anti-HIV-1 activity of SC35EK, we synthesized SC29EK and SC22EK (Figure 2) that contained four and three complete repeats of Z-EE-ZZ-KK, respectively. Because W117, W120 and I124 are located in the N terminus of C34 and are crucial for binding to N-HR [24,30], we deleted the C-terminal region of SC35EK to produce short peptides and observed the effect. Four complete Z-EE-ZZ-KK motifs in SC29EK provided sufficient inhibitory activity at a level comparable to SC34EK (Figure 2) [31]. As observed for SC34EK, SC29EK also maintained an inhibitory effect toward T-20-resistant HIV-1. SC22EK, comprising three Z-EE-ZZ-KK motifs, showed reduced anti-HIV-1 activity compared with SC29EK and SC34EK. By contrast, a C-HR-derived original (unmodified) peptide, C29 exhibited a >90-fold reduction in anti-HIV-1 activity. Moreover, C22 exhibited no anti-HIV-1 activity. These results suggested that four Z-EE-ZZ-KK motifs are required to maintain the inhibitory effect on membrane fusion for HIV-1 strains resistant to T-20, as well as wild-type HIV-1. Therefore, the introduction of the EK motif may be key for designing effective short peptides with anti-HIV-1 activity.
Fusion inhibitors with resistant mutations
T-20-resistance-associated mutation (S138A) introduced into T-20
In addition to engineering stabilizing electrostatic interactions, our laboratory has also established another strategy for the design of fusion inhibitors that can suppress the replication of T-20-resistant HIV-1. This strategy is based on the introduction of resistance-associated secondary mutations into the sequence of the original peptide inhibitor. Our studies revealed that primary mutations in the N-HR reduced binding affinity of not only T-20 and C34, but also the physiological or the intramolecular C-HR [21,32]. Therefore, HIV-1 develops the secondary or compensatory mutations, including N126K and S138A, to maintain/restore binding efficiency toward mutated N-HR. Such an effect may contribute to enhance the activity of fusion inhibitors. Indeed, T-20 containing a S138A substitution (T-20S138A), one of the secondary mutations observed in patients that fail to respond to T-20, restored anti-HIV-1 activity against T-20-resistant variants, as well as to wild type [32]. X-ray crystallographic and CD analyses revealed that the S138A substitution contributed to the stability of the N-HR:C-HR complex [32,33].
Another secondary mutation, N126K
As mentioned above, N126K, which is also a secondary mutation for T-20-resistant variants in vivo [8,34,35] and C34-resistant variants in vitro [32] are unfortunately located outside of the T-20 sequence that starts from position 127 of gp41. We have previously demonstrated that C34 with N126K (C34N126K) also maintains strong activity against a C34-resistant clone, HIV-1D36G/I37K/N126K/L204I [21,32]. Taken together, it is possible that the strategy to design peptide inhibitors utilizing resistance mutations has resulted in antivirals that can suppress variants resistant to the parental peptides.
Mutations for SC34EK resistance
Unfortunately, HIV-1 can develop resistance even to the SC34 and SC34EK next-generation fusion inhibitors with over 10 mutations in the gp41 coding region [36]. We examined whether our strategy mentioned above, was also applicable to SC34 and SC34EK mutants. This study first involved the synthesis of C34 peptides containing SC34 and SC34EK resistance mutations that emerged in the C-HR. Anti-HIV-1 activities of C34 with these mutations, which were observed in selection of either SC34 or SC34EK, were also restored, even against SC34EK-resistant HIV-1. These results further validate our strategy to overcome resistance to peptide fusion inhibitors by incorporating resistance mutations into the sequence of the original peptide inhibitor.
Other modified peptides for fusion inhibition
T-1249
T-1249 is a 39-amino-acid polypeptide composed of mixed sequences derived from HIV-1, HIV-2 and simian immunodeficiency virus (SIV; Figure 2) [37]. The peptide sequence was optimized by comparison of these viruses for efficient N-HR binding. T-1249 maintains antiretroviral activity against HIV-1, showing reduced susceptibility to T-20 [14,38]. In a clinical trial of patients previously treated with a T-20-containing regimen and who were treated with T-1249 for 48 weeks, gp41 commonly contained amino acid substitutions, G36D or N43K [38], which have also been observed for T-20 in vitro [10] and in vivo [39–41]. As selected by T-20-containing therapy, the N126K and S138A resistance-associated mutations in the C-HR were also introduced with the primary mutations. Therefore, although T-1249 has an extensively modified amino acid sequence, it only demonstrated a substantial genetic barrier and fitness cost compared with T-20. Consequently, development of T-1249 has been discontinued [42].
Sifuvirtide
Sifuvirtide (SFT; Figure 2) was designed based on the three-dimensional structure of the HIV-1 gp41 fuso-genic core conformation with a single ER motif [43] and showed potent activity against both primary and laboratory-adapted HIV-1 isolates. SFT is also highly effective against T-20-resistant HIV-1 variants. SFT blocked six-helix bundle formation in a dominant-negative manner, indicating that it has a similar mechanism of inhibition to C34 derivatives. In vitro selection of HIV-1 resistant to SFT with a dose-escalating method revealed that mutations were introduced into the gp41 (I37T, V38A/M, Q41R/K and N43K), either singly or in combination [44]. A compensatory mutation, N126K, was again observed. Therefore, mutant viruses acquired cross-resistance to T-20 to various extents. The in vivo efficacy of SFT will soon be revealed since Phase II clinical trials have been recently completed in China.
T-2410 and its derivatives
T-2410 is a C34-based 38-amino-acid polypeptide (Figure 2). Dwyer et al. [45] described the modification of T-2410 with an ER motif comprising glutamic acid (E) and arginine (R) substitutions that generate a single electrostatic interaction. A peptide with a single ER motif in a helix turn, which is also observed in SFT, showed enhanced stability of the six-helix bundle, extended in vivo half-life, and activity against wild-type HIV-1 and also HIV-1 resistant to T-20. In addition to the ER motif, the same study introduced alanine substitutions that theoretically enhance a-helicity of the peptides, even within the interactive site (T-2635 and T-2544), indicating that HIV-1 fusion inhibitors with potent antiviral activity have greater design flexibility (Figure 2). This study could not select resistant HIV for T-2544 after 70 days in culture, suggesting superior durability compared with T-20 [45] but these selection periods seemed to be relatively short and, in the case of SC34EK, selection of resistant variants took approximately 2 years [36]. Most recently, Eggink et al. [46] selected an HIV-1 variant resistant to T-2635 and revealed that multiple mutations within gp41 were required for resistance and this was accompanied with a dramatic loss of gp41 function, suggesting that properties of T-2635 and SC34EK are similar. One of the modifications at position 138 (Figure 2; for example T-2635 and T-2544) was incidentally the identical position of the secondary mutation site, indicating that the S138A substitution was elucidated by the enhancement of α-helicity not only by the compensatory effect (see Fusion inhibitors with resistant mutations) but also theoretically. These peptides maintained activity against T-20- and T-1249-resistant HIV-1, but exhibited little enhancement in activity itself compared with that of T-20.
Trimer of N-HR derived peptides
N-HR-derived peptides generally show only moderate activity compared with C-HR-derived peptides. The N-HR peptides form a well-characterized, trimeric coiled-coil bundle in the presence of C-HR peptides, but there is little physiological structural information on the N-HR trimer without C-HR due to their highly hydrophobic character that makes it difficult to synthesize in solution. Dwyer et al. [47] rationally designed synthetic N-HR peptides in soluble and thermostable N-HR trimers. The most effective peptide, T865AA, comprising a trimer of 51 amino acid residue N-HR derived peptides with alanine substitutions, showed enhanced anti-HIV-1 activity compared with wild-type peptides, with up to 20-fold greater potency. This in vitro study attempted to select resistant variants to another trimeric peptide, N13Δ trimer, but mutations were introduced in the C-HR. Some of these mutations, such as H132Y and E137K, were also observed in resistance to N- and C-HR-derived peptides [36,48,49]. However, activity of T-20 against N36Δ-trimer-resistant viruses was maintained or even improved up to fivefold. The structure of the polypeptide was solved in the 1.5 Å crystal structure of one of the subunits, T865RSV-AA, and showed that the isolated N-HR was very similar in conformation to the N-HR in the six-helix bundle. These results provide an initial and experimental model for the identification of novel N-HR-derived inhibitors.
Two distinct modes of action for fusion inhibitors
Enhancement of α-helicity is highly correlated with activity in almost all peptides when they interact with the N-HR, especially C34 derivatives. Some of C-HR-derived peptides, like T-20, do not interact with corresponding N-HR in vitro. At present, this discrepancy might be elucidated with a putative mechanism that the cellular membrane is essential for T-20 to exert its inhibitory effect against SIV fusion [50,51]. Peisajovich et al. [51] modified the C-terminal tryptophan-rich domain (C-TRD) of SIV-based T-20, which is believed to be important for association of the cell membrane with alanine (GNWF to ANAA). Surprisingly, the modified T-20 (SIV-T-20ANAA) did not show any inhibitory effect; however, SIV-T-20ANAA with C-terminal octylation that enables membrane association restored the activity. Therefore, to exert T-20 activity, its membrane association supports interaction of T-20 with the N-HR at the time of virus fusion. By contrast, in the case of C34 derivatives, these peptides can directly interact with the N-HR through N-TRD without cell membrane association in vitro. Hence, both TRDs located in the N- and C-terminal region of the C-HR are essential to exert their activity for C34 and T-20, respectively. However, the roles of two TRDs in the C-HR appear totally different.
Although T-20 and C34 contain distinct mechanisms of action, the emerging patterns of resistance mutations seem to be similar in derivatives generated between T-20 and C34. Most primary mutations are introduced in the 33–45 amino acid region with varied substitutions appearing, such as N43 to D, K or S; some additional mutations are also introduced at position 50. In contrast to the primary mutations in the N-HR, secondary mutations such as N126K, E137K and S138A, are common. Taken together, some cross-resistance is observed between C34- and T-20-resistant HIV-1 variants. Further studies are required to elucidate the common pattern of resistance despite distinct modes of action.
Conclusions
To date, several candidates for the next generation of fusion inhibitors have been developed and some are currently in clinical trials. To develop further potent inhibitors, C34 seems advantageous as a starting template. However, difficulties remain in the areas of pharmacokinetics and the administration route. Experimental small molecule fusion inhibitors have also been identified but show only a marginal effect [52–54], indicating that further development is needed. As more information becomes available on the use of peptide inhibitors and the development of resistance, the development of effective orally bioavailable small molecule fusion inhibitors may eventually lead to more effective HIV-1 therapies.
Footnotes
Acknowledgements
This work was supported in part by grants from the Promotion of AIDS Research from the Ministry of Health, Labour and Welfare of Japan, and a grant from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
The authors declare no competing interests.