
Abstract
The 24th ICAR meeting was held in Sofia, Bulgaria, 8–11 May 2011. This report summarizes the presentations by the ICAR award winners, Earl Kern and Brian Gowen; the keynote address by Albert (ADME) Osterhaus; the Plenary lectures by Raina Fichorova, Ralf Bartenschlager and Esteban Domingo; the invited speakers for the symposia; and a few of those by contributors.
This report aims to reflect the diversity of topics across different disciplines (chemistry to biology) discussed at ICAR: old viruses (smallpox), emerging viruses (SARS, new strains of influenza and flaviviruses), problematic viruses (HIV and HCV), sporadic viruses (arenaviruses), neglected viruses (enteroviruses), new research targets (for HCV) and new approaches (lethal mutagenesis). There were timely reports on promising compounds against adenoviruses, cytomegalovirus, HCV and HIV in clinical trials. This conference illuminated the constantly evolving field of antiviral chemotherapy by providing a forum to present and discuss new antiviral compounds, new uses for old compounds and exciting clinical results. This ICAR was a fitting testament to the ‘father of antiviral chemotherapy’, Bill Prusoff, who died aged 90 in April 2011.
Introduction
This review provides an overview of the conference highlights. As this is a research conference, any references to clinical results should not be taken as a recommendation for clinical use. I wish to thank all those authors who have kindly provided me with copies of their presentations.
This report does not follow the chronology of the meeting, but starts with the presentations by the recipients of the society's two major awards. These are followed by the keynote address, plenary lectures, two mini-symposia, the satellite clinical symposium and contributed presentations. A summary of this report is included within ISAR News found in this issue.
Gertrude Elion Memorial Award lecture: Why develop a drug for smallpox, a disease that has been eradicated?
Earl Kern (University of Alabama School of Medicine, Birmingham, AL, USA)
Variola virus is the agent of smallpox, which was successfully eradicated worldwide in 1977. Research on this virus is restricted to the CDC in USA and Novosibirsk in Russia, although several related viruses are used as surrogates. Monkeypox virus is indigenous in Africa with rodents being the natural reservoir. Cowpox has rodents as the main carrier in Europe. Camelpox virus has been used as a model virus in research; camels are the only known host. Vaccinia virus is used as the vaccine to provide protection against all these orthopoxviruses.
Smallpox (variola virus) is a large DNA virus that replicates in the cell cytoplasm and causes characteristic skin lesions. As it spreads from person to person by aerosol, and as predicted mortality rates of 30% to 40% have been reported, it has been considered as a potential agent of bioterrorism. A small aerosol release could start an epidemic by initially infecting 50–100 people. Although monkeypox disease is less contagious and less lethal than smallpox, it is also considered to be a potential bioterror agent. Although vaccination for variola used to be routine, there are problems, primarily in an immunodeficient vaccinee. One example was an accidental transfer of virus from a recently vaccinated individual to a susceptible host, a 2-year-old child with eczema. The life-threatening illness eventually resolved after treatment with vaccinia immune globulin, cidofovir and then ST-246.
There is a clear need for an antiviral agent to treat endemic monkeypox virus in Africa, to treat the zoonotic spread of cowpox in Europe and also to combat vaccination complications. However, the main driving force to develop antiviral therapies has been to protect against bioterrorism. The objective of the NIH proposal for biodefence was to develop two antiviral agents against orthopoxviruses, preferably the two agents having different mechanisms of action. Any potential agent should be active against variola, monkeypox and vaccinia viruses, it should be orally bioavailable, have a good safety profile, have long-term stability under various storage conditions and be cheap enough to stockpile in large amounts. Potential drug candidates were cidofovir (or its orally active analogue, CMX001) and ST-246. These two agents fulfill one of the objective criteria, having differing modes of action. CMX001 inhibits viral DNA replication, whereas ST-246 interferes with the envelopment and exit of mature virions. As it is not possible to do clinical studies with smallpox and as such studies with monkeypox would be logistically very difficult, the FDA have proposed the ‘animal rule’ whereby antiviral data in animals and safety data in healthy human subjects could be used for drug approval.
Both CMX001 and ST-246 showed good selective activity against all the orthopoxviruses in cell cultures and in a variety of animal models. As a bonus, the combination of these two compounds is synergistic in cell culture (vaccinia and cowpox) and in a mouse model (cowpox). ST-246 was well tolerated in Phase I clinical studies and is being added to the USA Strategic National Stockpile. CMX001, which has activity against various DNA viruses including the othopoxviruses, was also well tolerated in Phase I trials and is in Phase II trials against adenovirus, cytomegalovirus and BK virus infections. All these studies indicate that the NIH objective has been met successfully.
William Prusoff Young Investigator Award lecture: Development of countermeasures against pathogenic arenaviruses
Brian Gowen (Utah State University, Logan, UT, USA)
Arenaviruses form enveloped virions with a sandy appearance due to the packaging of host ribosomes, hence their name from the Latin for sand (arena). The genome is a bisegmented ssRNA (L and S), each encoding two proteins, viral polymerase (L), matrix protein (Z), nucleoprotein (N) and glycoprotein (GP), which is cleaved to form GP1 and GP2. L-mRNA and N-mRNA are transcribed from the vRNA genome of the L and S segments, respectively, whereas the Z-mRNA and GPC-mRNA are transcribed from the corresponding vcRNA antigenome.
For arenaviruses, the natural life cycle is a chronic infection in rodents with virus in excreta passing to humans. Limited person-to-person spread can occur through contact with body fluids. The acute human disease usually starts as a non-specific febrile illness. Later are seen the typical signs of haemorrhagic fever, profound inflammatory response, impaired coagulation and increased capillary permeability. Depending on the virus, the fatality rate is from 15% to 35%. Fortunately, outbreaks are uncommon but can cause serious disease in South America and Africa. Currently available therapies are limited. Ribavirin is used to treat Lassa fever and an attenuated virus vaccine (Candid #1) is being used in Argentina.
Research on highly pathogenic arenaviruses must be performed in BSL-3 or −4 containment but Pichinde virus (PICV) infections of hamsters and guinea pigs and Tacaribe virus infection of AG129 strain mice may be studied in BSL-2, making them convenient models for research.
In the hamster PICV model, interferon (IFN; Alfacon-1) gave good protection when treatment was started 4 h pre-infection and some protection when starting 24 h post-infection (versus placebo with usually no survivors). The combination of IFN and ribavirin was markedly more effective than either agent alone. Adenovirus-vectored IFN (DEF201) has potential advantages over administration of IFN. Following a single intranasal dose, steady expression of fully glycosylated IFN can be achieved, thereby avoiding daily injections and the bolus effect. With treatment starting 24 h pre-infection, there was a clear dose response with 1×108 PFU giving 100% survival. The same dose could be given 7 days prior to infection with retention of efficacy. Even with 14-day pre-treatment dose, there was significant, albeit reduced, protection. Dosing at 6, 24 and 48 h post-infection gave 100%, 90% and 50% survival, respectively. The presenter and others have completed efficacy studies with 10 viruses representing 7 different virus families. These studies have stimulated the start of IND-enabling safety studies with NIH support.
T-705 (favipiravir) is being progressed as an influenza therapy. In Japan, clinical trials are completed and NDA submitted. In the USA, Phase II trials are progressing. Its mechanism of action is via phosphorylation to the ribofuranosyl triphosphate which is a potent inhibitor of influenza viral polymerase. T-705 has been shown to be effective in various arenavirus cell culture assays. So far, all the data are consistent that T-705 acts against arenaviruses by targeting the viral polymerase. It is hoped that direct evidence will come from studies planned at Harvard, where T-705 R-TP will be tested in a Machupo arenavirus polymerase assay.
In the hamster PICV model, T-705 gave 100% protection when treatment was initiated 5 days after challenge. Even with treatment delayed to 6 days after infection, when the disease is well established, T-705 was shown to be active. Starting with a loading dose of 320 mg/kg twice in first day, then 100 mg/kg daily for 7 days, oral T-705 gave a 50% survival (versus 0% with placebo; P<0.001). In the treated group, the deaths were delayed, starting at approximately day 15; is it possible that a longer course of treatment may have prevented these deaths? When the same 8 day course of treatment was started on day 7 after infection, the deaths (60%) occurred as with placebo, but the remaining animals survived. The decreased efficacy when starting treatment on day 7 may be explained by pharmacokinetic studies in uninfected and infected hamsters. On day 7 relative to time of infection, the time (Tmax) of peak plasma concentrations of T-705 was much delayed in infected hamsters (from a few min to approximately 1 h) and the plasma concentrations were generally also reduced.
In the PICV guinea pig model, the disease can be followed by a rise in temperature from approximately day 2 to 10 and by loss of body weight from day 5 to 10. Measurement of virus titre in serum and various tissues showed the virus was widely distributed by day 7 when twice-daily treatment with T-705 was started and continued to day 20, with the study concluding on day 35. By various parameters, the higher dose of T-705 (300 mg/kg daily) gave excellent protection, with a beneficial yet reduced effect with the lower dose (150 mg/kg daily). The high dose gave 100% survival (versus 0% for placebo), a faster return to normal weight gain and a quicker normalization of temperature.
Although the activity of T-705 in both the hamster and guinea pig models were impressive, perhaps the most important information was given almost as an aside. So far, attempts to develop virus resistant to T-705 have been unsuccessful. With T-705 expected to be widely available for influenza therapy, the prospects for controlling the uncommon but serious arenavirus outbreaks have never looked more hopeful.
Keynote address: Emerging virus infections and intervention strategies
Albert (ADME) Osterhaus (Erasmus Medical Center, Rotterdam, the Netherlands)
In 1900, most deaths were due to infectious diseases. Now, virus infections, particularly newly emerging viruses, are still a major challenge. The worldwide effect of HIV is well known, with approximately 55 million people having been infected, there being more than 20 million deaths so far and at least a further 2 million deaths each year. Although animals are the source of most emerging viruses, newly identified viruses may already be well established in humans. For example, human metapneumonia virus (hMPV), was discovered in 2001. It was found in approximately 10% of young children with respiratory tract infections and in some fatal cases in immunocompromised patients. Then, it was shown that essentially everyone worldwide over 5 years old has been infected with hMPV.
Although smallpox has been eradicated, other animal poxviruses pose a threat, particularly monkeypox. Vaccine, given 24 h after infection, was not effective in a non-human primate model of monkeypox, but an antiviral compound, cidofovir, was protective (see Gertrude Elion Memorial Award lecture above). Measles infections have been vastly reduced by vaccination and there is the prospect of measles eradication. However, as there are other closely related animal morbilliviruses that could ‘jump’ to humans, it may be prudent to continue measles vaccinations indefinitely.
West Nile virus (WNV) first appeared in the USA in 1999 but by 2005 it had spread through the whole of the USA. By contrast, Europe has had only a few reported cases (2010). There are three subtypes of tick-borne encephalitis, each with well defined geographical borders. Chikungunya virus was detected in Italy in 2007. This virus seems to be expanding its range by adaptation to a different vector mosquito, although recent outbreaks have remained restricted to Italy. These geographical variations are some examples which we do not understand. A project, EMPERIE, has been set up to collect many samples for monitoring virus infections.
New technology has played an important role in managing new outbreaks of emerging viruses. For example, within 1 week of a sample of the severe acute respiratory syndrome (SARS) virus being received by the presenter's laboratory, the whole virus had been sequenced. Within 4 weeks, there was a declaration that a coronavirus, SARS-CoV, was the cause of the new outbreak. In contrast to influenza, virus was not excreted until approximately 3 days after symptoms, allowing patient isolation to be used effectively. IFN-α can be used both prophylactically and post-exposure. Candidate vaccines were developed but, fortunately, they were not needed. All the information from this cooperative research was put into an information pool and patented jointly.
Within the past 100 years, there have been four influenza A pandemics: 1918, H1N1 ‘Spanish flu’; 1957, H2N2 ‘Asian flu’; 1968, H3N2 ‘Hong Kong flu’; and 2009, H1N1 ‘Swine flu’. As influenza A is primarily an avian virus, 200,000 samples of bird feces have been tested. Recently, a new subtype, H16, was detected in black-headed gulls. New pathogenic influenza strains usually arise from reassortment of the eight RNA segments of the virus, often in chickens and pigs. There have been several small outbreaks recently: 1997, H5N1 in Hong Kong; 2003, H7N7 in the Netherlands; and 2003 to 2011, H5N1 in various sites with >500 hospitalized patients and >300 deaths. The antiviral drugs, oseltamivir and zanamivir have helped to control these outbreaks but resistance to both has been recognized. The newer adjuvanted vaccines appear to be effective up to 97% in those aged 15 to 65 years and 83% in those >65 years. Generally, data indicates that these vaccines are safe. One possible problem is still under investigation. There have been 81 reports of an association between vaccination and narcolepsy in Scandinavia but this association has not been reported in Canada, where the same vaccine has been used.
In conclusion, most emerging human viruses appear to originate from the animal world but new molecular techniques allow quicker and better identification of any new viruses.
Plenary lecture: vaginal microbicides
Raina Fichorova (Brigham & Woman's Hospital, Boston, MA, USA)
The HIV/AIDS pandemic has been continuing for 30 years, but a welcome decrease (approximately 15%) in new infections over the decade 1999 to 2009 may indicate that the pandemic has peaked. However, approximately 33 million people are living with HIV; of these, >20 million are in Africa. Preventing spread of HIV by using vaginal microbicides is seen as an important target. After several earlier failures, the first clinical trial to give a clinically useful reduction (39%) in HIV incidence was using 1% tenofovir gel (CAPRISA 004 trial) in South African women. Now it is time for a ‘change of tune’, to include more research, using a combination of approaches and adaptive clinical trial design.
Greater than 80% of HIV transmission is through mucosal epithelium. With hindsight, it is obvious that potential compounds should have been tested for their effect on HIV infectivity in epithelial cells cultures. Even 60 h after treating cells with nonoxynol-9, the epithelial cells were more able to be infected by HIV. Such research could have given a warning before undertaking the clinical trial, which showed that nonoxynol-9 was certainly not effective and may have been detrimental.
There are many gaps in our current knowledge; how is HIV infectivity altered by genetic variations, sexual practices, vaginal microbial flora, age and nutrition, for example? Cytokines are attractive as potential biomarkers because assays are easy, non-invasive and quantitative. A sign, reported to hang in Albert Einstein's office, warns of the difficulty: ‘Not everything that counts can be counted, not everything that can be counted counts’. Cytokine data so far have indicated that there are many baseline variations due to hormonal status, abstinence (lower cytokine levels) and microbial flora.
A highly effective vaginal microbicide remains an attractive target but many challenges lie ahead.
Plenary lecture: new insights into the hepatitis C virus (HCV) replication cycle and impact on known and novel drug targets
Ralf Bartenschlager (University of Heidelberg, Heidelberg, Germany)
With 7 genotypes, >100 subtypes and each existing as quasispecies, HCV presents a daunting challenge to any antiviral drug therapy. Encouragingly, several antiviral drugs are progressing through clinical trials. The first HCV protease inhibitor (PI) was voted by 18:1 for approval by the FDA 2 weeks (end April 2011) before this ICAR meeting. It was approved by the FDA 2 weeks after the meeting, on 23 May 2011.
Some potential novel targets include entry inhibitors, including erlotinib (a tyrosine kinase inhibitor already approved for treating cancer), host micro RNA (miR-122) and host protein, which binds to the HCV RNA between domains 1 and 2, thereby reducing HCV replication and HCV NS4A protease that degrades TLR3 and RIG-1 host proteins. Part of the mode of action of the current NS3/4A PIs, being evaluated in clinical trials, may be blocking the RIG-1 pathway and preventing HCV blocking IFN production. HCV NS4B is the main inducer of a membraneous web that forms approximately 48 h after HCV infection. Between 16 and 24 h after infection, there is rapid formation of double membranes, which then form a web-like structure in the cytoplasm of the infected cell. This web seems to be essential for HCV replication. HCV NS5A has been shown to bind to PI4 kinase IIIα, leading to activation of the kinase. The next steps are unknown but HCV replication is much reduced. NS5A has no known enzymatic function but domain 1 binds viral RNA and domain 3 is unfolded and essential for viral assembly. Possibly, the degree of phosphorylation guides its role towards RNA replication or of viral assembly. BMS 790052 was the first NS5A inhibitor to be evaluated in clinical trials, which validated NS5A as a target. Just one molecule of BMS 790052 inhibits approximately 10 to 100 NS5A molecules, possibly by blocking NS5A polymerization. NS5A needs host factor CypA; therefore, cyclosporine analogues are being evaluated in clinical trials. HCV NS5B is the viral RNA-dependent RNA polymerase. Several inhibitors are in clinical trials. HCV NS2 seems to be a central organizer of viral assembly.
In conclusion, in contrast with various other viruses, HCV has no latency, no integration, no covalently-closed circular DNA and resistant genomes are not archived. Therefore, therapy leading to a sustained virological response (SVR) has the potential for a complete cure. In clinical trials, combining an antiviral compound with standard of care, a SVR may be possible even when pre-existing resistant variants are present. Also, patients, who have not responded to standard of care, can have good SVR when an antiviral is added to their therapy.
Plenary lecture: Molecular mechanisms of viral resistance to nucleotide analogues and implications for lethal mutagenesis strategies
Esteban Domingo (Universidad Autonoma de Madrid, Madrid, Spain)
Several viruses seemed to have evolved with polymerases that do not copy the genetic information perfectly, and this has enabled these viruses to respond quickly to new external pressures on their replication, such as antiviral therapy. If the copying fidelity was to become too low, then the newly synthesized viral DNA or RNA would be almost random and no viable progeny would be formed. Therefore, for those viruses which already exist as quasispecies, this opens the possibility that a mutagenic agent would push the replication fidelity too low and so act as an antiviral agent.
A known mutagen, 5-fluorouracil was tried first, but it was then shown that ribavirin was mutagenic for viruses, first for polio and then for other viruses. With lymphocytic choriomeningitis virus (LCMV), ribavirin increased the mutation rate from 4.8×10–4 to 1.1×10–3.
Resistance of foot and mouth virus to ribavirin was investigated by passaging the virus with increasing drug concentrations, from 100 μM to 5,000 μM. Mutations first appeared in the polymerase, initially with an M296I transition, then M296I with P44S and next the triple mutant with P169S. These positions are usually conserved. There seem to be at least two mechanisms of virus resistance to ribavirin. The first two mutations seemed to reduce the ribavirin incorporation, the third restored the fidelity of the polymerase to approximately that prior to adding ribavirin.
With combination therapy, it is usually important to avoid sequential treatment with the drugs, but this is not necessarily so when one of the drugs is a mutagen. Computer modelling showed that there are some circumstances when combination therapy is better than sequential therapy but usually it is better to reduce the viral load with the inhibitor of virus replication and then sequentially treat with the mutagen.
Mini-symposium: Emerging diseases and antiviral therapy
Is there a role for antiviral therapy for the control of Japanese encephalitis? Ernest Gould (Université de la Mediterrraneé, Marseille, France) first considered the alternatives. Mosquito (vector) control is not feasible. A live attenuated vaccine is available in Asia but not FDA approved. A US version of the vaccine is in clinical trials. Although effective, these vaccines are not without side effects, need to be given in high dose and repeated each year. Also, there are four genotypes of the virus, and escape mutants are a potential threat. Therefore, there is a need for antivirals. As yet, none are available but the most promising in mouse models, so far, are an antisense RNA analogue and monoclonal antibodies. A candidate monoclonal antibody binds to the viral prM protein in an immature form, which then keeps the virus in a non-infectious state.
Onder Ergonul (Marmara University, Istanbul, Turkey) summarized the present situation with Crimean-Congo haemorrhagic fever (CCHF). The vector, a tick, is found only south of the 15th parallel and not in the American continent. Within Europe, CCHF is most common in Turkey, with >1,000 cases per year. There is a variation in fatality rates, which seems to be associated with the patients' ability to generate antibodies. Ribavirin efficacy is still debatable. Case studies have indicated that ribavirin is ineffective if treatment starts when the gastrointestinal tract is already bleeding. When given early, the data are not conclusive, although meta-analyses that focus on early ribavirin use seem to confirm its activity. Thus, controlled clinical trials are now seen as being unethical.
Although approximately 70% of infection can be related to tick bites, human-to-human transmission is possible and is an issue with healthcare workers. In high-risk situations, ribavirin should be used but in Turkey this has not always been the practice, leading to some deaths.
Bruno Canard (Université de la Mediterrranée, Marseille, France) considered the potential of RNA capping as a target for antivirals. (+)RNA viruses need to have their genomic RNA capped both to prevent the RNA from being attacked by 5′ to 3′ nucleases and to allow the viral RNA to bind to host ribosomes. Conceptually, there are three ways to get a cap: by entry of the viral RNA into the nucleus making use of the host capping process, by stealing a cap or by making a cap using viral enzymes. Viruses have adopted all three approaches. The NS5 protein of flaviviruses has a methyltransferase domain. This is able to catalyse two types of methylation, 7 Me on guanine and 2′ OMe on adenine. Within this domain, there is an essential pocket close to the active site that is highly conserved among flaviviruses but not present in eukaryotic enzymes.
The SARS episode stimulated a huge increase in our knowledge. For example, the NSp16 viral protein has apparently the correct sequence to be a capping enzyme but it has no activity. However, NSp10 binds to NSp16 to form a complex that is active. Curiously, a mutation in NSp10 (Y96F) promotes tighter binding of the complex and increased activity. So one wonders why the virus has not evolved with this change.
The use of filovirus-virus like particles (VLPs) was described by Stephen Becker (Philipps-Universität, Marburg, Germany). VLPs offer a safe alternative to working with viruses such as Marburg and Ebola. A mini-genome and several (approximately six) viral proteins can self-assemble to make VLPs. These can infect cells and allow the investigation of viral transcription. Ebola VP30, a part of the nucleocapsid, has been found to have a key role in viral transcription. The degree of phosphorylation is crucial to this role. Both fully phosphorylated and non-phosphorylated VP30 are inactive for allowing transcription from the first initiation site on the viral RNA but partially phosphorylated VP30 binds to the RNA and promotes transcription. Removal of phosphorus then allows VP30 to be released from the RNA, re-phosphorylation enables it to bind to the second site on the viral RNA and the second transcription can start. Phosphorylation of the VP30 serine 29 seems to be important. This VLP system is now being used for screening for potential antivirals.
The challenges posed by emerging viruses were summarized by Mike Bray (National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD, USA). Emerging viruses, which are able to spread from person to person, are a continuing potential public health threat in Africa and other ‘third world’ regions, whereas in the USA, they are seen as a biodefence threat. A lesser risk is from the zoonotic viruses for which humans are a dead-end host. Such infections are limited by the range of the host animal. For example, a vaccine against Argentine haemorrhagic fever has been effective because farm workers were easily identified as being ‘at risk’. Clearly, antivirals are needed for post-exposure therapy but maybe the only consensus is that antivirals should be discovered to treat researchers! Evaluating potential antivirals is difficult. Most models use mice – these often do not resemble the human disease. More realistic models need strict bio-containment, a serious bottleneck. Even with a widespread virus, such as WNV in the USA, it proved impossible to recruit more than a few patients in a trial to test an immunoglobulin. Ebola virus presents a much more difficult challenge. First discovered in 1976, there have been <3,000 cases identified to date, with only six outbreaks having >100 cases.
Two approaches seem to offer some promise. Sometimes, there are drugs being developed for one virus but they have useful activity against emerging viruses. For example, T-705 for influenza is also active against bunya and arenaviruses; CMX001 for herpes and adenoviruses is active against orthopoxviruses. The second approach is to educate people to lessen contact with the animal host. For example, simple messages to lessen the risk of Ebola infection include the following: ‘do not eat dead chimpanzees’ and ‘stay away from caves with bats’. To reduce the risk of catching Bolivian haemorrhagic fever from mice, buy a cat!
Mini-symposium: Medicinal chemistry and drug discovery
The cytidine nucleoside analogue, RG7128, and the uridine nucleotide analogue, PSI-7977 (Figure 1), are both in Phase IIb clinical trials for HCV therapy. Michael Sofia (Pharmasset Inc., Princeton, NJ, USA) said that a major long-term aim was to develop an antiviral combination that was effective without IFN being part of the therapy. A new compound would have to be potent, not just in cell culture, but also in patients. To obtain an SVR in various clinical trials, the time taken has fallen progressively from 48 weeks to 12 weeks. It would need to be active against all serotypes of HCV and against the S282T mutant, which was resistant to the previous pyrimidine nucleoside drugs in the whole cell replicon assay.
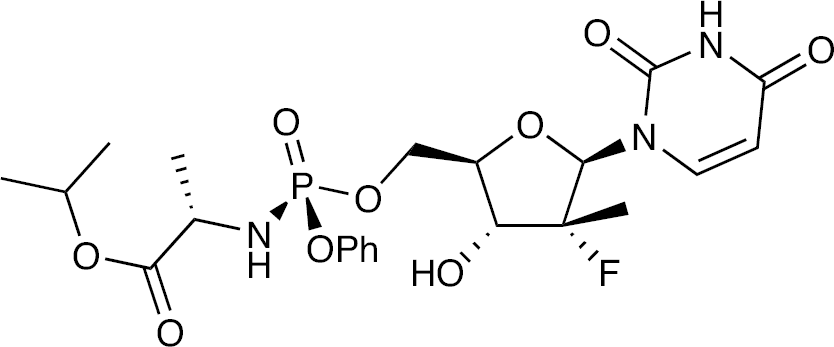
Structure of PSI-7977
RG7128 is a prodrug of PSI-6130. Making >100 modifications to PSI-6130 led to two guanosine analogues with modest activity, but the common triphosphate was a good inhibitor of HCV polymerase. A series of phosphoramidate and cyclic phosphate guanosine nucleotide prodrug analogues was synthesized and these compounds were shown to be equipotent against both the wild type and S282T mutant replicon. The diastereoisomers of the phosphoramidate derivative were separated and the more potent isomer, PSI-353661, gave higher levels of the diphosphate, this active form being a triphosphate analogue, than the other isomer. The cyclic phosphate prodrug version, PSI-352938 (Figure 2), produced exceptional levels of diphosphate (triphosphate analogue) in the liver when administered orally to rats and dogs. The latter compound, PSI-352938, was selected for development as it retained activity against the resistant mutant, S282T, and its metabolic pathway was complementary to that for PSI-7977.
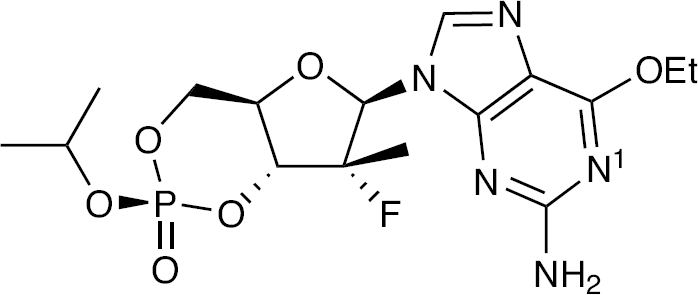
Structure of PSI-352938
In a Phase I trial, PSI-352938 (now called 938) was tested from 25 mg to 800 mg. Pharmacokinetic values were linear over this range. In a Phase II trial, 938 was dosed at 100 mg and 300 mg, each once daily for 7 days. HCV viral load decreased by approximately 4 log10 in both dosage groups. This was the best result seen so far with a nucleoside/nucleotide HCV antiviral and was unexpected, as 938 had not been the most potent compound in the HCV replicon assay. In a combination trial, PSI-7977 and 938 were dosed for 14 days either as monotherapies or as a combination. All groups gave a good reduction in HCV load (approximately 5 log10) but the viral load dropped slightly more rapidly with the combination therapy, the best seen so far in HCV clinical trials. As the combination of PSI-7977 and 938 is able to clear HCV in cell culture systems and shows promising preliminary human clinical results, it is hoped that further trials will show that it will result in SVRs in patients without using IFN.
The next presentation, by Rolf Hilgenfeld (University of Luebeck, Luebeck, Germany), was a complete change of topic: structure- and fragment-based discovery of antivirals against emerging and neglected RNA viruses. There are no effective therapies for enteroviruses, but these viruses can cause serious clinical diseases. The 3C protease of enterovirus 68 (EV68 3Cpro) was chosen as a prototype target.
Adapting Lipinski's rule of five to fragments, one gets a rule of three: ≤300 Da, ≤3 hydrogen bond donors and ≤3 hydrogen bond acceptors. Using a computer programme, find fragments that should bind to the substrate and join these fragments together to make one molecule (in silico). In practice, it is easier to look for an existing compound with similarities to the identified fragments. Following this approach, one compound, SG 74 was found by X-ray crystallography and NMR to bind to EV68 3Cpro but it was not an inhibitor. SG 82 was bound and an inhibitor, but toxic. SG 85 was an inhibitor and non-toxic. X-ray crystal structure has been obtained. SARS coronavirus has a protease that forms a dimer. The X-ray crystallography (at 1.65 Å resolution) confirmed that SG 85 does bind. In various assays with seven viruses, SG 85 was active (EC50≤5 μM).
The discovery of tegobuvir was presented by Gerhard Purstinger (Universitat Innsbruck, Innsbruck, Austria). This story started when a compound, being evaluated for HIV, was tested against bovine diarrhoea virus (BVDV) which was used as a model virus for HCV when no HCV models were available. SAR studies eventually led to GDJN-135 which was active in the HCV 1b replicon system (EC50 0.16 μM) and had a CC50 of 81 μM in a cytotoxicity assay.
At this time, Gilead Sciences Inc. joined the project. Mode of action studies showed that resistant mutants had changes in the HCV NS5B (polymerase). There was cross resistance with other GDJN compounds but not other known polymerase inhibitors. Further SAR studies led to GS 9190, tegobuvir. The synthesis of tegobuvir was shown. In a 4-week Phase IIa trial, the combination of tegobuvir and GS 9256 (PI) was compared with these two compounds combined with ribavirin, and this triple combination combined with IFN. The two compounds gave good initial reduction of HCV RNA which rebounded, the three compounds group had less rebound but the group with IFN added had 14/14 patients with HCV RNA levels <25 IU/ml. Side effects were those seen with ribavirin and IFN. In a Phase IIb trial, tegobuvir, which inhibits HCV polymerase, but not in the usual manner, is being combined with a PI, either GS 9256 or GS 9451.
When a compound has good activity in the HCV1b replicon assay but no activity in the HCV1a assay, the compound is likely to be an NS5A inhibitor. Leading up to BMS 790052, there is a great SAR story starting with a compound inactive against HCV1a and ending with BMS 790052, the first NS5A inhibitor, equally active against types 1a and 1b. John Link (Gilead Sciences Inc., Foster City, CA, USA) described how this work stimulated activity at Gilead Sciences, Inc. Starting with BMS 790052, SAR studies were described, using HCV1a as a screen and monitoring pharmacokinetic properties. This led, eventually, to GS 5885, with EC50=34 pM and 4 pM in type 1a and 1b, respectively. Taking into account the protein binding, the EC50 values are approximately 10-fold lower. The bioavailabilities (species) were 32% (rat), 53% (dog) and 41% (monkey). The serum half-lives were 4 h (rat), 7 h (dog) and 10 h (monkey).
In Phase I studies, once-daily dosing gave trough levels only approximately twofold lower than the peak levels. Over the dosing range from 1 mg to 90 mg, the pharmacokinetic parameters were near linear. In Phase II with 3 days dosing in HCV patients, the 1 mg dose gave a 2.5 log10 reduction in viral load, 3 mg gave a near optimum response and doses of 10 mg, 30 mg and 90 mg seemed to give similar responses with >2.5 log10 reduction in viral load after just the first dose. However, just as for the BMS drug, there were some partial responders linked to pre-existing mutations. Therefore, the intention is to use GS 5885 in combination. It has good potential for inclusion within a single-tablet regimen as drug dose is low and preclinical studies suggest that drug–drug interactions are unlikely.
In answer to a question, John Link said that GS 5885 has nM activities against HCV types 2, 3 and 4 (21 nM versus type 2a and 10 nM versus type 3a).
Chris Meier (University of Hamburg, Hamburg, Germany) presented much of the chemistry in this session. Carbocyclic nucleoside analogues have a number of potential advantages as drug candidates: improved enzymatic resistance to metabolism, enhanced hydrolytic stability, less cytotoxicity and enhanced ring flexibility, possibly enabling tighter drug–polymerase interactions.
The synthesis of a target compound was described. This illustrated the challenge facing chemists when the target compound has three chiral centres. In this instance, an enzyme was used to generate chirality from a racemic intermediate. The compound, a carba dT analogue, had a hydroxyl group that may allow DNA chain extension. Therefore, the triphosphate was prepared and tested in a chain extension assay using wild-type HIV reverse transcriptase, with the carba dT analogue and the natural dT in a 1:1 ratio. DNA chain extension was possible beyond a single carba dT analogue incorporation, but when there were two consecutive adenines in the template strand, two carba dT analogues were incorporated but no further DNA chain extension was detected.
Satellite: Clinical symposium
Clinical update on the activity of CMX001 against dsDNA viruses
Randall Lanier (Chimerix, Durham, NC, USA)
CMX001 is cidofovir with a phospholipid group attached. CMX001 has much enhanced oral bioavailability relative to cidofovir, and has a plasma half-life of 6.5 days. This prodrug delivers cidofovir into the cell. It is being developed for therapy of the DNA viruses, adenovirus and HCMV, and as a biodefence against poxviruses. It is also active against papillomaviruses, which do not encode a viral DNA polymerase, but viral resistance maps to the viral protein, large T antigen (LTAg), which is required for viral DNA replication.
So far, the clinical experience is based on trials with 440 subjects and a further 170 patients treated under emergency IND (E-IND). Children clear CMX001 at twice the rate of adults and therefore need twice the dose. There has been no evidence of nephrotoxicity characteristic of cidofovir. Gastrointestinal tolerance is expected to be the dose-limiting factor.
A Phase II trial (201), in stem cell transplant patients with CMV, is ongoing. Dosing with CMX001 is from 14 days post transplant and continues for 3 months. There are three dosing groups, 40 mg, 100 mg and 200 mg, each once weekly. A further cohort with twice-weekly dosing is starting. The end point is the proportion of patients with >200 copies/ml CMV at the end of the treatment period. As yet, all three dosage cohorts (50% on drug and 50% on placebo) have <50% patients with CMV. As the data are still blinded, one can only hope that the patients with CMV are all on placebo. However, experience with the patients treated under E-IND would suggest that CMX001 is an effective drug.
Enhanced clinical efficacy of tenofovir amidate prodrug GS-7340 compared to TDF in treatment-naive HIV-positive patients
Tomas Cihlar (Gilead Sciences Inc., Foster City, CA, USA)
The currently approved prodrug of tenofovir (TFV) is tenofovir disoproxil fumarate (TDF), which is widely used in HIV therapy, mainly as part of the single-tablet regimen Atripla. TDF improves the oral bioavailability of TFV in plasma, but targeted delivery of TFV into cells could further enhance treatment efficacy, yet may reduce the risk of renal and bone effects by lowering the plasma concentrations of TFV. A series of amidate prodrugs of TFV was synthesized and GS-7340 was selected (Figure 3).
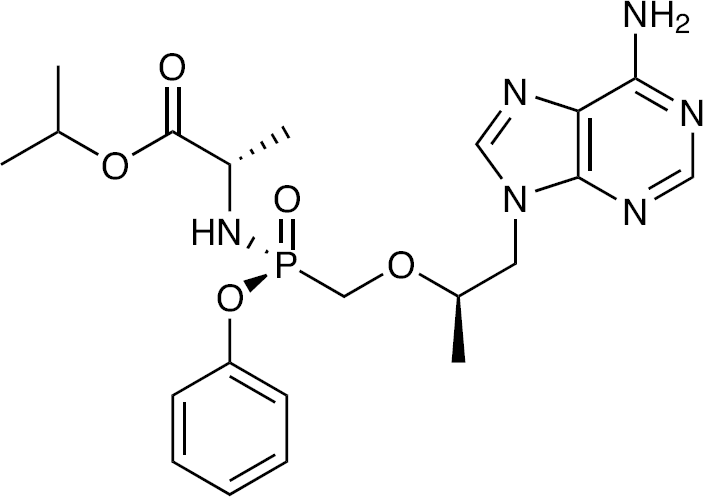
Structure of GS-7340
In cell culture HIV assays, GS-7340 was approximately fivefold more active than TDF, but their cytotoxicities were similar – thus the selectivity index was improved by approximately fivefold. The initial activation step of GS-7340 is within the cell and mediated by cathepsin A. In dogs, dosed with either TDF or GS-7340, the plasma:intracellular concentration ratios were 1:5 and 1:140, respectively. More importantly, the active metabolite, TFV-diphosphate (TFV-DP), is formed rapidly and at high concentrations from GS-7340.
GS-7340 was tested in a Phase Ib randomized double-blind 14-day monotherapy trial (GS-120–1101) in treatment-naive HIV patients. The reductions in HIV RNA load were 0.94, 1.51 and 1.71 log10 in the 300 mg TDF, 50 mg GS-7340 and 150 mg GS-7340 arms, respectively. As expected, the plasma concentrations of TFV from GS-7340 were lower than from TDF, but the TFV-DP levels in patients' PBMCs were enhanced. Relative to the TFV-DP from TDF, the levels for the two dosage groups of GS-7340 were higher by 7- and 18-fold on day 3 and by 4- and 30-fold by day 14, respectively. There were no serious adverse events; mild headache and nausea occurred in a few patients in each arm. No resistance mutations were found at the end of the dosing period.
In summary, short-term monotherapy with GS-7340 (either 50 mg or 150 mg) was more effective than TDF (300 mg) in treatment-naive HIV-infected patients. Reduced systemic exposure is likely to reduce the risk of TDF-associated toxicities. A lower dose of GS-7340 will be useful when considering drug combination pills and reduced cost of goods may enable wider use. As TDF is part of combination regimens used extensively as the backbone for HIV therapy, GS-7340 is well positioned to potentially replace TDF as the standard prodrug of TFV.
Phase I results for the novel and potent HCV NS5A inhibitor GS-5885
John Link (Gilead Sciences Inc., Foster City, CA, USA)
For an introduction to GS-5885 and a summary of the Phase I and early Phase II results, see the presentation given by John Link within the mini-symposium Medicinal chemistry and drug discovery.
AIC246: A new option for HCMV treatment – overview on preclinical virology and Phase IIa proof-of-concept data
Holger Zimmermann (AiCuris GmbH & Co., Wuppertal, Germany)
AIC246 (Figure 4) was initially discovered as a hit in a high throughput screen. The activity (EC50 approximately 5 nM) is highly specific for HCMV, whilst not active against murine CMV. The dose–response curve is unusually steep, the EC90 being approximately 7 nM. The mechanism of action is via inhibiting the HCMV terminase complex, consisting of UL56, UL89 and UL104, which cleaves HCMV progeny DNA into unit genome lengths as the viral DNA is packaged into virions. There is no comparable human cell complex. As current HCMV therapies all target the viral polymerase, no cross-resistance would be expected.
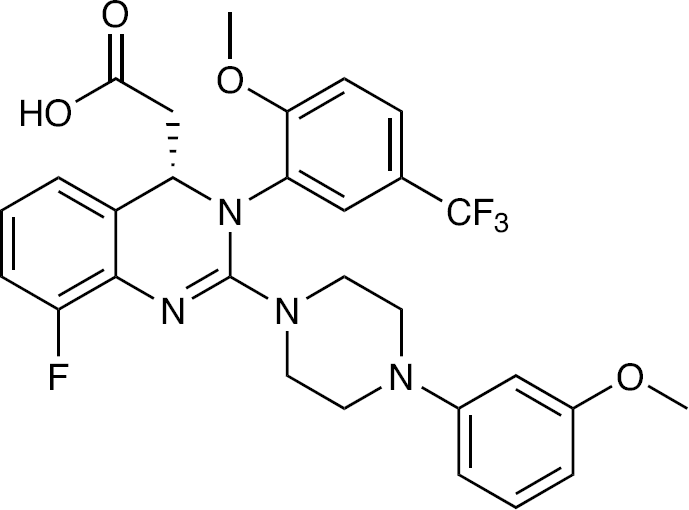
Structure of AIC246, a Quinazoline derivative
Of 10 Phase I trials, 9 have been completed. So far, >250 subjects have been dosed with AIC246, single doses up to 320 mg, multiple doses up to 240 mg twice daily for 2 weeks. To date, there have been no safety issues. Pharmacokinetic studies showed that Tmax was approximately 1 h, terminal half-life was 10 h and <5% metabolized by CYP450. The oral bioavailability was approximately 50%. Once-daily dosing should be possible.
In a Phase II randomized open-label dose-ranging proof-of-concept trial, AIC246 was dosed at 40 mg twice daily or 80 mg once daily for 14 days. The observational control was standard of care using valganciclovir (VGCV). The transplant patients could have any level of positive HCMV viraemia and were eligible for preemptive therapy to prevent HCMV disease. Although the number of patients per group was small (n=7, 9 and 9, respectively), AIC246 seemed to be at least as effective as VGCV. The proportions of patients achieving complete virus clearance by day 14 were 71%, 55% and 33%, respectively (virus in plasma) and 86%, 89% and 56% (virus in whole blood). In this trial, AIC246 was well tolerated at both doses, mean trough levels were higher than EC90 values, no major adjustments to immunosuppressive medication were required, all patients achieved a reduction in viral load and VGCV-resistant virus was successfully treated with AIC246.
Under an E-IND, AIC246 was used to treat a patient with multi-resistant HCMV (ganciclovir, foscarnet and cidofovir) with multi-organ disease. Treatment with AIC246 was started at 120 mg once daily, and later increased to 240 mg once daily, in total for 49 days. Viraemia resolved on day 28 and clinical disease regressed. Following therapy, there was no virological relapse for up to 4 months.
An ongoing Phase IIb trial is evaluating three different doses given once daily for 84 days as prophylactic treatment. The primary end point is ‘prevention of an active replication in HCMV positive patients’. So far, it seems that AIC246 is highly effective against HCMV viraemia using doses up to 80 mg once daily and against HCMV disease at doses up to 240 mg once daily.
Update on ATC
Susan Cox (Avexa, Richmond, Victoria, Australia)
In a Phase II/III trial, apricitabine (ATC) has been evaluated in HIV patients failing lamivudine/emtricitabine therapies, all with the M184V mutation. After week 24, all patients were put on open-label ATC (800 mg twice daily). At ICAR 2009, Susan Cox reported that, with a few patients then reaching 96 weeks, the key results were that the M184V strain remained in most patients and no HIV resistance to ATC had been detected. At this ICAR, the results to week 144 were reported to have remained similar. As for earlier timepoints, the M184V mutant, with which patients entered the trial, is still maintained. Similarly the TAMS are the same as before with no new TAMS appearing. One new mutant, L74V, has been detected but this did not change the EC50 for ATC. Of the two dosing groups in this trial, 800 mg and 1200 mg, the 800 mg dose has now been selected as the dose to use.
Safety issues have not yet emerged. Remarkably, viral resistance to ATC has not been seen in any patient.
At a recent meeting with the FDA, it was agreed that a single confirmatory 14-day study needed to be undertaken prior to filing for approval. The centres for this study have been identified. Avexa are now looking for a partner to progress ATC through to approval.
Contributor presentations
Susceptibility to neuraminidase inhibitors and M2 blockers of some seasonal influenza strains isolated in Bulgaria 2004–2007
Lora Simeonova (The Stephan Angeloff Institute of Microbiology, Sofia, Bulgaria)
To investigate resistance in the seasonal strains of influenza, the sensitivities against rimantadine, oseltamivir and zanamivir were determined in antiviral assays [1]. Also, the gene segments encoding the HA, NA and M2 proteins were sequenced to detect known resistance mutations. Of the 26 strains (H1N1 and H3N2), 22 were sensitive to rimantadine and two H1N1 and two H3N2 strains were resistant. Sequencing of an H3N2 virus (A/Sofia/1250) detected S31N and V27T in the transmembrane region of the M2 protein; these are known resistance mutations. Known resistance mutations were not found in the other three resistant strains. All 26 strains were sensitive to oseltamivir and zanamivir.
A viable human influenza A virus lacking neuraminidase (NA) activity: Isolation and characterization
Martina Richter (Jena University Hospital, Jena, Germany)
From a collection of 25 influenza clinical isolates (Germany 2002–2006), one clinical isolate had a mixture of full-length and defective NA genes. In this study [2], 20 plaques were picked randomly. Of these, 12 contained virus with defective NA genes and 8 with full-length gene. Further plaque purification and gene sequence showed the defective NA gene contained three deletions and encoded a 25-amino-acid protein that lacked the active centre of the enzyme. The NA-lacking virus was able to replicate in cell culture although the titres were approximately 5,000× lower than wild-type virus. Electron microscopy confirmed that virus release from cells was less efficient with the NA defective virus than the wild-type virus. Sequencing of NA gene from the original clinical isolate indicated that the full-length NA gene was present only as a small proportion. Presumably, the virus with the defective NA gene had a replicative advantage so long as some wild type virus was present to help with release of the NA defective virus from the cell.
Human papillomavirus genotype distribution in women in Montenegro
Danijela Vujošević (Institute of Public Health, Podgorica, Yugoslavia)
The purpose of this study [3] was to determine the range and frequency of different papilloma genotypes in women in Montenegro. Of 189 participants, 38 (20%) were positive for papilloma. HPV genotype 16 was the predominant type (14/38). The next most frequent types were 58 (4/38), 31 and 6 (each 2/38). Other types were detected in individual participants. Mixed infections were detected in 7/38 participants, mainly in younger women, aged 25 to 30 years. The conclusion from this study was that, to determine which women are at risk of developing cervical cancer, it is necessary to use a test which is able to detect a broader range of high-risk HPV types than just the HPV types 16 and 18 in the vaccines.
Nitazoxanide is an indirect inhibitor of HCV replication through modulation of cellular kinase CK1 alpha to enhance HCV NS5A hyperphosphorylation
Abigail Montero (Georgetown University Medical Center, Washington, DC, USA)
Nitazoxanide (NTZ) is a licensed compound being investigated clinically against HCV. This follows the discovery that NTZ and its metabolite, tizoxanide (TIZ), inhibit HCV in cell culture assays. Previously the mechanism of action had been shown to be indirect; therefore, the purpose of this study [4] was to further investigate the mechanism. In enzyme assays, TIZ was inactive against HCV polymerase, protease and helicase. As HCV replication is known to be localized on an intracellular web, intracellular membrane preparations were used to study the effect of NTZ. After 48–72 h of NTZ treatment, there was a four- to sixfold enhancement of hyperphosphorylation of HCV NS5A (p58) and a corresponding decrease in basally phosphorylated NS5A (p56). Phosphorylation of NS5A is known to act as a switch from active viral genome replication to RNA packaging and virion assembly. The hyperphosphorylation of NS5A was associated with a three- to fourfold higher enzymatic activity of casein kinase 1-α (CK1a) which is known to phosphorylate NS5A. However, TIZ had no direct effect on the enzymatic activity of CK1a. These data provide a primary mechanism of action of NTZ and its metabolite TIZ. Treatment leads to an overproduction of hyperphosphorylated form of HCV NS5A, causing a premature switch from viral genome replication to virus assembly.
Targeting the flavivirus helicase
Eloise Mastrangelo (University of Milan, Milan, Italy)
Flaviviruses are ssRNA viruses such as Dengue (4 serotypes), yellow fever (YFV), WNV, Japanese encephalitis (JEV) and tick borne encephalitis (TBEV) viruses. During viral replication, the newly synthesized dsRNA is unwound by the viral helicase. The flaviviral helicase is a monomeric protein with three domains. Based on the crystal structure of Kunjin viral helicase [5], the authors hypothesized that the ssRNA entrance site (α-helical gate) was between domains II and III and could be a drug target [6]. An in silico docking search led to the identification of some candidate compounds, which were tested in a helicase unwinding assay and in antiviral assays. Of the identified compounds, three were effective in the dsRNA unwinding assay but only Ivermectin was active against several viruses (YFV, Dengue, JEV and TBEV), with EC50 values <1 μM. Time-of-addition experiments confirmed that Ivermectin is effective in inhibiting the flaviviral replication cycle in which the viral helicase is functionally active. As Ivermectin has been used clinically for the treatment of various parasitic diseases for >20 years, there may already be some data from tropical areas indicating activity of this old drug against flaviviruses.
Conclusions
It is impossible to record all the interesting presentations in this review, but it is hoped that this report is indicative of the range of topics covered. A great strength of ICAR meetings is the potential for cross-fertilization between differing areas; for example the use of a compound for another virus such as favipiravir (T705) being developed for influenza but now reported to be effective against arenaviruses. There were reports on old compounds with potential new uses: one was a better understanding of the mode of action of Nitazoxanide (NTZ) against HCV, another, Ivermectin, currently used as an anti-parasitic agent, was reported to have good activity against four flaviviruses.
Personally, I found the major award lectures, the mini-symposia and plenary lectures provided a good overview of important areas of antiviral research. The satellite clinical symposium proved beyond doubt that antiviral chemotherapy is continuing to make important new progress. In the HIV field, GS-7340 looks well placed to replace TDF as the prodrug of TFV. By contrast, apricitabine seems to be an excellent compound, with no patients having resistant HIV after 144 weeks on therapy, but needing a partner to get it through the last stage of approval. AIC246 is a promising therapy for HCMV, for which valganciclovir has essentially been the only available drug for years. For HCV treatment, a new inhibitor of HCV NS5 was reported and expected to be used in combination therapy. A major advancement in HCV therapy would be to have an effective drug regimen that does not include IFN. Such a prospect now seems more likely with the report on RG7977 and PSI-352938.
Footnotes
Acknowledgements
AVH would like to thank the ISAR Officers and Conference Committee for organizing another successful meeting and to Angel Galabov for such a warm welcome to his city, Sofia. Lastly, this ICAR was a fitting testament to the ‘father of antiviral chemotherapy’, Bill Prusoff.
The author declares no competing interests.