
Abstract
Background:
In addition to activities needed to catalyse integration, retroviral integrases exhibit non-specific endonuclease activity that is enhanced by certain small compounds, suggesting that integrase could be stimulated to damage viral DNA before integration occurs.
Methods:
A non-radioactive, plate-based, solution phase, fluorescence assay was used to screen a library of 50,080 drug-like chemicals for stimulation of non-specific DNA nicking by HIV-1 integrase.
Results:
A semi-automated workflow was established and primary hits were readily identified from a graphic output. Overall, 0.6% of the chemicals caused a large increase in fluorescence (the primary hit rate) without also having visible colour that could have artifactually caused this result. None of the potential stimulators from this moderate-size library, however, passed a secondary test that included an inactive integrase mutant that assessed whether the increased fluorescence depended on the endonuclease activity of integrase.
Conclusions:
This first attempt at identifying integrase stimulator compounds establishes the necessary logistics and workflow required. The results from this study should encourage larger scale high-throughput screening to advance the novel antiviral strategy of stimulating integrase to damage retroviral DNA.
Introduction
Treatment of HIV-1 infection is a major success story in clinical medicine and provides a model for antiviral therapy. However, development of drug resistance necessitates the continuing identification of new antiretroviral drugs as well as exploration of novel antiviral strategies to prevent or treat AIDS. One important target for antiretroviral agents is the viral integrase protein, which is present in all infectious retroviruses and must be enzymatically active for virus transmission, replication and pathogenesis [1]. Integrase is an endonuclease that acts by nicking the ends of viral DNA at a specific site to prepare the viral DNA for integration, and then inserting those ends non-specifically into cellular DNA [2]. After years of research, blocking the action of integrase was shown to have significant clinical benefits [3–5], and an integrase inhibitor is now commonly included in combination antiretroviral drug regimens.
In addition to the activities needed to catalyse integration, retroviral integrases have a non-specific endonuclease activity that can nick any DNA sequence [6], and this activity is markedly stimulated by certain small compounds, including 1,2-ethanediol (ED), 1,2-propanediol, 1,3-propanediol and 1,2,3-propanetriol (glycerol) [7]. In the case of ED, kinetic studies showed that the mechanism of stimulation involves an increase in Vmax [8]. It has been suggested by several investigators that stimulating or unregulating integrase-mediated DNA nicking could cause this enzyme to damage viral DNA before integration occurs and abort the infection before it becomes established [1,6,9], thus supplementing the host-mediated degradation of viral DNA that protects cells from integration [10]. The demonstration that viral DNA in HIV-1 pre-integration complexes (PICs) is susceptible to nucleases [11,12] supports this novel idea, and studies of pre-integration latency suggest that viral DNA could be vulnerable for several days [13]. In fact, the documented antiviral effect of packaging non-specific bacterial nucleases into retroviruses by linking them to the Gag protein [14–17] provides experimental precedent for an antiretroviral strategy that involves a strategically located nuclease. Thus, the idea of stimulating integrase – which is packaged into every infectious retrovirion as part of the Gag–Pol polyprotein – can be considered a natural version of capsid-targeted viral inactivation [18]. If the increased nicking activity were restricted to the PIC while still in the cytoplasm, which is likely if dissolution of the PIC exposes integrase to protein degradation [19], then toxicity to cellular DNA would be avoided. Moreover, any collateral damage to cellular DNA would likely also be clinically beneficial because it would be limited to newly infected cells that had just been entered by HIV [20].
Development of a pharmacological agent that stimulates integrase to damage DNA is a logical extension of the theoretical and experimental foundation described above. Although the four integrase stimulator (IS) compounds listed earlier can cause integrase to nick 50% of a DNA substrate [7], high concentrations of those chemicals are required for stimulation. Thus, to assist efforts to develop more potent IS compounds for preclinical evaluation, we recently described a non-radioactive, plate-based, solution-phase assay for non-specific DNA nicking [8]. We now report the first use of this assay to screen a library of drug-like chemicals for stimulators of the non-specific DNA nicking activity of HIV-1 integrase.
Methods
HIV-1 integrase
HIV-1 integrase was expressed in bacteria and purified under native conditions as described previously [21,22]. The purified protein was dialysed against storage buffer [8], diluted to 4 pmol/μl, and stored in aliquots at −70°C. An active-site mutant of HIV-1 integrase that contains a D116I amino acid substitution and lacks enzyme activity [23] was purified and handled similarly.
Chemical library
The DIVERSet collection of compounds was purchased from ChemBridge Corporation (San Diego, CA, USA). The 50,080 chemicals in this library have molecular weights ranging from 200 to 540 Da and stock concentrations of 5 μg/μl in dimethylsulfoxide (DMSO). The chemicals were supplied as sets of 80 compounds in 96-well plates, which were stored at −20°C. Each chemical was screened at a final concentration of 1 μg/μl.
Plate-based fluorescence assay for DNA nicking
The dual-tagged 49-mer oligodeoxynucleotide in Figure 1 was purchased from Integrated DNA Technologies, Inc. (Coralville, IA, USA). The 5′-end of this DNA is labelled with a fluorophore (fluorescein [FAM]) and the 3′-end is linked to a quencher (black hole quencher-1 [BHQ-1]). The oligonucleotide was dissolved in 10 mM Tris-HCl (pH 8.0) and 1 mM EDTA, quantified by spectrophotometry at 260 nm [8], and stored in aliquots at −70°C. Reactions were assembled in 384-well plates on a cooling block with the aid of an Eppendorf epMotion 5070 Workstation (Mississauga, ON, Canada). Each reaction had a final volume of 10 μl and contained 2 μl of test compound (or water, DMSO or ED as controls, with the final concentrations of DMSO or ED being 20%), 5 pmol of substrate DNA (final concentration 500 nM), 25 mM Tris-HCl (pH 8.0), 10 mM dithiothreitol (DTT), 10 mM MnCl2 and 1 μl (4 pmol) of HIV-1 integrase. ED was used as a control because it is as active as other available IS compounds [7] but does not cause pipetting difficulties from high viscosity. Reagents were delivered in the following order: 2 μl of test compound or controls, 6 μl of a master mix that contained substrate DNA plus Tris-HCl, DTT and integrase, and 2 μl of MnCl2 (the divalent metal was added last to delay the start of the reaction and assembling the mixtures on a cooling block also helped synchronize all reactions). DNase I (1 μl of a 1 μg/μl stock solution, for a final concentration of 0.1 μg/μl) was then added manually to two control wells to assess the maximum signal for each plate. Plates were sealed, briefly mixed by placing them for 10 s on a vibrating Pipet-Aid (Drummond Scientific Company, Broomall, PA, USA), spun for 30 s at 4°C, then placed in an incubator at 37°C for 90 min. Reactions were stopped by transferring the plates to a 65°C oven for 10 min to inactivate integrase and then spun again.
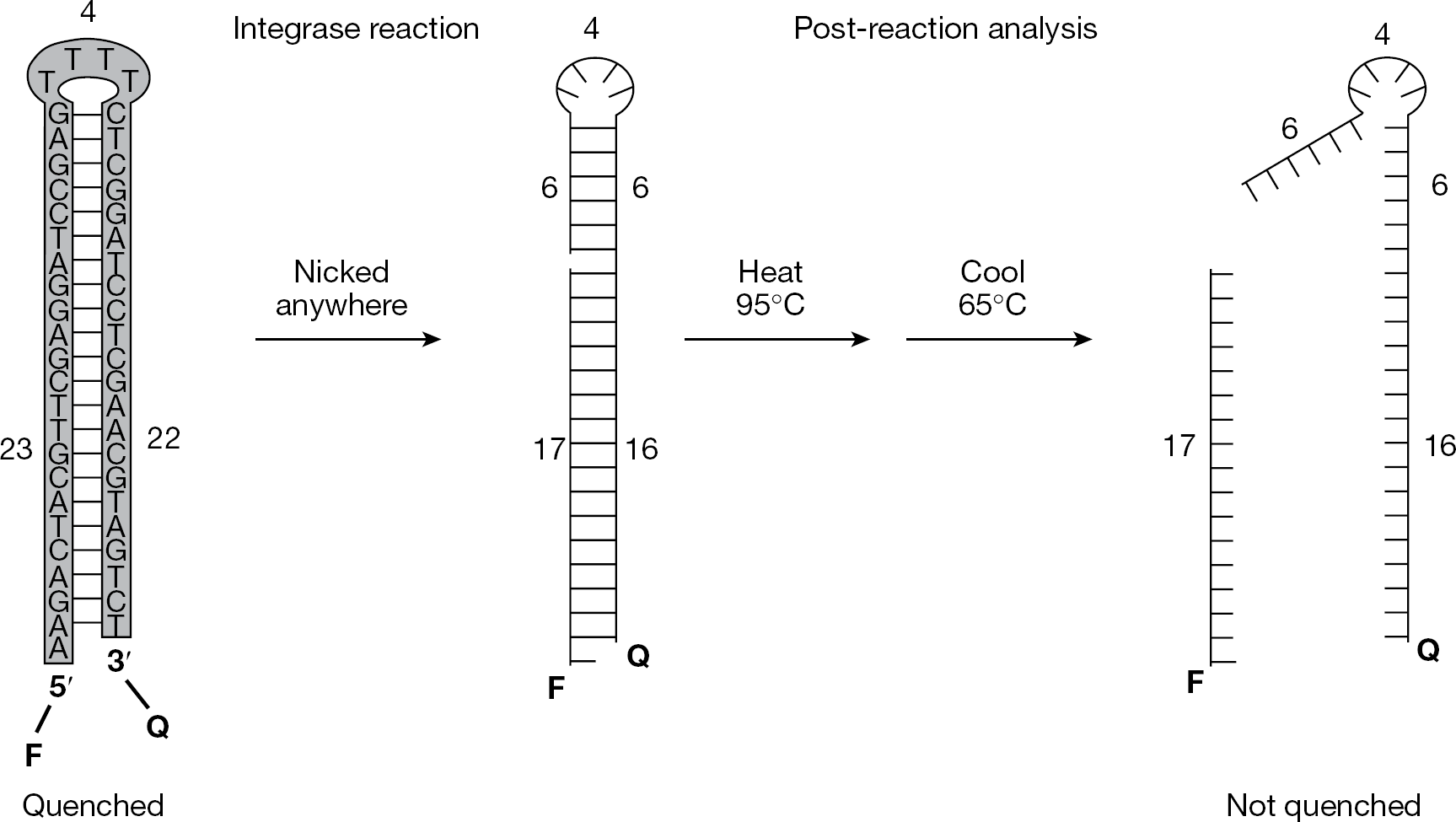
Non-radioactive, plate-based, solution-phase assay for non-specific DNA nicking
Analysis of completed reactions
Fluorescence of completed reactions was measured using an Applied Biosystems (Foster City, CA, USA) 7900HT Real-Time PCR System in the ‘Absolute Quantitation’ mode for FAM with the passive reference set to ‘none’. The machine was programmed for a single denaturation at 95°C for 1 min and renaturation at 65°C for 1 min, followed by 2 min 59 s at 65°C during which fluorescence of each well was read 21 times (when we tried a data collection phase of exactly 3 min, the machine started another pass across the plate and read one of the columns of wells an extra time). Data were exported as a Microsoft Excel file, then copied and pasted into a customized Excel file that automatically averages the final 10 reads for each well, calculates the control data and displays the results on bar graphs [8].
Results
Non-specific nicking assay for the primary screen The development and validation of a solution phase assay for non-specific DNA nicking (Figure 1) was described previously [8]. Briefly, the assay uses a 49-mer oligonucleotide that is 5′-labelled with a fluorophore, 3′-tagged with a quencher, and designed to form a hairpin that mimics the double-stranded radioactive substrates commonly used in gel-based nicking assays [6]. Reactions were conducted in 384-well plates during a 90 min incubation at 37°C. Nicking anywhere in the sequence will unlink the fluorophore and quencher groups and yield fluorescence after heat denaturation and subsequent cooling to a point above the melting temperatures of nicked products (Figure 1, right). By contrast, the unnicked hairpin should reform and quench fluorescence at temperatures below the melting temperature of the original stem (Figure 1, left). Post-reaction analysis is facilitated by programming a real-time PCR machine to perform a single heat denaturation followed by cooling to 65°C (which is between the melting temperatures of unnicked substrate and nicked products), and taking advantage of the ability of such machines to read fluorescence at a high temperature. No cycling is performed, and the PCR machine can process each plate and measure the fluorescence of every well in <10 min. The assay was shown to be linear with time, amount of integrase and concentration of a known IS, and the reaction conditions described in the Methods are within the linear portions of these curves; additionally, the presence of 20% DMSO did not unquench the fluorescence of unnicked substrate or mask the signal from nicked DNA [8].
Screening a chemical library: logistics and control data We screened the 50,080-member DIVERSet collection of drug-like chemicals for stimulation of non-specific DNA nicking in reactions with HIV-1 integrase. This library is supplied as sets of 80 compounds in 96-well plates, and a robotic workstation was used to deliver chemicals from four source plates to one 384-well assay plate. Thus, 320 chemicals were tested in each assay, and the entire library was screened on 157 assay plates (the last plate tested the final 160 compounds). Each assay plate also included 16 controls: 2 wells with water and 6 wells with DMSO as negative controls for baseline nicking, 6 wells with ED as an example of a known IS compound, and 2 wells to which DNase I was added as positive controls to indicate the maximum signal from extensive nicking of the DNA substrate.
The mean fluorescence for the DMSO, ED and DNase controls from each assay plate are shown in Figure 2A. As might be expected, the values for baseline nicking in reactions with DMSO (Figure 2A, circles) and stimulated nicking in reactions with the known IS ED (triangles) varied slightly with each preparation of purified integrase (the vertical lines in Figure 2A indicate different integrase preparations). Note that baseline nicking in the absence of ED reflects unstimulated nicking plus a small amount of stimulation by the 1% final concentration of glycerol (another IS compound) that was provided by the integrase storage buffer [8,21]. Similarly, maximum signal after exposure of the substrate to DNase varied with each lot of fluorescent oligonucleotide, although these values were always very high (Figure 2A, squares, where the four arrows mark new stocks of the 49-mer substrate).
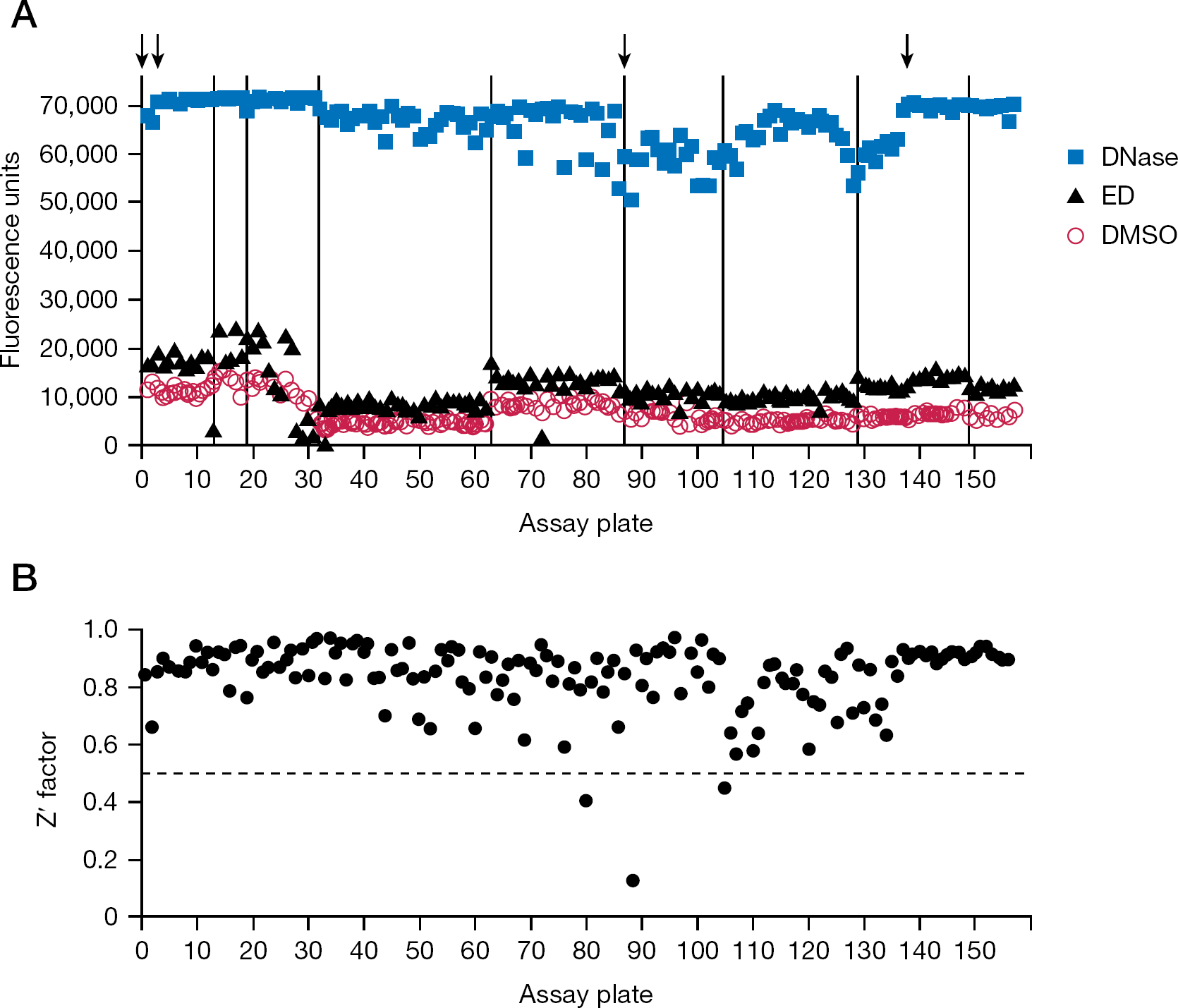
Quality control data from screening the full library
Stimulation of nicking by ED was observed on 145 (92%) of 157 plates, as indicated by clear separation (and an absolute difference of at least 2,100 fluorescence units) between reactions with ED and concurrent reactions with DMSO (Figure 2A, compare triangles to circles; the 12 plates that did not pass this quality test [for example, plate 13] were subsequently retested). Overall, stimulation by ED relative to the amount of nicking in the DMSO controls (omitting data for the 12 initially failed plates) averaged 1.8-fold, with an absolute difference that averaged 5,354 fluorescence units (these values were 1.9-fold and 5,400 units, respectively, when subsequent data from the 12 repeated plates were included).
We also calculated the Z' factor for each assay; this parameter is a function of the range of an assay (the difference between positive and negative controls) and the variation of the controls, with values ≥0.5 considered optimal for high-throughput screening assays [24]. The Z' factor for each assay plate (Figure 2B) averaged 0.84 (with or without inclusion of the repeated plates), and only 3 times was it <0.5 (the low Z' factors for plates 80, 88 and 105 were due to variation in the reactions with DNase, but even on these plates the DNase controls averaged >50,000 fluorescence units). As in the original description of this assay [8], reactions with DNase were used as the positive controls for these calculations because a potent IS is not yet available. In the absence of an optimal reference compound, others have also used reagents as positive controls (such as antibodies or other proteins) that were different from the test chemicals being screened [25].
Results from the primary screen
Each assay plate was processed as in the Methods to yield four bar graphs; Figure 3 shows an example of one of these graphs, which was modified slightly to include all four types of controls in the figure. Each graph presents data from one-fourth of an assay plate, representing four rows of 21 reactions (a total of 84 reactions). Each set of 21 reactions includes 1 of the controls (Figure 3, the grey bars) plus 20 assays with chemicals from the library (the black bars, testing 10 compounds from each of 2 source plates). In the example shown, reactions 1 and 22 (with water and DMSO, respectively) yielded <13,000 fluorescence units, reaction 43 (with ED) yielded approximately 19,000 units, and reaction 64 (with DNase I) had approximately 71,000 units. The fluorescence of the reaction that contained ED was greater than the mean + 3 SD of the concurrent negative controls that contained DMSO (as indicated by the horizontal line in Figure 3); in fact, 84% of the almost 900 individual ED reactions across the 145 plates in Figure 2A (excluding the 12 plates that had to be repeated) had fluorescence above the mean + 3 SD of concurrent DMSO controls (and 95% were above the mean + 2 SD).
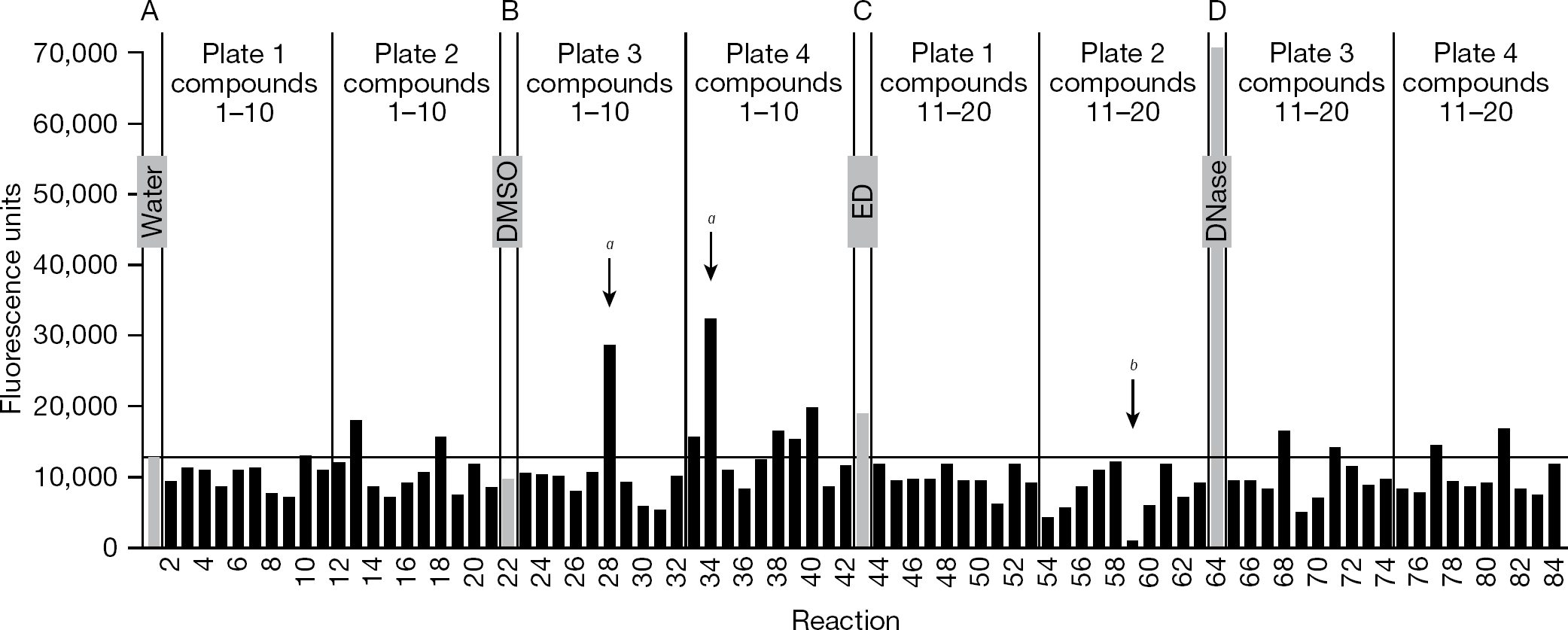
Example of output from the assay
Several of the test chemicals in Figure 3 also yielded fluorescence above the controls, sometimes in excess of that from the ED reaction (for example, reaction 40). However, to have a manageable hit rate in the primary screen, we focused on chemicals that greatly exceeded the controls. Two such chemicals that yielded markedly increased fluorescence (reactions 28 and 34) are indicated by ‘a’ in Figure 3. Given the higher values for the DMSO control reactions on plates 1–31 compared with subsequent plates (which was related to the integrase preparation, as seen in Figure 2A), we defined potential stimulators as exceeding 25,000 fluorescence units on assay plates 1-31 or exceeding 20,000 units on plates 32–157. Overall, 292 of the 50,080 chemicals (a primary hit rate of 0.6%) exceeded these thresholds without also having a visible colour that could have artifactually caused this result. We should note that 12.3% of the chemicals in the library caused discernible colour on the assay plate (most often yellow or orange, which could increase or decrease fluorescence without a predictable pattern) and were excluded from further analysis; another 0.3% of the compounds yielded a negative value and also were excluded. We also observed that 0.7% of the chemicals in the library yielded markedly diminished fluorescence (<1,000 fluorescence units), an example of which is indicated by ‘b’ in Figure 3 (reaction 59); although such chemicals might be worthy of further analysis, they were not the focus of this project.
Secondary assays for hits from the primary screen Because many – if not most – hits from a chemical screen may be due to mechanisms other than the desired one, secondary (or counter) assays are required to validate primary hits. We initially used a gel-based assay with radioactive double-stranded oligonucleotide substrates [8] to retest potential stimulators identified in the primary screen. In fact, neither of the potential stimulators identified in Figure 3 was found to stimulate integrase in radioactive assays (data not shown). Similarly, no hits from any of the first 12 plates were validated as stimulators in radioactive assays. Given the large number of potential stimulators identified by the primary screen (and many others that were close to the defined threshold), the plate-based assay was adapted for an automated and more efficient counter screen, starting with assay plate 13. For this purpose, potential stimulators (primary hits) were retested in two parallel reactions, one using wild-type integrase (to confirm the initial results) and one using an inactive integrase mutant (to validate that the increased fluorescence depended on the action of integrase). Importantly, the known IS ED increased the fluorescence of reactions with wild-type integrase but not reactions with the inactive integrase (data not shown).
Many chemicals that increased fluorescence in the primary screen also yielded increased fluorescence upon retesting (for example, Figure 4A, where the black bars indicate primary hits from the initial screen). Overall, 38% of these chemicals when retested exceeded the mean + 3 SD of the concurrent controls (as indicated by the horizontal line in Figure 4A), 20% caused a signal above 15,000 fluorescence units, and 13% were above 20,000 units. Similar confirmation rates upon retesting have been reported in the literature for other high-throughput assays and underscore the importance of excluding false-positive hits from initial screens [26–29]. Moreover, none of the primary hits passed the predefined secondary test of causing greatly increased signal in reactions with wild-type integrase but not in reactions with an inactive mutant (that is, the heights of the bars for each chemical were comparable between Figures 4A and 4B). Of note, we observed visible colour on the source plate or assay plate for 18% of the primary hits during set up of the secondary assays (as indicated by ‘a’ in Figure 4; that these colours had not been recorded during the primary screen reflects greater scrutiny during the counter screen, including examination of the source plates for any colour). We also retested approximately 300 compounds that had caused more moderately increased fluorescence in the primary screen (for example, 15,000–20,000 units for assay plates 32–157). These chemicals often yielded results just above or below the mean + 3 SD of the concurrent DMSO controls upon retesting (for example, Figure 4A, the grey bars), but none clearly increased signal with wild-type but not with an inactive integrase (Figure 4A compared with Figure 4B).
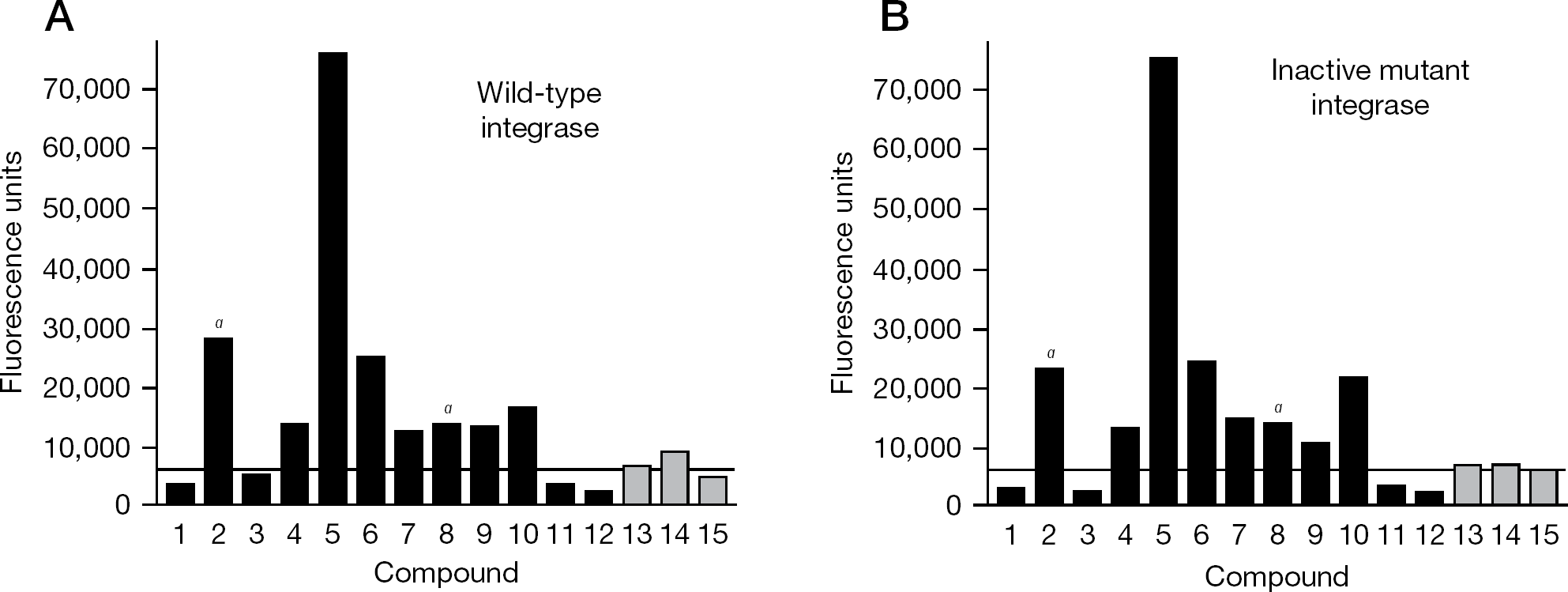
Examples of secondary assay
Discussion
New strategies to attack human retrovirus infections are needed. There is precedent in clinical medicine for stimulating the function of other proteins, such as the cystic fibrosis transmembrane conductance regulator [30], and successful chemical screens have been reported for stimulators of other proteins and enzymes, including the human homologous recombination protein RAD51 [31] and glucokinase [32]. To date, four chemicals have been shown to stimulate HIV-1 integrase to nick DNA non-specifically [6,7], but potent and relatively non-toxic IS compounds are needed for further preclinical development. Ultimately, a useful agent would have to perturb integrase in such a way that newly synthesized viral DNA (or cellular DNA) is sufficiently damaged to overcome DNA repair mechanisms and prevent the permanent integration of viral DNA.
The recent development of a plate-based assay for non-specific DNA nicking that is suitable for high-throughput screening (Figure 1) made it possible to begin efforts to identify IS compounds. In the current report, we describe the first attempt at such screening, using a moderate-sized library of approximately 50,000 chemicals. Importantly for future efforts, a semi-automated workflow was established for the primary and secondary screens, primary hits were readily identified from a graphic output, and a logical counter screen was developed. Thus, the approach and techniques were shown to be logistically feasible (Figures 2 and 3).
In this initial screening effort, none of the hits from the primary screen were validated using a secondary assay designed to exclude artifactual results that did not depend on the action of integrase (Figure 4). Increased fluorescence that was independent of integrase function may have been due to autofluorescence of the test compounds. Although pre-screening the chemical library for autofluorescence might have excluded such chemicals from the secondary screen (but not from the primary screen given the characteristics of our robotic workstation), it still would have been uncertain whether autofluorescence of stock compounds on the source plates would be relevant to diluted samples under reaction conditions; thus, testing all chemicals in the primary assay was appropriate. It also is possible that the increased signal from the false-positives resulted from the compound directly damaging the DNA substrate or otherwise dissociating the hairpin, perhaps by affecting the pH or by chelating the divalent metal that we have found to be necessary to maintain the hairpin. Any of these possible explanations underscores the importance of performing an appropriate counter assay.
It should be noted that a reported screen for stimulation of another enzyme tested 120,000 compounds to identify one stimulator of glucokinase [32]. Thus, although the experiments described in the current report did not identify any new IS from a much smaller chemical collection, the techniques and approaches described here can now be applied to larger scale high-throughput screening to advance the novel antiviral strategy of stimulating integrase to damage retroviral DNA.
Footnotes
Acknowledgements
We thank the Drug Discovery, Development, and Delivery Core of the Department of Pharmacology and the Penn State Hershey Cancer Institute for access to the chemical library, and Rob Brucklacher and Georgina Bixler of the Penn State Hershey Genome Sciences Core Facility for assistance with the PCR machine (all at the Penn State College of Medicine, Milton S Hershey Medical Center). We also thank Fred Krebs (Drexel University College of Medicine) for support and encouragement. This work was supported by Public Health Service grant R21AI075929 from the Microbicide Innovation Program of the Office of Women's Health and National Institute of Allergy and Infectious Diseases (to MK), and by an American Recovery and Reinvestment Act Administrative Supplement Providing Summer Research Experiences for Students and Science Educators (to MK to support JBE).
The authors declare no competing interests.