
Abstract
Background:
Novel indolylarylsulfones (lASs), designed through rational structure-based molecular modelling and docking approaches, have been recently characterized as effective inhibitors of the wild-type and drug-resistant mutant HIV-1 reverse transcriptase (RT).
Methods:
Here, we studied the interaction of selected halo- and nitra-substituted IAS derivatives, with the RT enzyme carrying the single resistance mutations K103N and Y181I through steady-state kinetic experiments. Results: The studied compounds exhibited high selectivity to the mutant RT in complex with its substrates, behaving as uncompetitive inhibitors. The presence of the K103N mutation, and to a lesser extent the Y181I, stabilized the drug interactions with the viral RT, when both its substrates were bound.
Conclusions:
The characterization of these mutation-specific effects on inhibitor binding might be relevant to the design of more effective new generation non-nucleoside reverse transcriptase inhibitors, with better resilience towards drug resistant mutants.
Introduction
Drugs used to treat the infection of HIV, the aetiological agent of AIDS, fall into six classes: nucleoside reverse transcriptase inhibitors, nucleotide reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors, entry inhibitors including the CCR5 coreceptor antagonist maraviroc and the integrase inhibitor raltegravir.
At the present time, NNRTIs are standard drugs used in active HAART, consisting of the combination of three drugs (recommended) from at least two different classes. However, the rapid emergence of resistance mutations, yielding drug-resistant mutant HIV-1 strains, and the occurrence of adverse side effects, frequently impair the beneficial effects of these treatments [1,2]. Therefore, improving the therapeutic profile of NNRTIs towards the resistance mutations is a key challenge for the development of novel effective HIV therapies.
Crystal structures of reverse transcriptase (RT)/ NNRTI complexes showed that the drug interacts with a hydrophobic pocket (non-nucleoside binding site [NNBS]) of the RT. Binding of the NNRTI induces conformational changes of RT, leading to enzyme inactivation. Resistance is caused by mutations of some amino acids into the NNBS, which ultimately result in reduced affinities towards the inhibitors [3–5].
The HIV-1 RT itself undergoes a conformational reorganization upon interaction with its substrates template-primer (TP) and deoxynucleoside triphosphate (dNTP), so that three structurally distinct mechanistic forms can be recognized in the reaction pathway catalyzed by HIV-1 RT: the free enzyme, the binary complex of RT with the TP (RT/TP), and the catalytically competent ternary complex of RT with both nucleic acid and dNTP (RT/TP/dNTP) [6,7]. Several kinetic studies have shown that, contrary to the majority of non-competitive first and second generation NNRTIs, some novel NNRTIs showed different binding affinities to the enzyme in complex with its TP and/or dNTP substrates, behaving as mixed type or uncompetitive inhibitors [8,–12].
We have previously reported the identification of novel effective indolylarylsulfones (IASs), whose development was based on the derivative L-737,126 (Merck Co., Inc., Readington, NJ, USA), obtained by coupling natural and unnatural amino acids to the 2-carboxamide functionality and introducing different electron-withdrawing substituents at positions 4 and/or 5 of the indole nucleus [13–16]. A series of new highly flexible molecules was derived, and found to inhibit HIV-1 replication at nM concentrations, in a fashion similar to efavirenz.
However, since the K103N and Y181I substitutions still significantly reduced the potency of inhibition of most IAS derivatives, we started the synthesis of a novel series of IAS analogues in order to optimize these molecules in their interactions with the mutated RT. Here, we present the characterization of the binding affinity and the inhibition kinetics of five novel halo- and nitro-substituted IAS derivatives (RS compounds listed in Figure 1), bearing an N-(cycloalkylamino)methyl moiety at the 2-carboxamide nitrogen, with respect to the three different mechanistic forms of HIV-1 RT, either wild type (WT) or carrying the K103N or Y181I mutation. These mutations were selected because they can induce highlevel resistance towards all clinically approved NNRTIs, including etravirine (TMC-125) [17,18]. Our data show that some of these molecules achieve a more stable binding to the mutated RT in complex with its substrates, with respect to the WT enzyme.
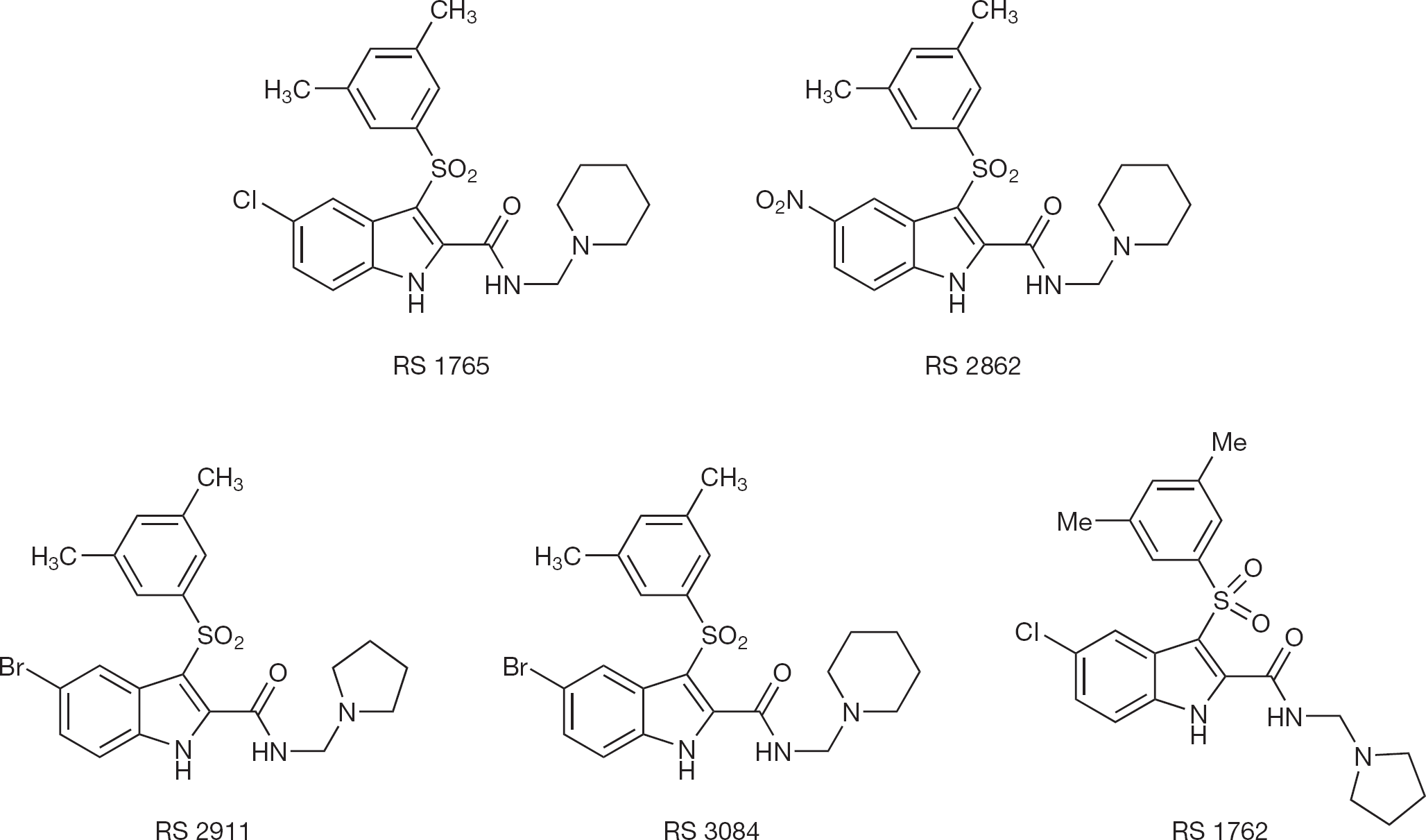
Structures of the novel halo- and nitro-indolylarylsulfone derivatives studied
Materials and methods
Chemicals
All the reagents were of analytical grade and purchased from Sigma-Aldrich (St Louis, MO, USA), Merck Sharp & Dohme (Readington, NJ, USA), ICN (Research Products Division, Costa Mesa, CA USA) or AppliChem GmbH (Darmstadt, Germany). Radioactive 2′ deoxy-thymidine 5′-triphosphate [3H]dTTP (40 Ci/mmol) was purchased from Perkin Elmer (Waltham, MA, USA), while unlabelled dNTPs were from Boehringer Ingelheim GmbH (Ingelheim, Germany). GF/C filters were provided by Whatman International, Ltd (Maidstone, UK).
Chemistry
IAS derivatives RS 2911, 1765, 2862, 3084 and 1762 were prepared by Mannich reaction of 5-chloro-, 5-nitro- or 5-bromo-3-[(3,5-dimethylphenyl) sulfonyl]-1H-indole-2-carboxamide with piperidine or pyrrolidine in tert-butanol at reflux for 7 h in the presence of 37% formaldehyde. Full experimental description of preparation and characterization of IAS derivatives has been reported elsewhere [19].
Nucleic acid substrates
The homopolymer poly(rA) and the oligomer oligo(dT)12–18 (Pharmacia & Upjohn Inc., Pfizer, Peapack, NJ, USA) were mixed at weight ratios in nucleotides of 10:1 with 25 mM Tris HCl (pH 8.0) containing 22 mM KCl, heated at 70°C for 5 min and then slowly cooled to room temperature.
Expression, purification and cloning of recombinant HIV-1 RT forms
Recombinant heterodimeric RT, either WT or the K103N, L100I and Y181I variants, were expressed and purified as briefly described below. The HIV-1 RT gene fragment spanning codons 2 to 261 carrying the K103N, L100I and Y181I mutations was amplified by PCR, digested with Acc1 and Pvu2 restriction enzymes and cloned into the expression plasmid p6HRT (δXho1/Bgl2). The resulting constructs were used for the production in Escherichia coli (BL21) and purification of recombinant His-tagged RT enzymes in a fast protein liquid chromatography system using Ni-NTA superflow column (Qiagen, Venlo, the Netherlands) and Q-Sepharose column (GE Healthcare, Little Chalfont, UK). All the enzymes were purified to a >95% purity, as confirmed by SDS-PAGE and Gelcode Blue stain, and had a specific activity on poly(rA):oligo(dT). One unit of DNA polymerase activity corresponds to the incorporation of 1 nanomol of dNMP into acid-precipitable material in 60 min at 37°C. Western blotting confirmed the identity of the purified proteins by the means of anti-RT or anti-His monoclonal antibodies.
HIV-1 RT RNA-dependent DNA polymerase activity assay
Poly(rA)/oligo(dT) was used as a template for the RNA-dependent DNA polymerase reaction by HIV-1 RT, either WT or carrying the resistance mutations. For the activity assay, a 25 μl final reaction volume containing TDB buffer (50 mM Tris-HCl [pH 8.0], 1 mM dithiothreitol, 0.2 mg/ml bovine serum albumine and 2% glycerol), 10 mM MgCl2, 0.5 mg of poly(rA):oligo(dT)10:1 (0.3 mM 3′-OH ends) and 10 mM 3[H]dTTP 1 Ci/mmol was finally introduced into tubes containing aliquots of different enzyme concentrations (5–10 nM RT). After incubation at 37°C for the indicated time, 20 μl from each reaction tube was spiked on glass fibre filters GF/C and immediately immersed in 5% ice-cold trichloroacetic acid (AppliChem GmbH, Darmstadt, Germany). Filters were washed three times with 5% trichloroacetic acid and once with ethanol for 5 min, then dried and, finally, added with EcoLume® Scintillation cocktail (ICN, Research Products Division, Costa Mesa, CA USA), to detect the acid-precipitable radioactivity by PerkinElmer® Trilux MicroBeta 1450 Counter (Perkin Elmer Inc., Waltham, MA, USA).
Steady-state kinetic assays
Steady-state kinetic assays were also performed to evaluate the activity of HIV-1 RT in the presence of fixed concentrations (50% inhibitory dose [ID50]) of selected inhibitors and variable concentrations of one of the two substrates, either poly(rA)/oligo(dT) or [3H]TTP, while the other was maintained at saturating doses. [3H]TTP concentrations varied between 0.2–20 mM, while poly(rA)/oligo(dT) doses ranged from 10–400 nM. These experiments led to the determination of maximal velocity (Vmax), Michaelis constant (Km) and catalytic rate (kcat) parameters from Michaelis-Menten curves, as mentioned in the Kinetic parameter calculation section. The true inhibitor dissociation constant (Ki) values were derived as described.
Kinetic model
The mechanisms of action for the examined compounds were derived by the analysis of the effects observed on the different equilibriums involved in the enzymatic reaction catalyzed by the HIV-1 RT, as previously described [20]. According to the ordered mechanism of the polymerization reaction, whereby the TP binds first, followed by the addition of dNTP, HIV-1 RT can be present in three different catalytic forms: as a free enzyme (RT), in a binary complex with the TP (RT/TP) and in a ternary complex with TP and dNTP (RT/TP/dNTP). The resulting rate equation for such a system is very complex and impractical to use. For these reasons, the general steady-state kinetic analysis was simplified by varying one of the substrates (either TP or dNTP), while the other was kept constant. When the TP substrate was held constant at saturating concentration and the inhibition process at various concentrations of dNTPs was analysed, at the steady-state all of the input RT was in the form of the RT/TP binary complex and only two forms of the enzyme (the RT/TP and the RT/TP/dNTP) could react with the inhibitor. Similarly, when the dNTP concentration was kept constant at saturating levels and the inhibition at various TP concentrations was analysed, RT was present either as a free enzyme or in the RT/TP/dNTP complex.
Kinetic parameter calculation
Values were calculated by non-least-squares computer fitting of the experimental data to the appropriate rate equations. Steady-state inhibitor binding was analysed according to a non-competitive inhibition mechanism (Equation 1):
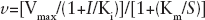
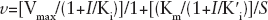
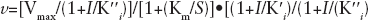
As previously reported, when TP was saturating, K' i .= the affinity for the free enzyme (K i free), whereas at saturating dNTP, K' i = the affinity for the binary complex (K i bin). In all cases, K“ i = the affinity for the ternary complex (K i ter). It follows that, if K' i >K“ i , then both Km and Vmax values will decrease at increasing inhibitor concentrations and the rate equation can be simplified to Equation 1.
The true inhibition constant, K
i
, was calculated for each inhibitor from the different enzymatic forms along the reaction pathway, including free enzyme, binary complex with the TP and ternary complex with both the TP and the dNTP substrates; it was derived from the variations of the apparent K
i
(K
i
app) values as a function of varying concentrations of the TP or dNTP substrates, respectively, as shown in Equation 4:
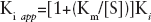
where [S] is the concentration of the variable substrate and Km is the Michaelis constant.
The apparent binding rate values (k
app
) were also evaluated from experimental data through this following exponential equation (Equation 5):
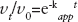
Being [E]0 equal to the input enzyme concentration, [E]t to the enzyme able to react at the time point t, whereas [E:I]
t
is the enzyme bound to the inhibitor at the time point t, it follows that (Equation 6):
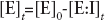
The kon and koff parameters, indicating the association and the dissociation rates of the examined compounds towards the enzyme, respectively, were calculated according to Equations 7 and 8:
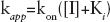
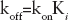
Data analysis and statistics
Data obtained were analysed by linear and/or non-linear regression analysis using GraphPad Software (San Diego, CA, USA).
Antiviral assays
Human CEM cell cultures (3×105 cells/ml) were infected with 100 50% cell culture infectious dose HIV-1 (IIIB) or HIV-2 (ROD) per ml and seeded in 200 μl well microtitre plates, containing appropriate dilutions of the test compounds. After 4 days of incubation at 37°C, syncytia cell formation was examined microscopically in the CEM cell cultures. Cytotoxicity was measured after 4 days of incubation at 37°C in mock-infected cell cultures, by determining the number of viable cells using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide method.
Results
Novel halo- and nitro-IASs are potent inhibitors of HIV-1 in both RT and CEM cells assays
Compounds RS 2911, 1765, 2862, 3084 and 1762 (Figure 1) were tested against HIV-1 RT WT and mutant RTs carrying common drug resistance mutations, such as K103N, L100I and Y181I. In general, the compounds showed reduced potency towards the mutants. However, the loss of potency for compounds RS 2911 and RS 1762 towards the RT mutants K103N and L100I was comparable to the clinically approved efavirenz and TMC-125. Compound RS 3084 showed a resistance profile towards all three mutants comparable to TMC-125 (Table 1). The mutation most affecting the IAS derivatives was the Y181I substitution.
Inhibitory potencies of indolylarylsulfone derivatives against HIV-1 RT WT or carrying drug resistance mutations
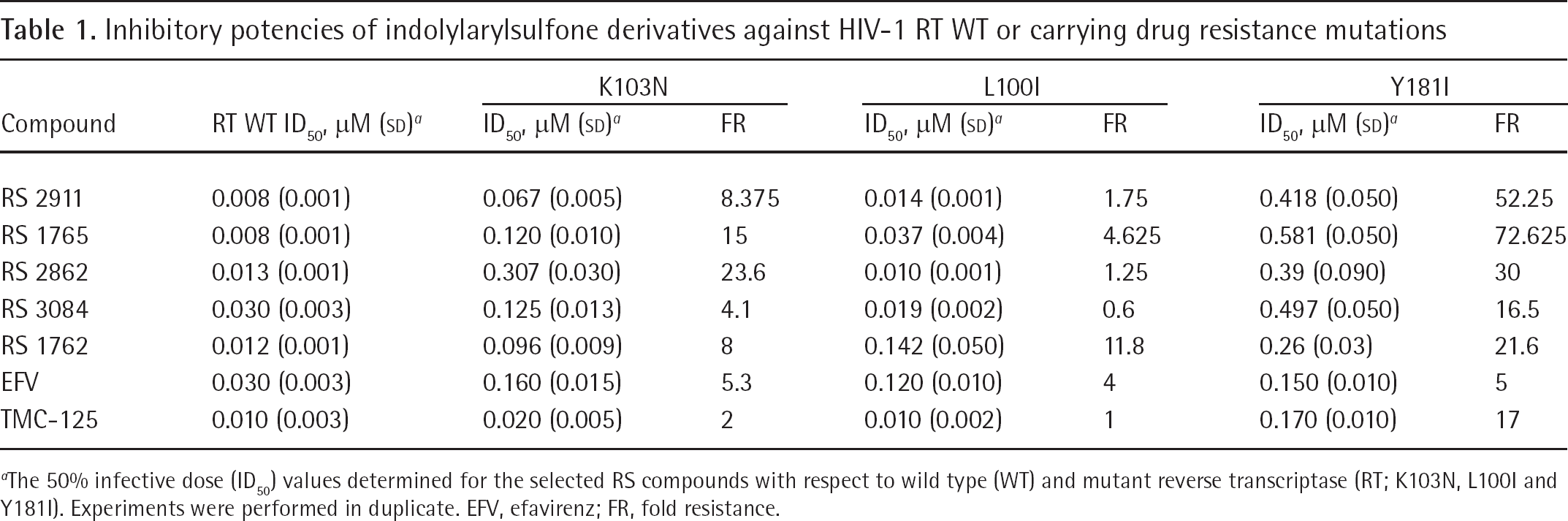
The 50% infective dose (ID50) values determined for the selected RS compounds with respect to wild type (WT) and mutant reverse transcriptase (RT; K103N, L100I and Y181I). Experiments were performed in duplicate. EFV, efavirenz; FR, fold resistance.
Such compounds were also potent inhibitors to HIV-1 (IIIB) in CEM cells, with potencies and selectivity superior to nevirapine and comparable to efavirenz (Table 2).
Antiviral activity of the halo- and nitro-indolylarylsulfone compounds
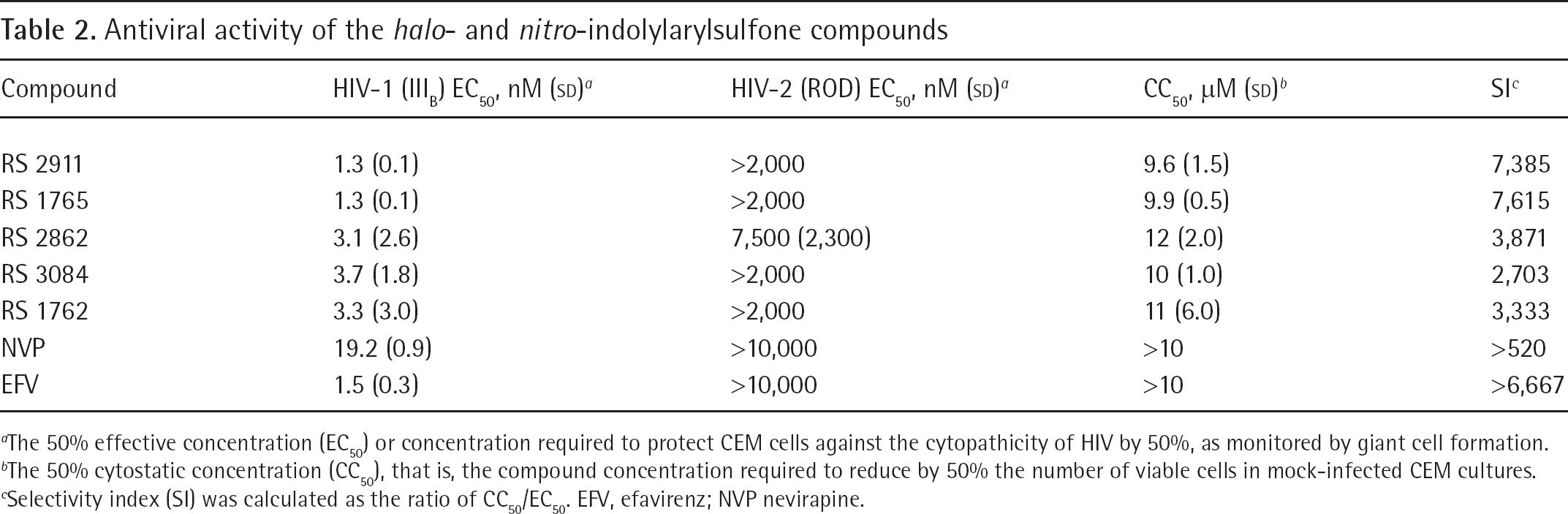
The 50% effective concentration (EC50) or concentration required to protect CEM cells against the cytopathicity of HIV by 50%, as monitored by giant cell formation.
The 50% cytostatic concentration (CC50), that is, the compound concentration required to reduce by 50% the number of viable cells in mock-infected CEM cultures.
Selectivity index (SI) was calculated as the ratio of CC50/EC50. EFV, efavirenz; NVP nevirapine.
Novel halo-IASs show different mechanisms of inhibition towards HIV-1 RT WT, K103N and Y181I
In order to better characterize the mechanism of interaction of the IAS derivatives with HIV-1 RT, the RNA dependent DNA polymerase activity of HIV-1 RT, either WT or mutated (K103N and Y181I), was measured in the presence of fixed concentrations of the inhibitors and variable concentrations of nucleic acid or nucleotide substrate, respectively (Figure 2), in agreement with the simplified kinetic pathway described in Materials and methods. The Km, and Vmax values calculated for each enzyme, are listed in Additional files 1, 2 and 3.
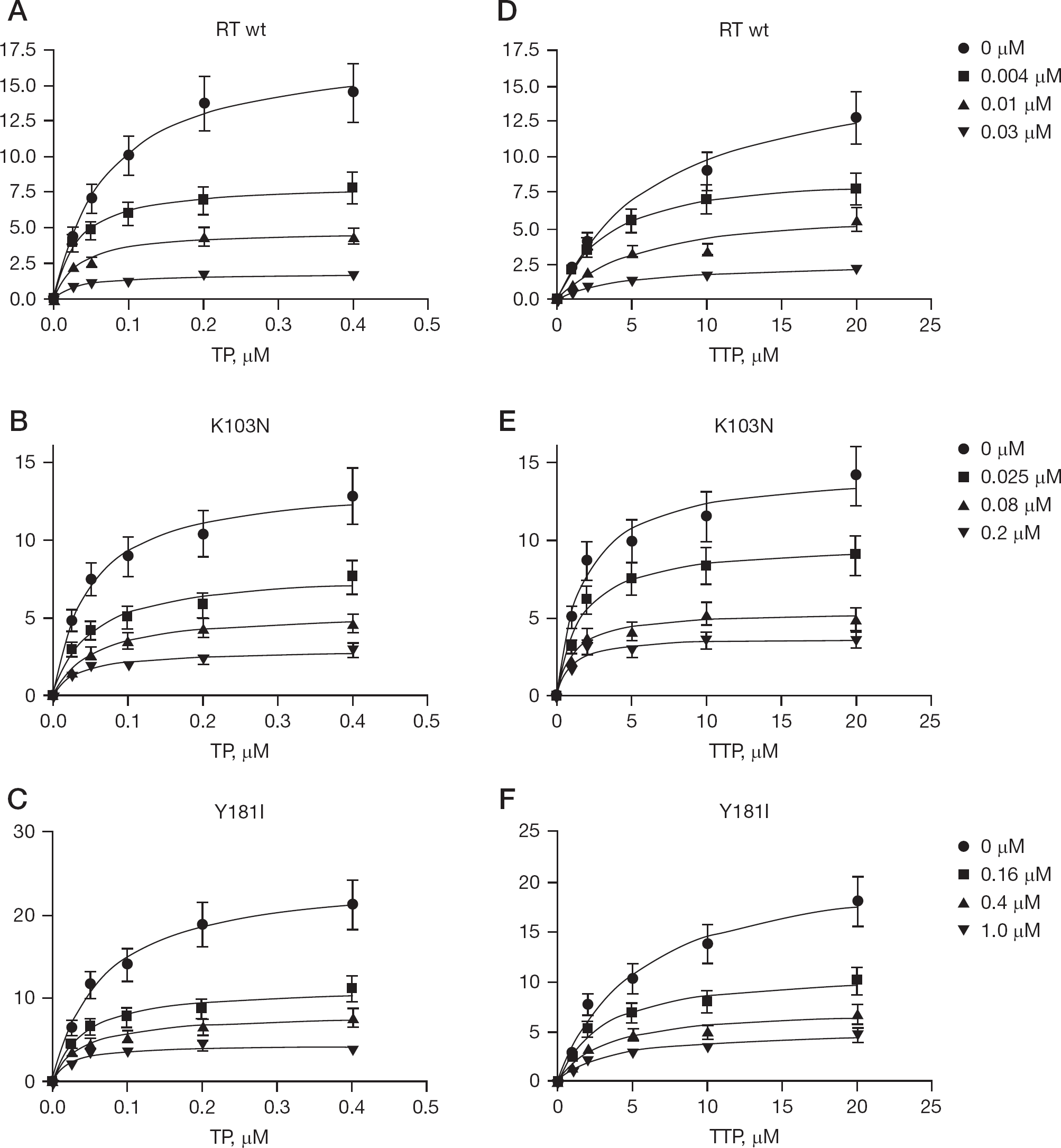
Steady-state kinetic analysis of the inhibition of HIV-1 RT by compound RS 1762
In the case of the WT RT (Additional file 1) the V values, obtained for all the compounds tested, significantly decreased as a function of the inhibitor concentrations, whereas the Km values appeared unaffected, indicating a fully non-competitive mechanism of action, by which the affinity of each inhibitor for the non-nucleoside binding pocket was not influenced by the binding of substrates to the vicinal catalytic site.
The inhibitors exhibited a similar non-competitive behaviour when the enzyme carried the K103N mutation (Additional file 2), with the exception of the compounds RS 2862 and RS 3084. In the latter case, both the Vmax and the Km values significantly decreased with varying either nucleic acid (TP) or nucleotide substrates. The same was observed in the presence of the compound RS 2862, but only as a function of the TP substrate. A decrease in both the Vmax and the Km kinetic parameters, is diagnostic of a so-called uncompetitive mechanism of inhibition. In the most common and simplest case, an uncompetitive inhibitor binds preferentially to one (or more in case of multisubstrate reactions) enzymesubstrate complex along the reaction pathway. This is different from non-competitive inhibition, where the compound binds with equal affinity the enzyme alone or in complex with its substrates. This mechanism was visualized by analysing the data with Lineweaver-Burk double reciprocal plots, where a set of parallel lines was obtained, as expected (Additional file 4).
In the case of the mutant Y181I (Additional file 3), all the inhibitors showed a non-competitive behaviour, with a reduction of the Vmax levels, while the Km values were mostly unaffected, with the exception of RS 1765, which showed a decrease of both parameters as the nucleotide concentrations were varied.
By applying the appropriate rate equations (see Materials and methods), the equilibrium dissociation constant (Ki) values for all the inhibitors were also calculated with respect to the three different enzymatic forms along the reaction pathway. As reported in Table 3, in the case of RT WT, these values were equal among the three catalytic forms of RT (K i fee=K i bin=K i ter), according to the non-competitive mechanism of action shown by all the compounds Concerning the K103N mutant, in the case of RS 2862 we found that K i bin<<K i ter<K i free, indicating a preferential affinity for this inhibitor to the binary RT/TP complex, which appeared approximately 8-fold higher than to the ternary RT/TP/TTP complex and approximately 10-fold higher compared to the free enzyme, clearly suggestive for non-competitive mechanism of inhibition. Similarly, for the compound RS 3084 we observed that K i ter<<K i free<K i bin, suggesting that this the free enzyme and ternary complex (K i ter≈K i free) was calculated by the decrease of Km values as a function of increasing doses of the compound (Figure 3A), while the K i bin was derived by the variations of the apparent equilibrium dissociation constant (K i app ) values as a function of TTP concentrations (Figure 3B). Thus, the presence of the K103N mutation - moreso than Y181I - rendered the three enzymatic forms along the reaction pathway non-equivalent in terms of inhibitor binding.
Kinetic parameters for the interaction of the halo- and nitro-indolylarylsulfone derivatives with the different HIV-1 RT substrate complexes
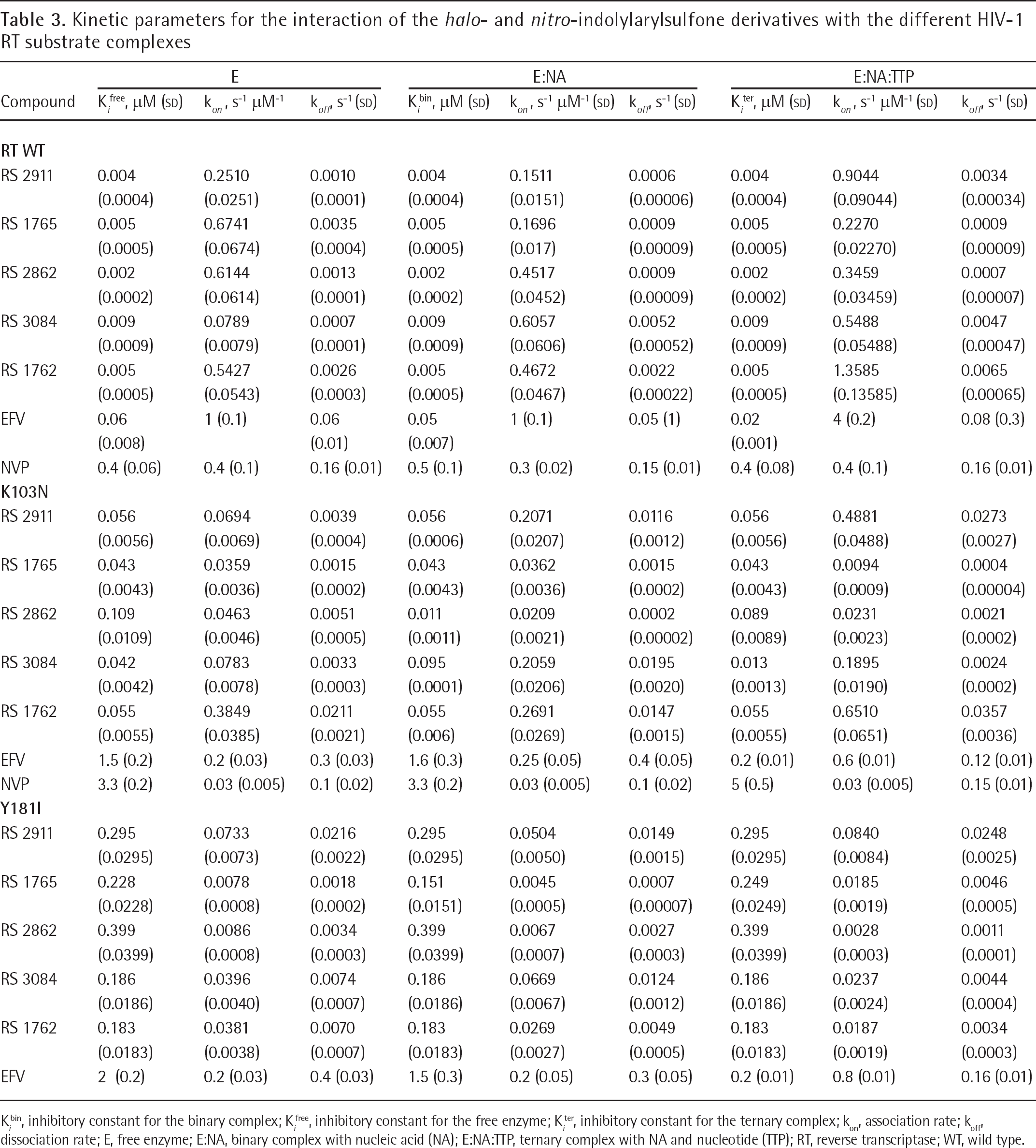
K i bin, inhibitory constant for the binary complex; K i free, inhibitory constant for the free enzyme; K i ter, inhibitory constant for the ternary complex; kon, association rate; koff, dissociation rate; E, free enzyme; E:NA, binary complex with nucleic acid (NA); E:NA:TTP, ternary complex with NA and nucleotide (TTP); RT, reverse transcriptase; WT, wild type.
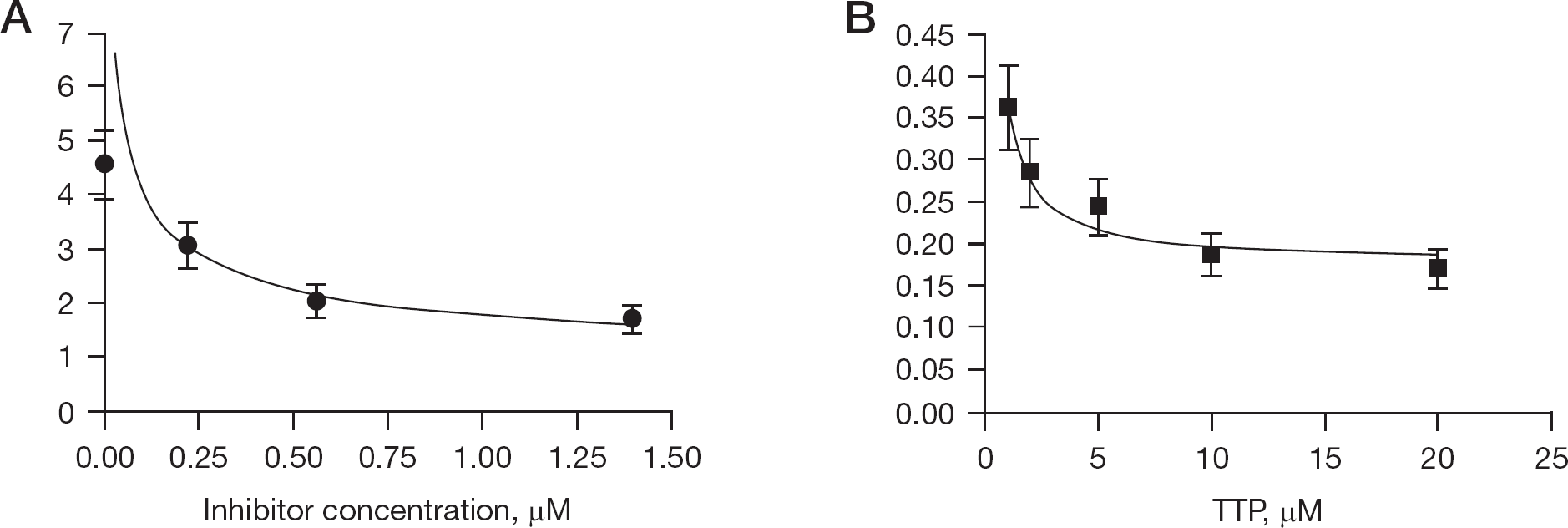
RS1765 is an uncompetitive inhibitor of Y181I HIV-1 reverse transcriptase
Effect of K103N and Y181I resistance mutations on IAS inhibition activity
The relative resistance index (RRI) was calculated as the ratio of the inhibitor dissociation constant toward the mutated enzyme (K i mut) to that of the RT WT (K i WT) for both the K103N and Y181I mutations (Figure 4).
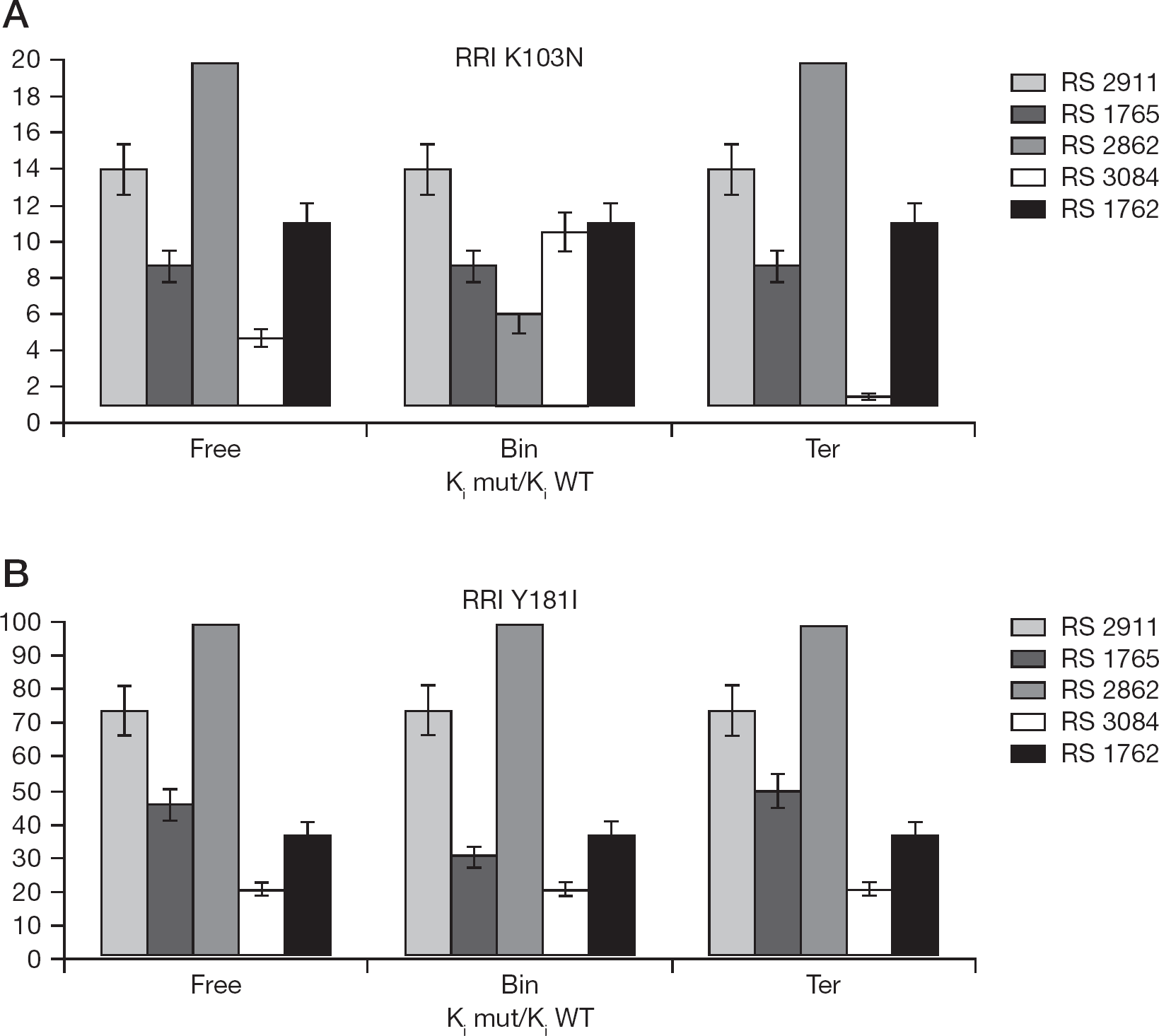
RRIs expressed as the ratio between the equilibrium dissociation constant towards to the mutated enzyme and the one evaluated to RT WT
The higher the RRI value is the lower the inhibitor affinity for the mutant with respect to the WT enzyme. As shown in Figure 4A, the mutation K103N generally reduced the affinities of the compounds tested, as indicated by all RRI>1, but with a different degree of resistance for the various enzymatic forms along the reaction pathway. In particular, compound RS 3084 showed an RRI close to 1 for the interaction with the ternary complex of the K103N mutant. In the case of the Y181I resistance mutation, all the RRI values were significantly higher compared to K103N, although the inhibitor RS 3084 showed the lowest indexes, without any distinction among the three catalytic forms of the enzyme, whereas the compound RS 1765 was more active against the mutant RT combined in the binary complex (Figure 4B).
The K103N and Y181I resistance mutations induce slow binding: Tight binding interactions with the IAS derivatives to the viral RT
We next evaluated the association (kon) and dissociation (koff) rates of the different inhibitors towards the free enzyme, the binary complex with the nucleic acid and the ternary complex with both the nucleic acid and the nucleotide substrates, for WT RT, K103N and Y181I (Table 3). Since Ki =kof/kon an increase of the K i value (resistance to inhibition) may be associated with a decrease in kon values (slower association) or to an increase in the koff values (faster dissociation) of the inhibitor. The relative association index (RAI; RAI=K on WT/k on mut) or relative dissociation index (RDI; RDI=K off mut/k off WT) was thus calculated. With RAI or RDI values >1, the inhibitor either binds slower to the mutated enzyme or dissociates from it more rapidly than in the case of WT RT. Conversely, RAI or RDI values <1 indicate a major selectivity (faster binding and/or slower dissociation) of the inhibitor to the mutant enzyme with respect to the WT.
As shown in Figure 5, both the K103N and the Y181I mutations generally decrease the association rates of most of the compounds to the enzyme, being the RAI>1. However, the inhibitor RS 2911 showed RAI<1 for the binary complex of K103N and modestly reduced association velocities (two- to threefold) with respect to the ternary and free enzyme. Compound RS 1762 also showed RAI close to 1 towards all the enzymatic forms, while compound RS 3084 showed association rates to the substrate-conjugated forms of the enzyme only approximately two- to threefold lower as compared to the correspondent WT forms.
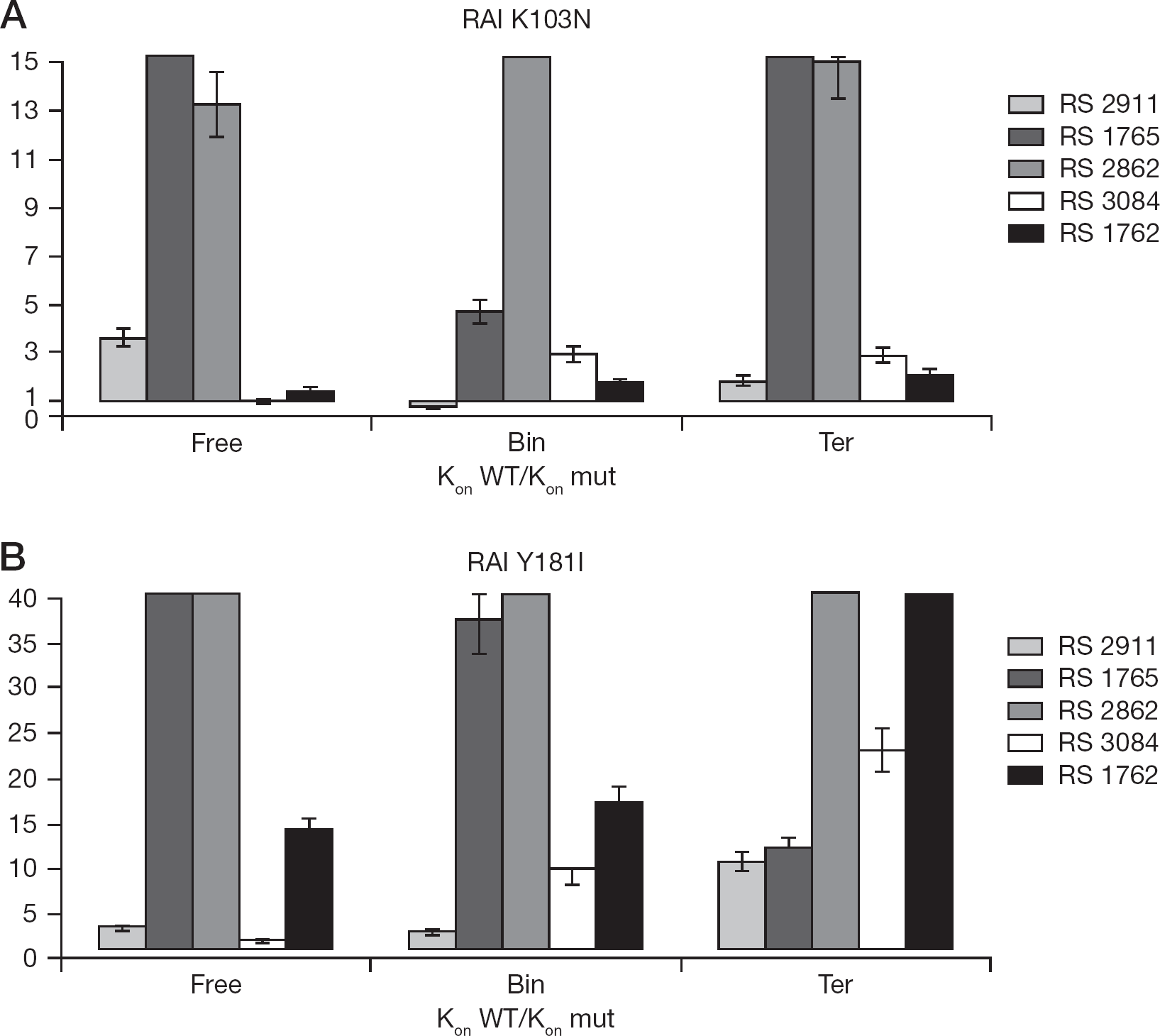
RAIs expressed as the ratio between the association rate towards RT WT and the one evaluated to the mutated enzyme calculated for the examined compounds
In the case of the Y181I mutant, compound RS 2911 showed higher association rates to the binary and the free mutated enzyme, while inhibitor RS 3084 seemed to preferentially associate to the free enzyme, with respect to the binary and ternary complexes.
When the dissociation rates from both the mutated enzymes were determined and compared to the corresponding values for RT WT (Figure 6), in the presence of the K103N mutation, compound RS 1765 showed RDI<1 towards to the free and ternary forms and RAI close to 1 with the binary enzymatic form. Inhibitor RS 3084 seemed to bind more stably to the ternary complex of K103N, with its RDI<1, and showed four- to fivefold increased dissociation rates from the corresponding free and binary enzyme. Compound RS 2862, which had previously shown higher association rates to WT RT than to both the mutants with respect to all the enzymatic forms, appeared to dissociate slower from K103N in the binary complex than from WT (RDI<1).
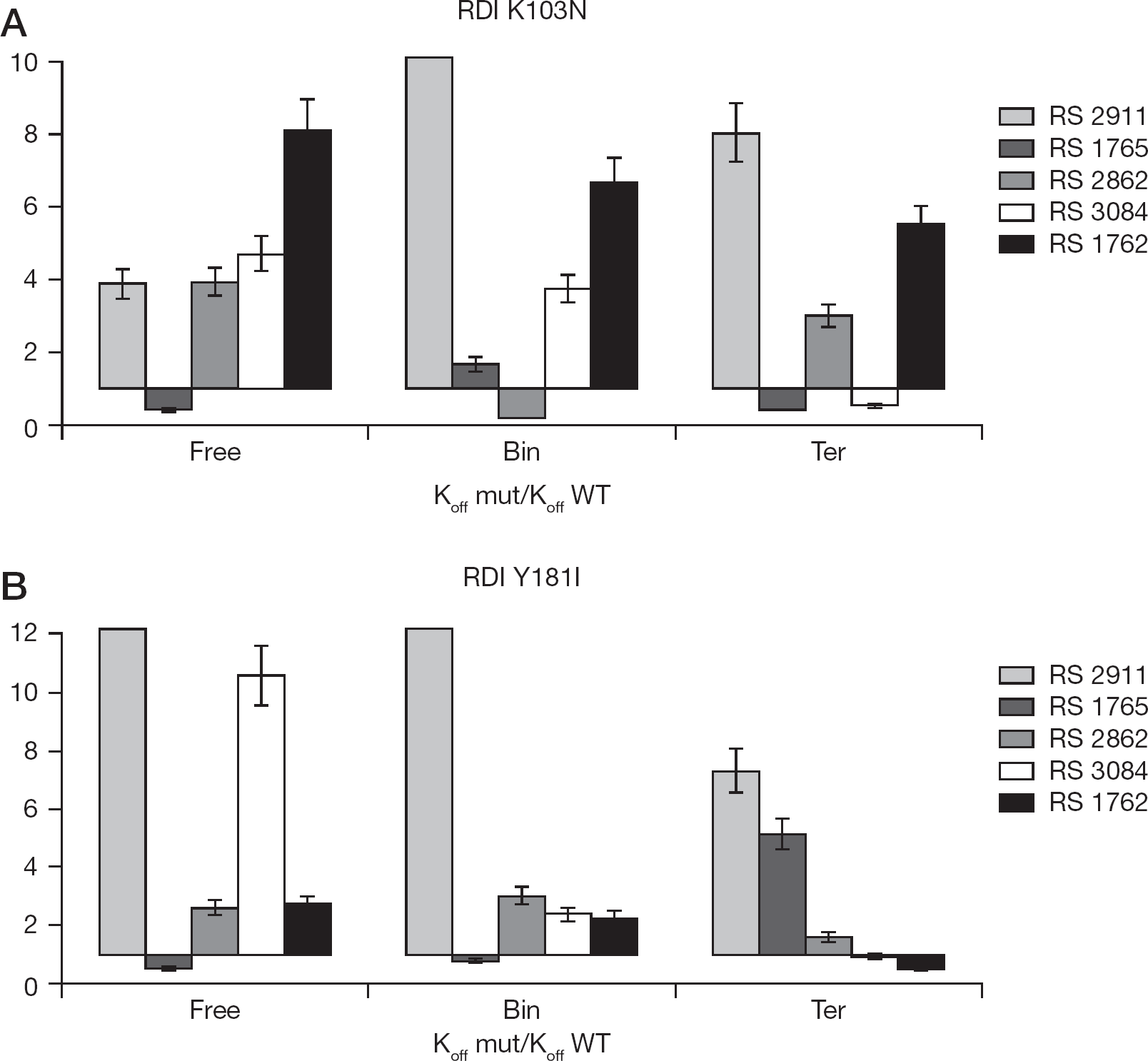
RDIs expressed as the ratio between the dissociation rate towards to the mutated enzyme and the one to RT WT
When the RDIs for the Y181I mutant were calculated, compound RS 1765 showed RDI<1 for both the free and binary enzymatic forms, while the RDI value for the ternary enzyme was only slightly increased. The dissociation rates for the inhibitor RS 2862 did not significantly differ among the three catalytic forms of the Y181I mutant and all the RDI values appeared very close to RT WT (RDI⩽2). Interestingly, the inhibitor RS 1762 showed comparable dissociation velocities (RDI⩽2) between RT WT and Y181I, in case of the free enzyme and the binary complex, while its RDI value was <1 when both substrates were bound (ternary complex). Collectively, these data indicate that the Y181I mutation, more clearly than the K103N, causes resistance to the IAS derivatives by reducing their association rates (slow binding), while these compounds were able to partially compensate this reduced binding by making very stable interactions with the mutated NNRTI-binding pockets (tight binding), even stronger than with the WT enzyme. This dual slow binding-tight mode of interaction was specific for these inhibitors.
Discussion
IAS NNRTIs have proven to be very effective in suppressing viral replication of HIV-1 WT and some mutated viral strains, even better than efavirenz [13,15,16,19]. In the present study we characterized the mechanisms of inhibition of five novel halo- and nitro-substituted IAS derivatives (RS compounds; Figure 1) bearing a (cycloalkylamino)methyl group at the indole-2-carboxamide nitrogen, with respect to the three different mechanistic forms of the HIV-1 WT and the mutated RTs carrying K103N and Y181I mutations.
The binding kinetic analysis revealed that, while the association rates of these compounds to the viral RT were reduced by the K103N mutant, the IAS derivatives were nonetheless able to achieve a stable binding thanks to slower dissociation rates from the mutated RT with respect to the WT enzyme. Such a reduction, however, was strongly influenced by the presence of the bound substrates. For example, compound RS 2862 showed a fivefold slower dissociation rate of the inhibitor from the mutant enzyme in complex with the TP substrate, with respect to the equivalent complex with RT WT. Similarly, RS 3084 showed a twofold slower dissociation rate of this compound from the ternary complex of the mutated enzyme with respect to RT WT. In the case of the Y181I mutant, compound RS 1765 showed a twofold reduced dissociation rate from the mutant than from the WT when bound with the TP substrate.
It is known, thanks to several crystal structures, that binding to RT of either NNRTIs or the nucleic acid and nucleotide substrates, can induce conformational changes in the enzyme [3,–7]. The NNBS itself is not accessible in the apoenzyme structure (‘closed pocket form’). Binding of NNRTIs induce flipping of the aromatic sidechains of Y181 and Y188, along with displacement of the β9-β10 loop in the palm and the β12-β13 hairpin in the fingers. In particular, NNRTI binding seems to stabilize an extended conformation of the fingers and thumb subdomains [6]. Binding of the nucleic acid induces a shift in the p66 thumb subdomain, from a closed into an open conformation. Subsequent binding of the nucleotide causes the fingers to close down on the active site. Thus, substrate binding could influence the architecture of the NNBS. If an ‘open conformation’ of the NNRTI binding site is present in one of the intermediate structural states of RT during the polymerization process, it is possible that some NNRTIs can show preferential interaction with such an intermediate [21]. Our kinetic data suggest that the conformation of the binary RT-TP and ternary RT-TP-dNTP complexes, with the thumb subdomain already in an open conformation, might present a more favourable network of interactions with the IAS derivatives at the level of the NNBS.
In summary, the novel IAS derivatives described here showed nanomolar inhibitory potencies against the NNRTI-resistant K103N and Y181I HIV-1 RT mutants. In addition, they showed slower substrate-dependent dissociation rates from these mutated enzymes than from WT RT, suggesting that they can achieve more stable binding interactions to the mutated NNBS site with respect to the WT form. These results provide the proof-of-principle for the design of new mutant-specific NNRTIs.
Footnotes
Acknowledgements:
This work has been partially supported by the ISS AIDS Grant Contract number 40H26 to GM and by ‘Franca Rame and Dario Fo’ Nobel Foundation grant in aid to AS. Authors from Sapienza University thank Istituto Pasteur - Fondazione Cenci Bolognetti (Grant 2009) and FILAS (Finanziaria Laziale di Sviluppo, Grant 2010) for financial support.
The authors declare no competing interests.