
Abstract
HCV infection is a significant worldwide health problem and is a major cause of hepatocellular carcinoma. The current standard of care, interferon and ribavirin, is only effective against a proportion of the patient population infected with HCV. To address the shortcomings of existing therapy, the development of direct acting antiviral agents is under investigation. The HCV RNA dependent RNA polymerase is an essential enzyme for viral replication and is therefore a logical target against which to develop novel anti-HCV agents. Nucleosides have been shown to be effective as antiviral agents for other viral diseases and therefore, have been investigated as inhibitors of HCV replication. The development of prodrugs of nucleoside 5′-monophosphates has been pursued to address limitations associated with poor nucleoside phosphorylation. This is required to produce the nucleoside 5′-triphosphate which is the anabolite that is the actual inhibitor of the polymerase enzyme. Prodrugs of nucleoside 5′-monophosphates have been developed that enable their delivery into cells and in vivo into the liver. The implementation of these prodrug strategies has ultimately led to the identification of several prodrugs of nucleoside 5′-monophosphates that are potent inhibitors of HCV replication in vitro. They have progressed into the clinic and the early data demonstrate greatly reduced viral load levels in HCV-infected patients. This review will survey the state of nucleotide prodrugs for the treatment of HCV.
Introduction
HCV is known to have infected approximately 180 million individuals worldwide [1]. It is estimated that of those infected with HCV approximately 80% will develop chronic liver disease and a significant proportion of those infected will eventually develop liver cirrhosis and subsequently hepatocellular carcinoma [2]. HCV is a single-stranded, positive sense RNA virus of the Flaviviridae. Six major viral genotypes with over 100 viral subtypes have been identified for HCV. Genotypes 1a and 1b are the most prevalent genotypes in the western world with genotypes 2 and 3 comprising 20–30% of this population. HCV genotypes 2–6 predominate in the developing world [3,4]. Because HCV replicates in the cytoplasm of infected cells by a membrane associated replication complex and the virus has an RNA genome with no DNA intermediate during replication, no genomic templates are stably integrated into the host genome. Therefore, virological cures are possible for HCV patients. This is in contrast to other viruses such as HIV and HBV where the viral genome is integrated into the host DNA and a virological cure is considered remote. However, for HCV-infected patients virological cures are made difficult due to the high rate of HCV viral replication and by the high spontaneous mutation rate of the virus. This high mutation rate is a result of poor replication fidelity exhibited by the HCV polymerase and an apparent lack of proof reading [4].
The current therapy for treating chronic HCV infection consists of regular injections of α-interferon (IFN) with daily oral administration of ribavirin (RBV). This standard of care (SOC) regimen does not act by directly attacking the virus but functions by boosting the host immune response. For genotype 1 patients regular IFN/RBV treatments for 48 weeks result in only 40–50% of patients achieving a sustained virological response (SVR) indicative of a cure [5,6]. However, for genotype 2 and 3 patients the SVR rates can be as high as 75%. It is also known that subpopulations which include individuals of African ancestry tend to respond less well to IFN/RBV treatments [7]. Recent genome-wide association studies have shown that a single nucleotide polymorphism 3kb upstream of the IL28B gene correlates with a significant difference in response to IFN therapy [8]. IL28B which encodes the type III interferon IFN-λ—3, is known to be upregulated by IFNs and by RNA viral infections. It has been shown that HCV patients who harbour a TT or TC allele in their IL28B gene tend to respond less well to IFN/RBV treatment than do those having the CC genotype.
Patients who choose to undergo IFN/RBV therapy face not only the possibility of not responding to treatment but also must contend with the potential for multiple and sometimes serious side-effects that include influenza-like symptoms, fatigue, hemolytic anaemia and depression. The intolerable side effects can result in a high rate of drug discontinuations. Consequently, the modest cure rates and subpopulation differences combined with the side-effect profile for SOC have prompted an urgency to develop alternative novel, safe and effective therapies. As in the case of other viral diseases, the development of direct acting antivirals (DAAs) has become a focus. Development of small molecule agents to attack essential viral proteins has the potential benefit of reducing toxicities and side effects associated with manipulating host functions and hopefully such DAA therapies will not have the intolerable side effects exhibited by current SOC.
The push to identify small molecule DAAs has also prompted the discussion around the possibility of eliminating or at least reducing the use of IFN/RBV from treatment regimens. Although clinical development of first generation DAAs has focused on combinations with SOC in the hope of both shortening duration of treatment and increasing the cure rate, the long-term desire is to completely eliminate the use of IFN/RBV from treatment regimens. Clearly this is an aspirational goal and a goal that can only be achieved if an immune component of therapy is not absolutely required to eliminate those vestiges of undetectable virus [9]. In addition, such a lofty goal can be realised if small molecule DAAs either alone or more probably in combination can drive viral loads to undetectable limits and maintain those undetectable levels over the necessary course of therapy and after cessation of treatment without having viral breakthrough resulting from the emergence of resistant virus. As has been shown with HIV highly active antiretroviral therapy (HAART), the HCV treatment paradigm will likely require combinations of anti-HCV agents [7]. The desire is to suppress virus as rapidly and completely as possible in order to give the body's natural immune system the opportunity to clear residual virus and to hold back emergence of resistant virus. However, what will comprise those ideal combinations of DAAs is yet to be determined and studies to clarify this question are under active discussion and investigation.
HCV has a 9.6 kb genome of positive-stranded RNA. This genome encodes a precursor polyprotein that is processed into 10 functional proteins: three structural proteins and seven non-structural proteins [10]. Several of the non-structural proteins have been the focus of intensive efforts to identify small molecule DAA agents as inhibitors of HCV replication. Of the seven non-structural proteins, molecules that inhibit the functions of the NS3/4 protease, NS4A, NS4B, NS5A and NS5B RNA dependent RNA polymerase (RdRp) have advanced to the clinic [11,–15]. The most advanced agents are the NS3/4 protease inhibitors telaprevir and boceprevir. Each of these compounds has completed Phase III clinical investigation and both have been shown to be efficacious in treating HCV infection when given in combination with SOC. However, each of these first generation protease inhibitors suffers from the lack of genotype coverage, undesired side effects, that may limit their usage, and the early emergence of resistant virus. It is therefore not surprising that even with the potential benefits of these first generation DAAs, warehousing of patients by physicians occurs in order to wait for approval of more effective and tolerable agents.
The HCV RNA dependent RNA polymerase is an ∼68 kd protein that has the typical palm-finger-thumb structural motif found in many viral polymerases (Figure 1) [16,17]. HCV polymerase is an essential enzyme involved in RNA replication. Phylogenetic analysis shows a 65% homology of HCV RdRp across genotypes and an 80% homology within a particular genotype [3]. The HCV polymerase active site is located in the palm domain where the conserved aspartic acid residue-containing GDD motif is located [18]. This conserved GDD motif is common to viral polymerases in general [18]. Through a divalent metal ion (Mg++ or Mn++) the GDD motif functions to coordinate the binding of the ribonucleoside triphosphate. The HCV polymerase catalyzes the addition of a single ribonucleoside triphosphate monomer to the 3′-end of the growing RNA chain by the formation of a 3′,5′-phosphodiester linkage. To accomplish this process, the polymerase must simultaneously bind a template RNA strand, a primer RNA strand and a ribonucleoside triphosphate monomer [19,20]. Therefore, investigation of nucleoside analogues is a rational choice for the development of inhibitors of the HCV NS5B polymerase.
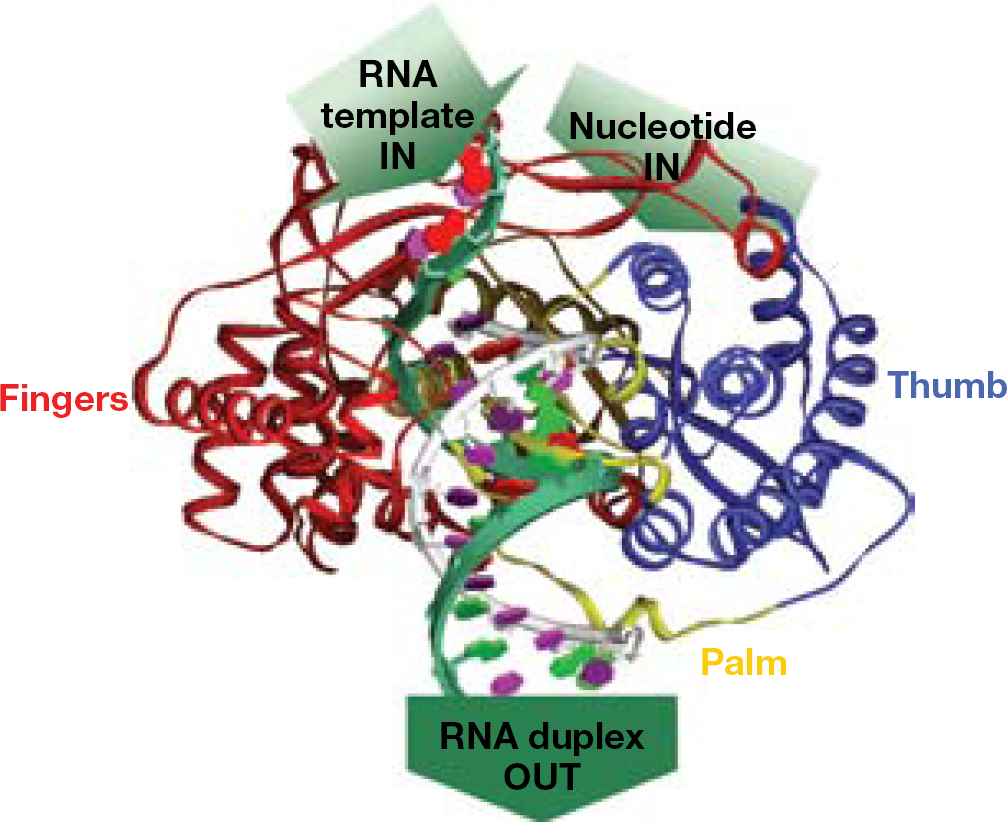
Model of HCV NS5B RNA complex
Of all the DAAs under clinical investigation, nucleoside/nucleotide NS5B polymerase inhibitors hold the promise of pan-genotype coverage and a high barrier to development of resistant virus. As in the case of HIV infection where nucleosides have become the backbone of therapy (for example, TRUVADA® and Combivir®), HCV nucleosides/nucleotides are positioned to assume a similar role. To date, only nucleosides/nucleotides have demonstrated broad genotype coverage both in the laboratory and in human clinical studies [21]. In addition, to date, no pre-emergent resistant virus has been detected in clinical studies [22]. It is for these reasons that nucleosides/nucleotides are positioned to play a prominent role in developing HCV treatment paradigms.
The HCV polymerase has been shown to be a uniquely selective polymerase as it relates to the development of nucleoside/nucleotide inhibitors. Over the last 10 years only two broad classes of nucleosides have emerged as inhibitors of this polymerase [23–25]. These include the 2′-methyl and the 4′-azido classes (Figure 2). However, these classes and subgroups within these classes have clearly differentiated themselves in preclinical and clinical studies. This differentiation is exemplified in their viral selectivity, viral resistance, overall safety and clinical efficacy profiles. Resistance associated with the 2′-methyl class of nucleosides is associated with the S282T amino acid alteration located in the finger domain of the HCV polymerase [26–29]. This mutation has been shown to be difficult to raise in vitro and has not been detected as a pre-existing mutation in clinical isolates [22]. Similarly, for the 4′-azido class, the S96T amino acid alteration has been identified in vitro but has not been observed in the clinic [27].
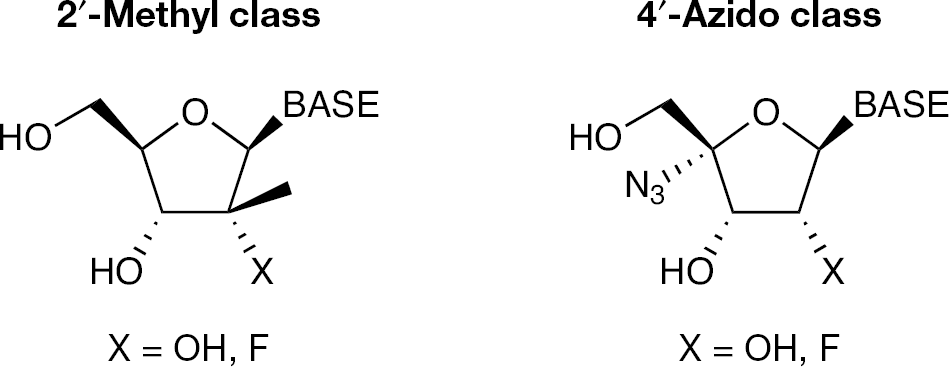
Two major classes of nucleosides that are known to be inhibitors of HCV polymerase.
Implementation of a prodrug strategy has played a prominent role in the development of a number of nucleosides and nucleotides for the treatment of HCV infection [23,30]. These prodrugs have been developed to overcome, not only, bioavailability and stability issues but also to address key anabolism limitations important to nucleoside activation. Simple ester prodrugs of both the 2′-methylcytidine, 2′-α-fluoro-2′-β-C-methylcytidine and 4′-azidocytidine nucleosides were developed to overcome both bioavailability issues and to curb undesirable metabolism [21,31,32]. Prodrugs of the phosphate group of nucleoside 5′-monophosphates were developed to address not only bioavailability issues but also poor in vitro and in vivo conversion of the parent nucleoside to the active nucleoside 5′-triphosphate [33]. Because nucleosides must be converted to their 5′-triphospates to be active as inhibitors of the HCV polymerase, they need to undergo a series of phosphorylation steps catalyzed by three separate kinases (Figure 3). These kinases convert the nucleoside first to the monophosphate, then to the diphosphate and finally to the active triphosphate. However, it is not uncommon that in the phosphorylation cascade, a nucleoside or its corresponding mono- or diphosphate is a poor substrate for one of the kinases. In particular, it is the first kinase in the phosphorylation cascade that is generally the most substrate selective. Therefore, it is not unusual that bypassing the first kinase results in achieving high levels of the active triphosphate. Because nucleoside monophosphates are enzymatically dephosphorylated and negatively charged, they do not readily enter cells and therefore are not desirable as drug candidates. To overcome the limitations of administering a nucleoside monophosphate-containing agent, prodrugs of the 5′-monophosphate nucleoside have been employed. Prodrugs of nucleoside monophosphates have been known for many years and a number of phosphate prodrug strategies have been developed to address the need to deliver a 5′-monophosphate nucleoside into the cell [33–35]. However, there have been few examples where a nucleotide prodrug has been shown to deliver the corresponding 5′-monophosphate in vivo to the desired site of action [33]. Often the prodrug moiety decomposes prior to achieving its objective because of either chemical or enzymatic instability in the gastrointestinal tract and/or plasma.
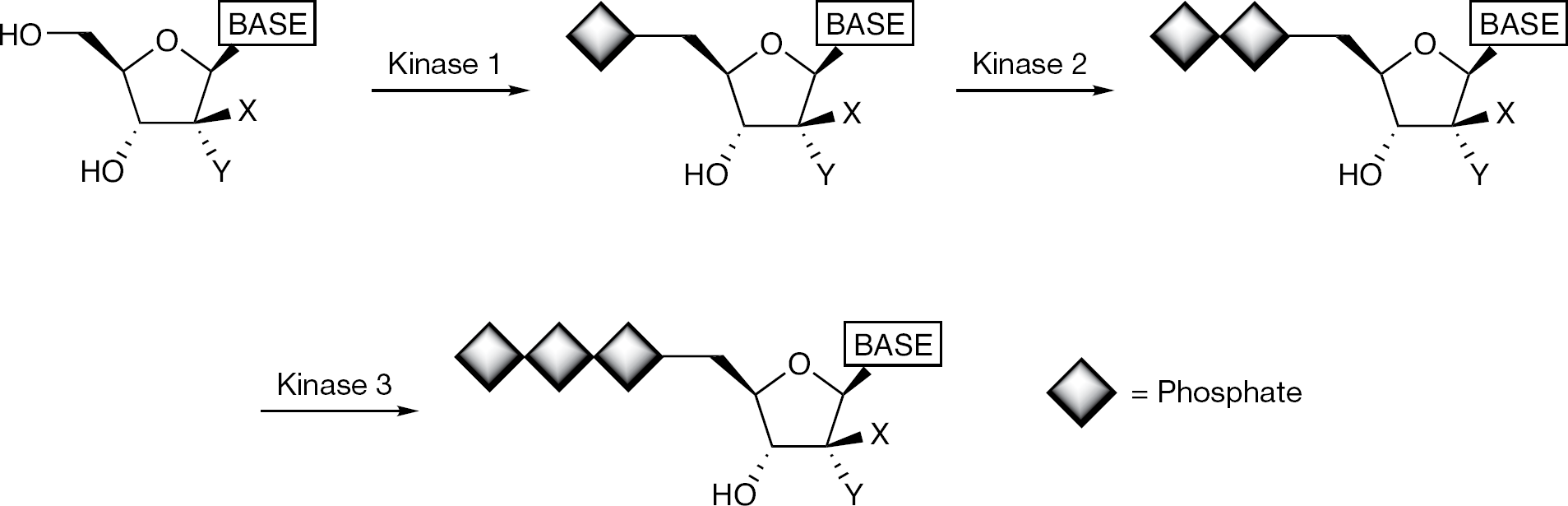
Nucleoside kinase activation pathway
The development of a nucleoside phosphate prodrug useful for the treatment of HCV faces several challenges. The nucleoside phosphate prodrug must have sufficient chemical stability to be formulated for oral administration. It must be stable to conditions of the gastrointestinal tract such that the prodrug reaches the site of absorption intact. The prodrug must have good absorption properties and must not undergo appreciable enzymatic degradation during the absorption phase. Once absorbed, the prodrug needs to have sufficient stability in the blood in order to reach the target organ: for example the liver in the case of HCV. The prodrug must then be transported into hepatocytes and release the free 5′-monophosphate nucleoside which can subsequently be converted to the active triphosphate derivative. Since HCV is a disease of the liver, and the liver is the first organ the prodrug encounters after absorption, HCV is an ideal disease for which to develop a targeted nucleotide prodrug strategy. Consequently, several of these nucleoside monophosphate prodrugs have advanced to the clinic and have demonstrated proof of concept for treating HCV. Here, the application of phosphate prodrugs in the development of nucleotide inhibitors of HCV and the status of nucleotide prodrugs under investigation for the treatment of HCV infection will be reviewed.
Nucleotide phosphoramidates
Nucleotide phosphoramidates were first disclosed by McGuigan et al. [36] as a prodrug strategy to deliver a nucleoside 5′-monophosphate for the treatment of HIV and cancer. The structure of a nucleotide phosphoramidate typically consists of a nucleoside 5′-monophosphate where the phosphate group is masked by appending an aryloxy group (usually a phenol) and an α-amino acid ester (Figure 4); however, other related constructs have also appeared. The phosphate group is ultimately revealed by a sequence of enzymatic and chemical steps that requires either carboxyesterase or cathepsin A to cleave the terminal amino acid ester, intramolecular displacement of the phosphate phenol and then enyzmatic cleavage of the amino acid moiety by a phosphoramidase or histidine triad nucleotide-binding protein 1 (HINT 1) [37–39]. It is believed that the phosphoramidate prodrug construct increases lipophilicity of the nucleoside 5′-monophosphate and therefore increases cellular permeability and ultimately intracellular nucleotide concentrations. Since the phosphoramidate prodrug moiety contains a chiral phosphorus centre, issues arise with regard to development of a compound that consists of a mixture of isomers with implications arising from differential activity of each of the isomers, pharmacokinetics and manufacturing optimization, etc. In addition, the typical phosphoramidate contains a phenolic substituent that is released during metabolism to the free monophosphate. Successful development also considers the metabolic release of this phenolic substituent. Selective examples employing the phosphoramidate strategy to achieve kinase bypass for nucleoside inhibitors of HIV reverse transcriptase (RT) inhibitors, for example, ddA, d4T and d4A, showed that in vitro whole cell enhancement in potency could be achieved [40–42]. Although the phosphoramidate strategy was explored extensively to deliver nucleotides for the treatment of HIV and colon cancer [33,36], proof of concept in the clinic has yet to be reported. However, the phosphoramidate prodrug approach has proven to be a valuable strategy in the development of HCV nucleotide therapy.
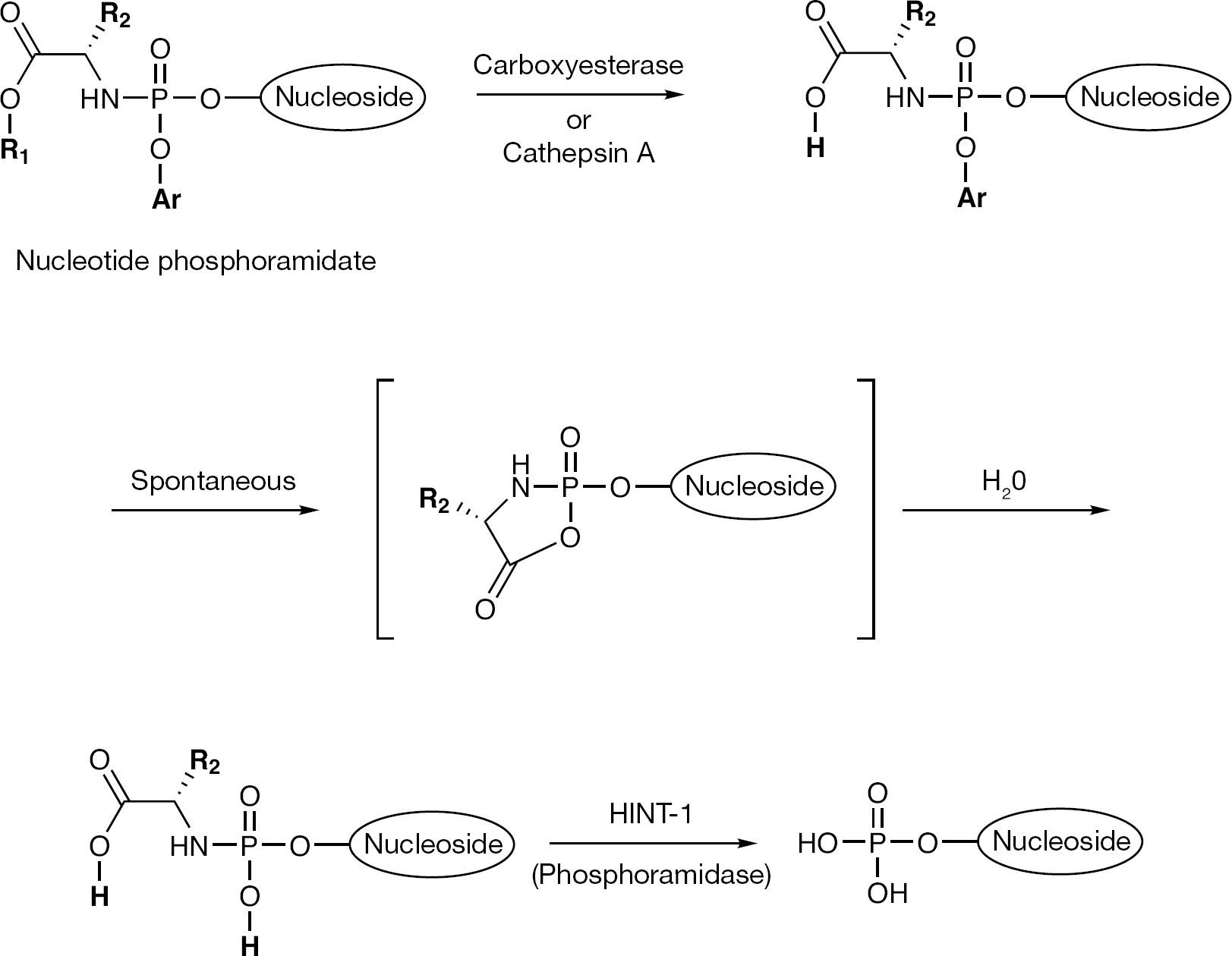
The phosphoramidate prodrug decomposition pathway that results in the release of the nucleoside 5′-monophosphate
2′-C-Methyl ribonucleotide phosphoramidates
The 2′-C-methylcytidine nucleoside, NM107 (
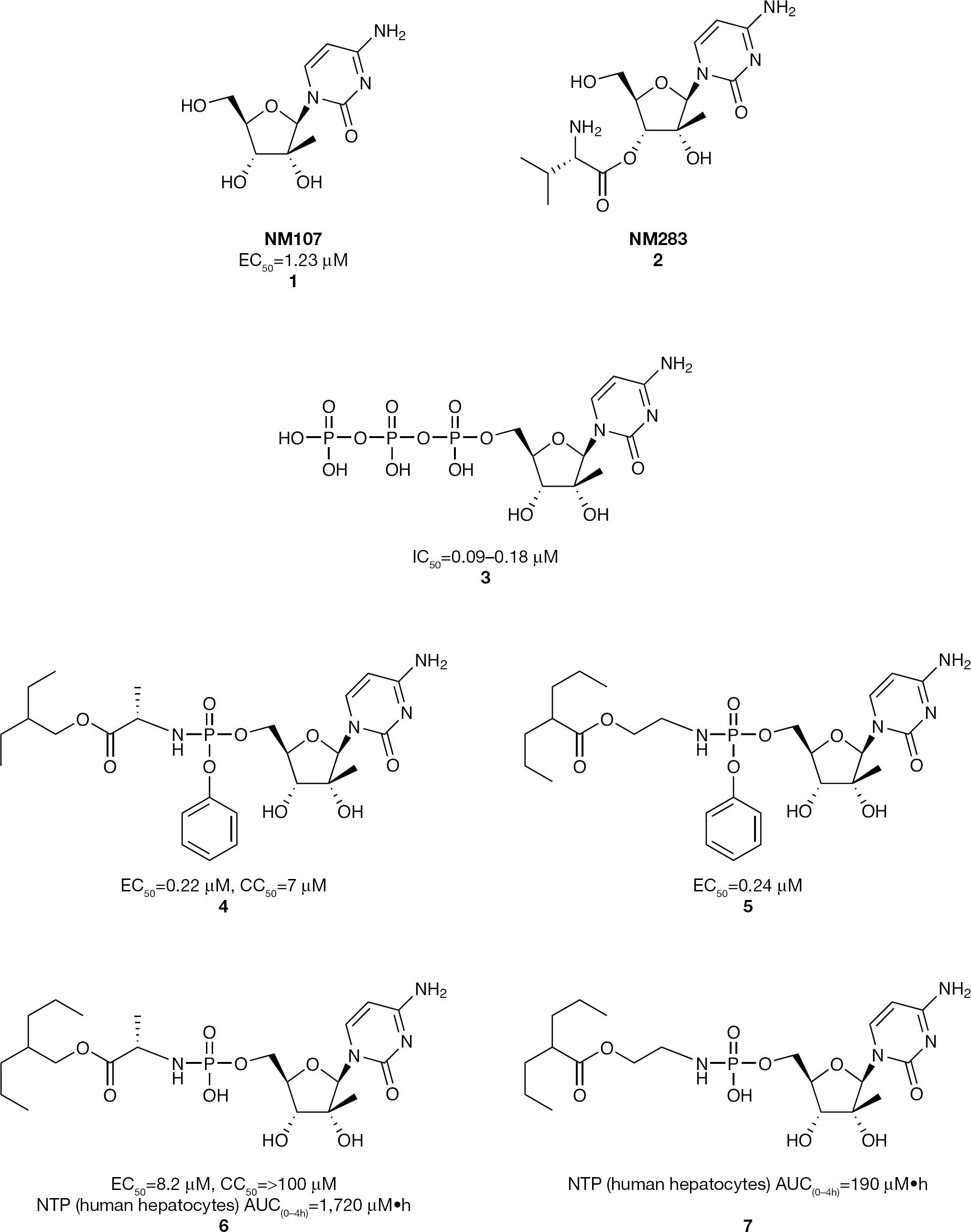
2′-C-Methylcytidine nucleosides and nucleotide phosphoramidate prodrug inhibitors of HCV replication
Subsequent work on the development of the 2′-C-methyl class of nucleosides focused on 5′-phosphate nucleotide prodrugs. It was observed that the 2′-C-methylcytidine triphosphate (
Another series of 2′-C-methylcytidine phosphoramidate prodrugs having an acyloxyethylamino phosphoramidate promoiety was studied (Figure 5) [46]. The phosphoramidate prodrug
Phosphoramidate monoesters of 2′-C-methylcytidine were also explored (Figure 5) [45]. In this case the ami-date moiety was either an α-amino acid (
Other 2′-C-methyl nucleosides containing purine bases were also investigated as inhibitors of HCV, including the 7-deaza-2′-C-methyladenosine derivative MK0608 (
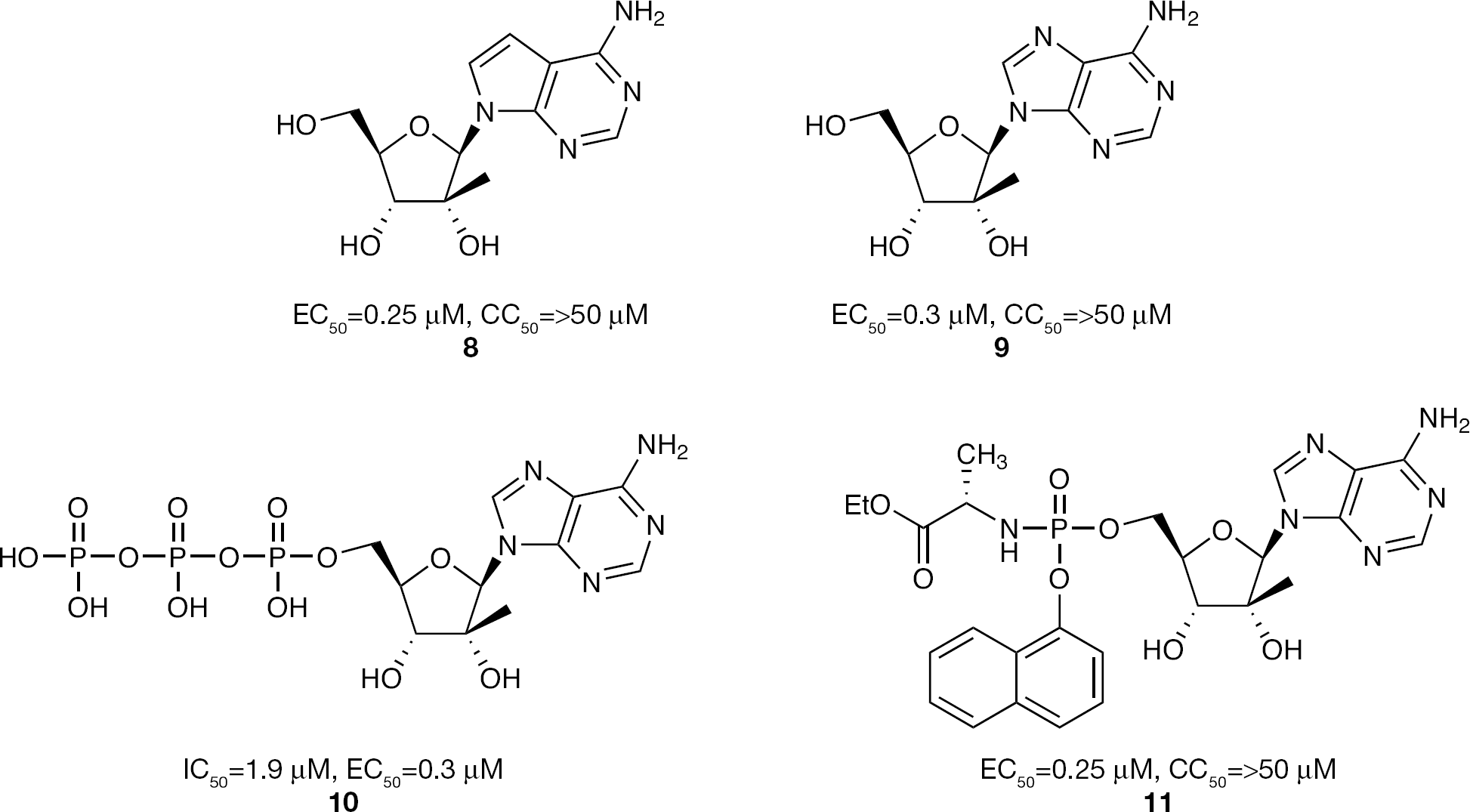
2′-C-Methyladenosine nucleoside and nucleotide phosphoramidate prodrug inhibitors of HCV replication
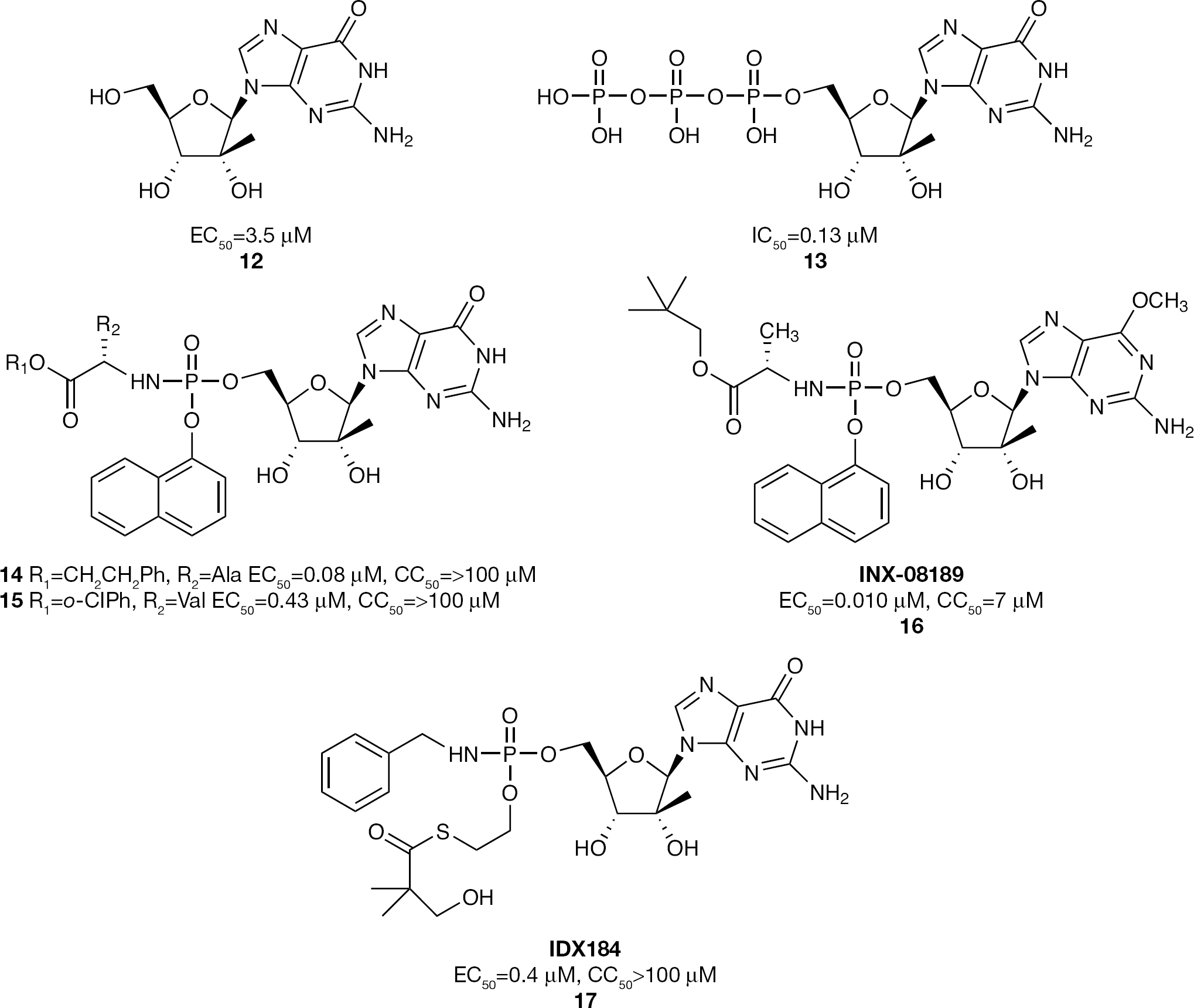
2′-C-Methylguanosine nucleoside and nucleotide phosphoramidate prodrug inhibitors of HCV replication
Further investigation of the 2′-C-methylguanosine phosphoramidate series led to the evaluation of substitution at the C-6-position of the guanosine base with the intention of increasing lipophilicity and thus improving cellular uptake relative to the natural guanosine derivative
In a single-ascending-dose Phase Ia study in healthy volunteers administered doses ranging from 3 mg to 100 mg, INX-08189 (
Another application of phosphoramidate prodrug technology for delivering a 2′-C-methylguanosine 5′-monophosphate is exemplified by IDX184 (
4′-Azido ribonucleotide phosphoramidates
The 4′-C-azidocytidine nucleoside R1479 (
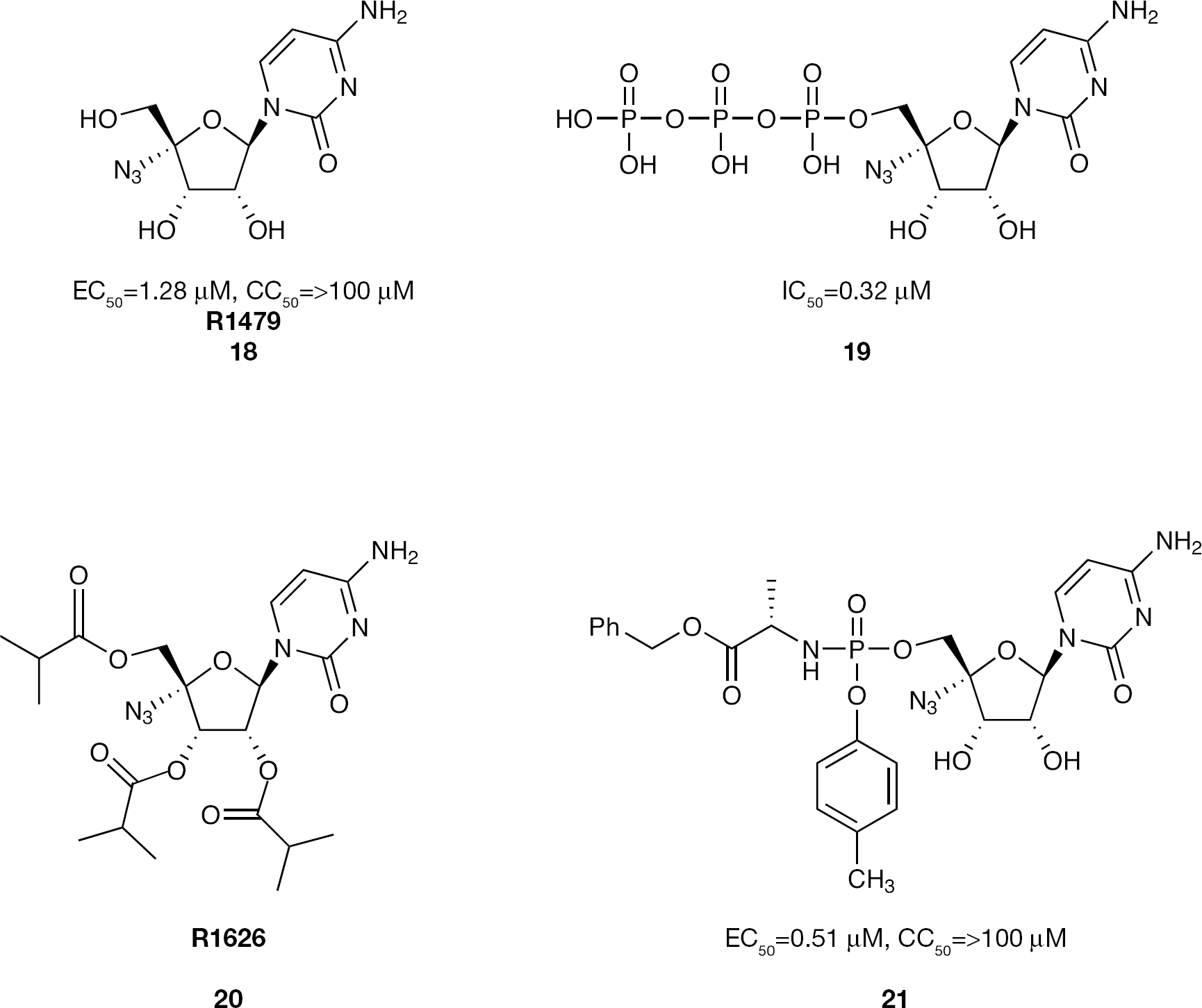
4′-Azido cytidine nucleoside and nucleotide phosphoramidate prodrug inhibitors of HCV replication
In an effort to increase the potency of R1479 (
Further evaluation of the 4′-azido pyrimidine class of nucleosides investigated the 4′-C-azidouridine derivative
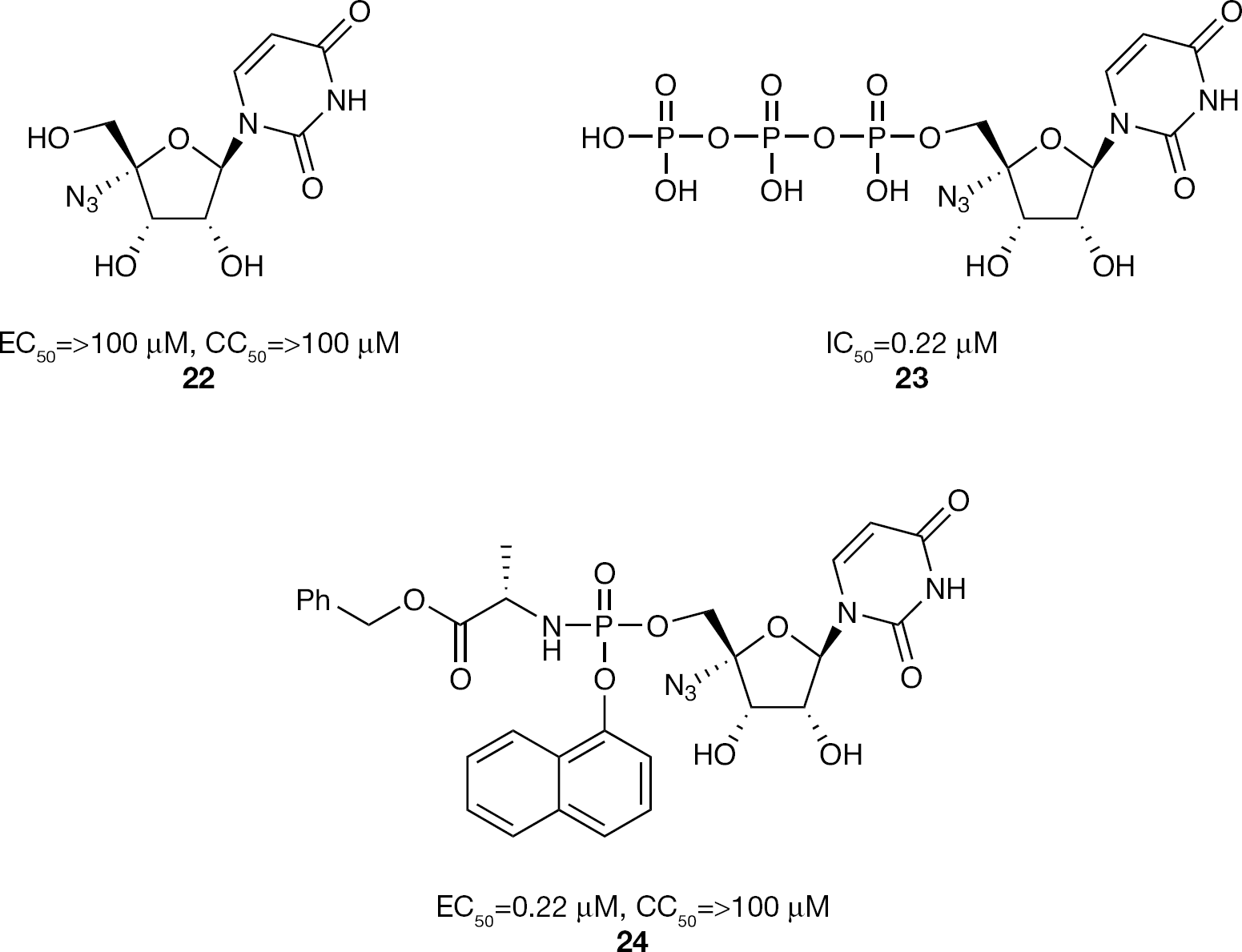
4′-Azido uridine nucleoside and nucleotide phosphoramidate prodrug inhibitors of HCV replication
In the 4′-azido riboside class of nucleosides, the phosphoramidate prodrug strategy was also applied to the adenosine derivative
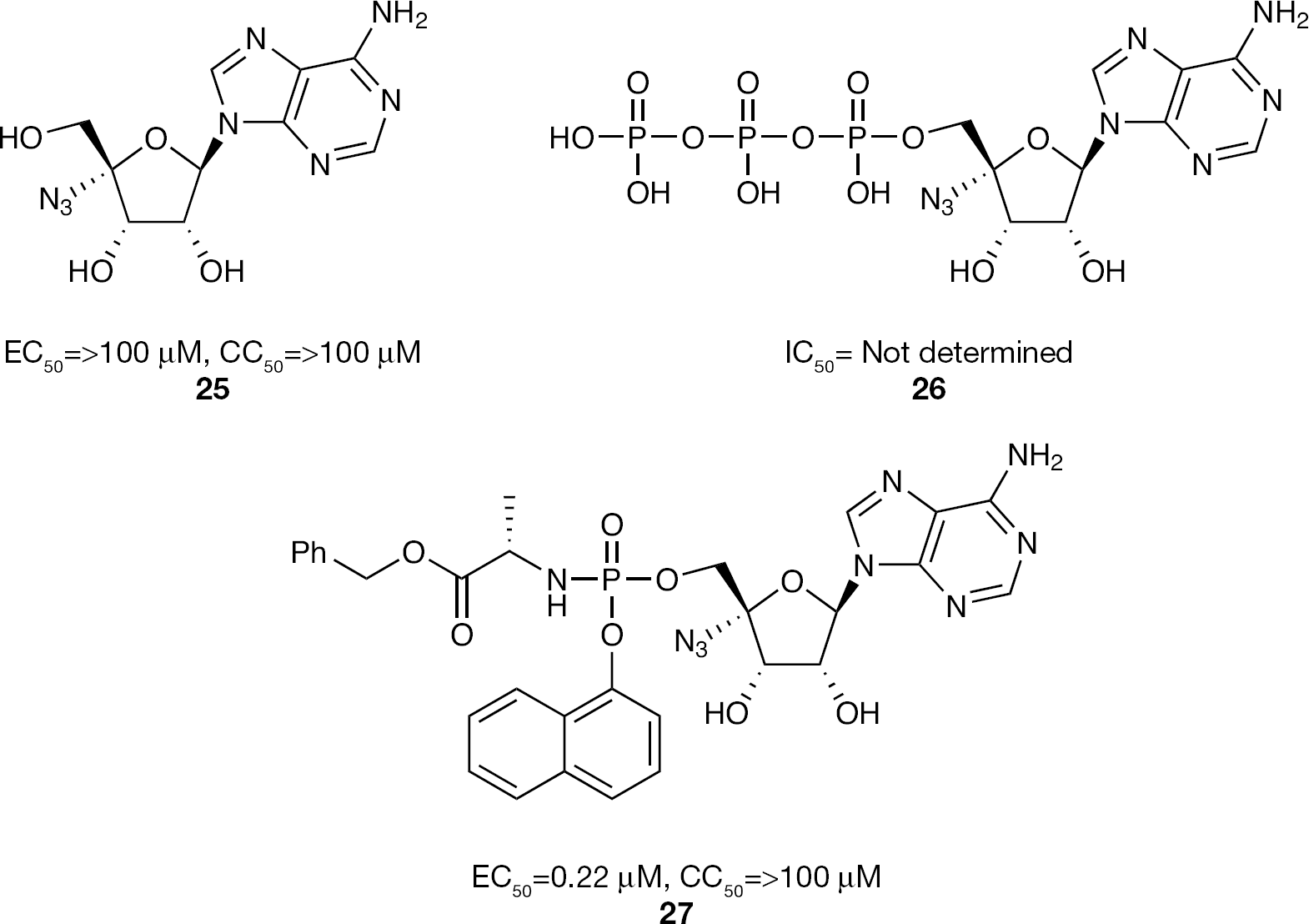
4′-Azido adenosine nucleoside and nucleotide phosphoramidate prodrug inhibitors of HCV replication
2′-α-Fluoro-2′-β-C-methyl nucleotide phosphoramidate prodrugs
In 2005, the first disclosure of a 2′-α-fluoro-2′-β-C-methylcytidine nucleoside was made with the publication of PSI-6130 (
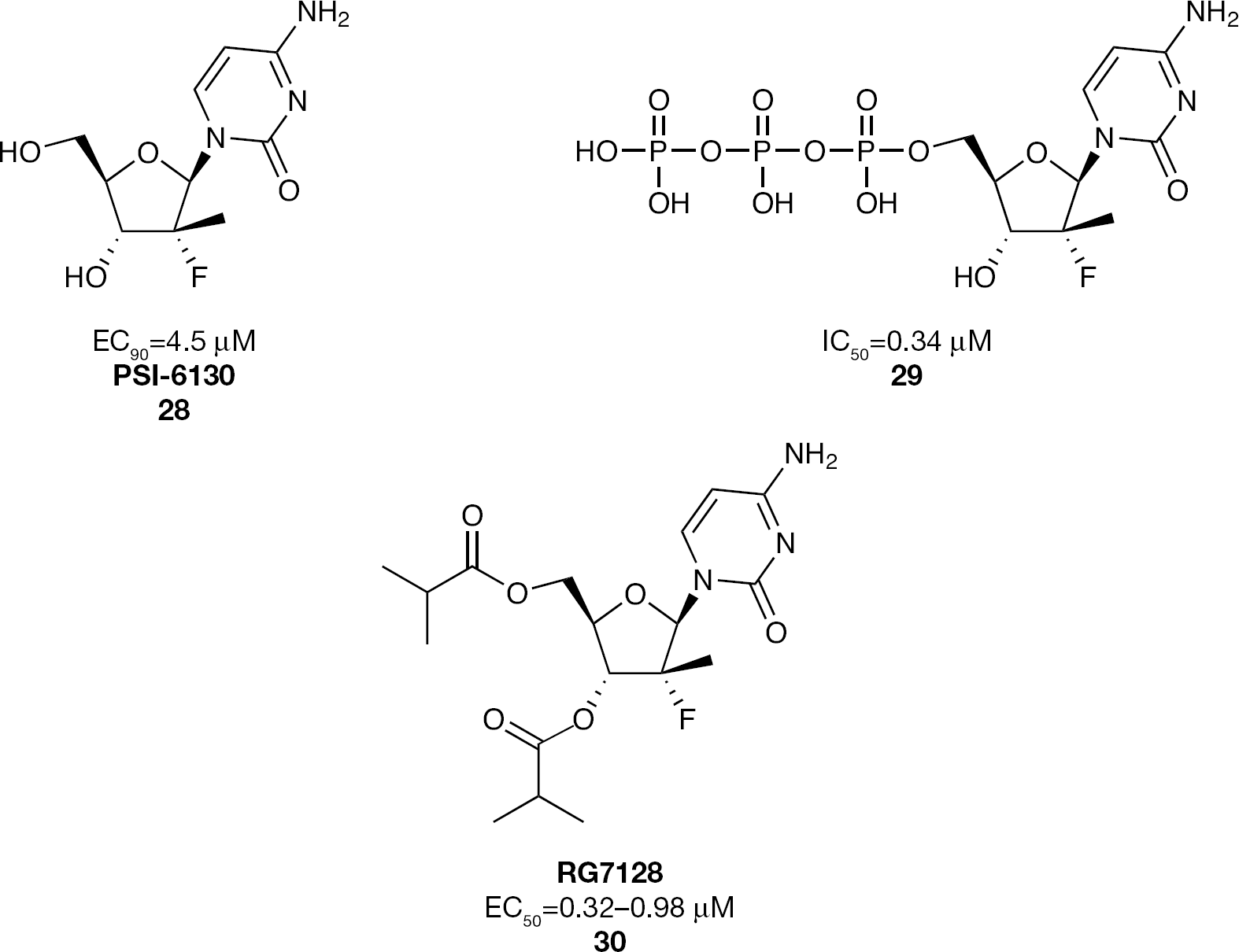
2′-α-F-2′-β-C-Methylcytidine nucleoside inhibitors of HCV replication
RG7128 (
In both the genotype 1 and genotype 2/3 Phase IIa clinical studies where RG7128 (
RG7128 (
With the hope of demonstrating proof of concept that the combination of two DAAs could lead to an IFN-sparing clinical regimen, RG7128 (
Metabolism studies on the cytidine nucleoside PSI-6130 (
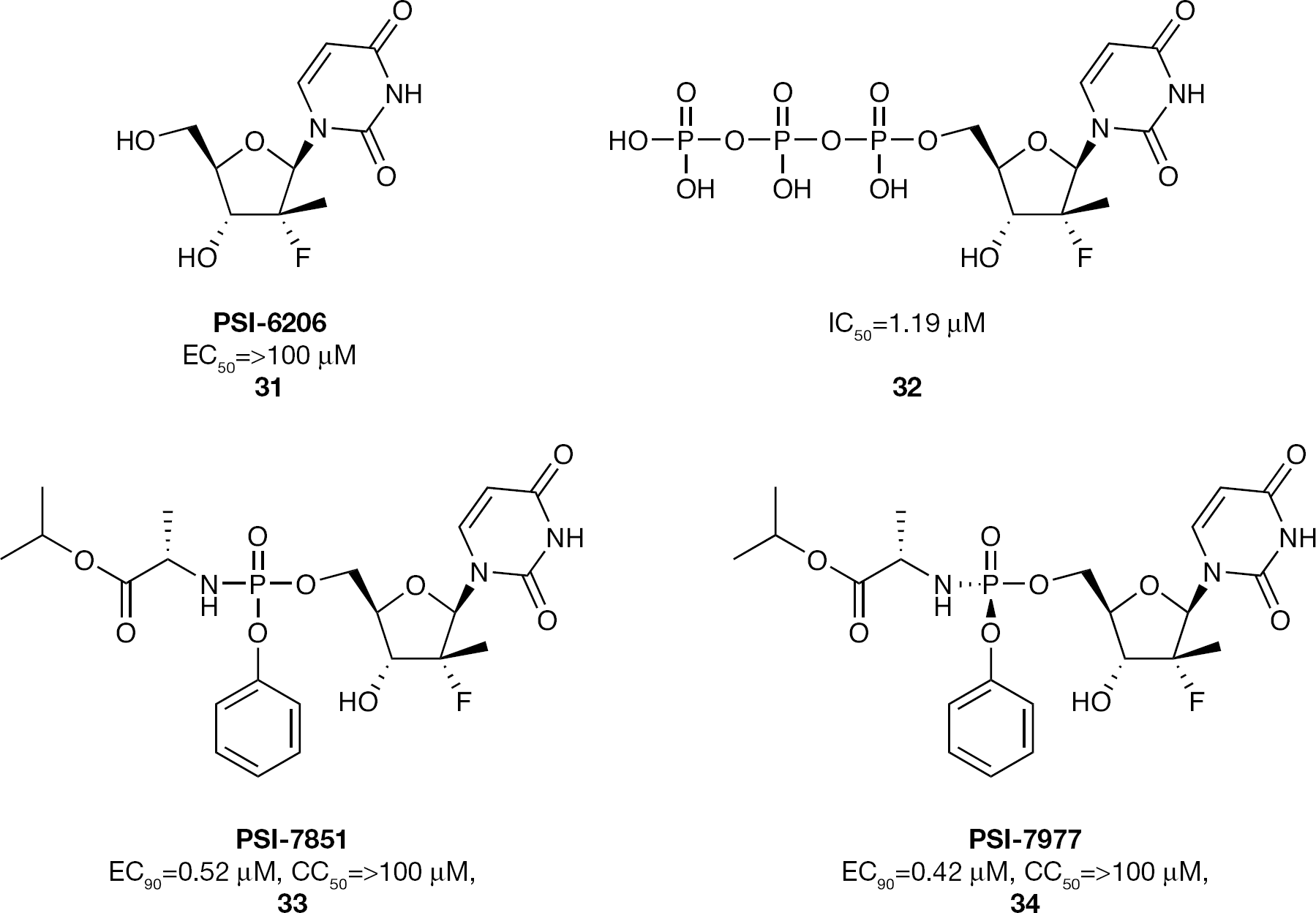
2′-α-F-2′-β-C-Methyluridine nucleoside and nucleotide phosphoramidate prodrug inhibitors of HCV replication
In a single ascending dose clinical study of healthy volunteers receiving doses up to 800 mg once daily, PSI-7851 (
With proof of concept demonstrated in HCV patients, further development of the 2′-α-F-2′-β-C-methyluridne phosphoramidate proceeded using PSI-7977 (
Further structure-activity relationship studies around the 2′-α-F-2′-β-C-methyl class of nucleosides had shown that the guanosine derivative
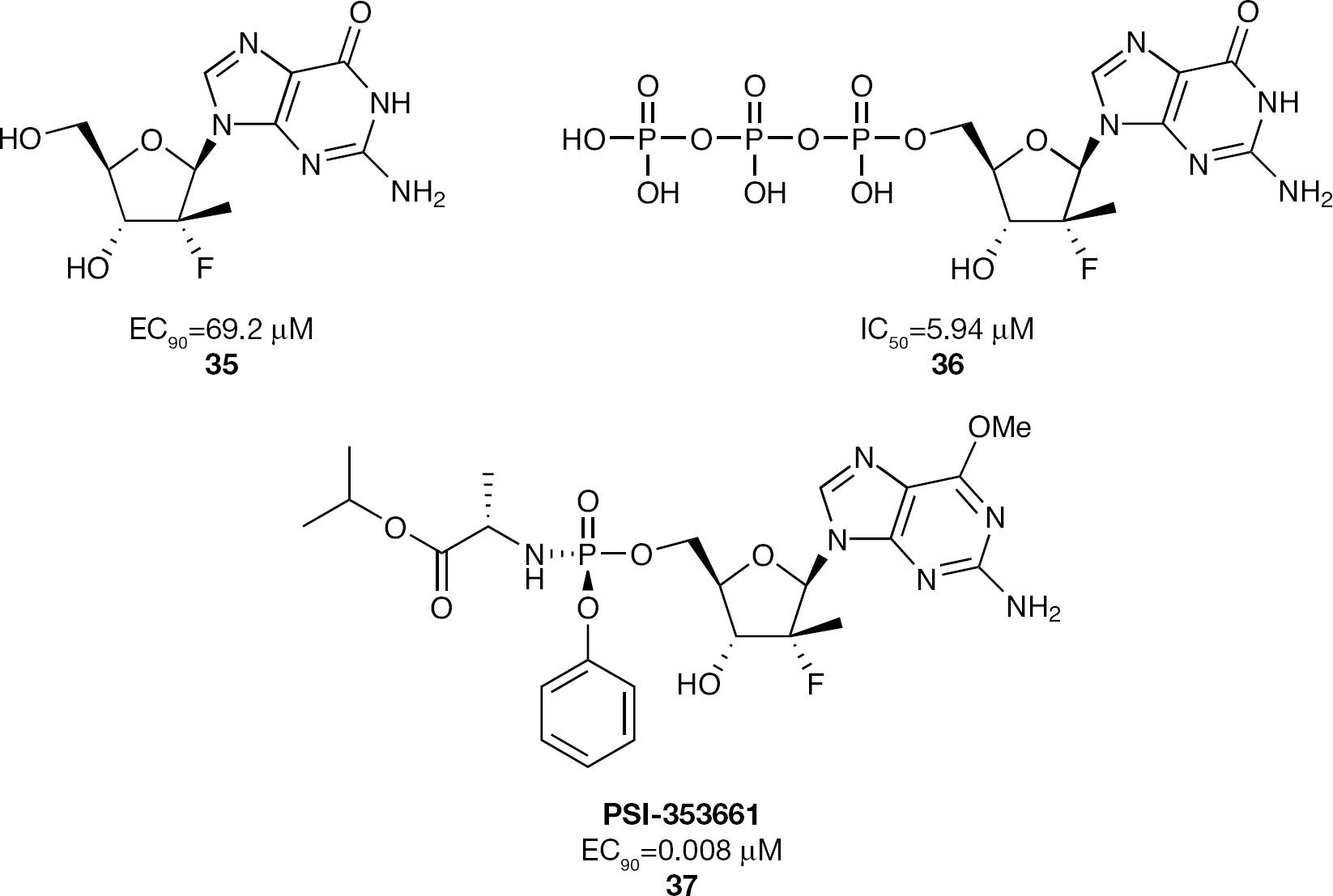
2′-α-F-2′-β-C-Methylguanosine nucleoside and nucleotide phosphoramidate prodrug inhibitors of HCV replication
Another 2′-α-F-2′-β-C-methyl nucleoside having a 7-ethynyl-7-deaza adenosine base unit (
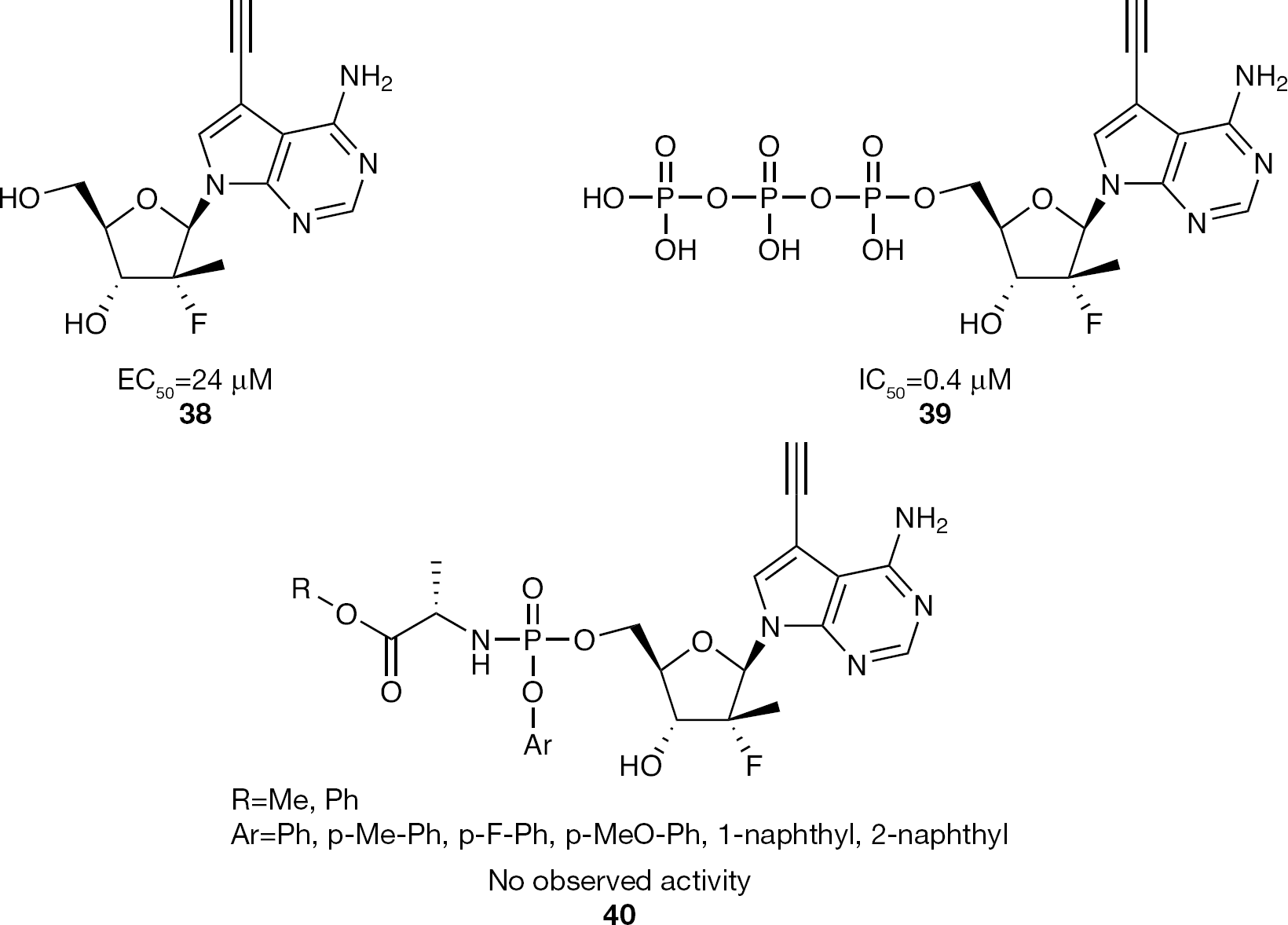
7-Deaza-7-ethynyl-2′-α-F-2′-β-C-methyladenosine nucleoside and nucleotide phosphoramidate prodrug inhibitors of HCV replication
3′,5′-Cyclic phosphate nucleotide prodrugs The use of a 3′,5′-cyclic phosphate prodrug construct for delivering a nucleoside 5′-monophosphate into cells is a relatively uncommon prodrug motif [95–97]. Its application has only recently been selectively applied in the development of nucleotide inhibitors for HCV polymerase. Little is known about the mechanism by which these cyclic phosphates are metabolized to the desired 5′-monophosphates; however, based on the reported HCV data and recent clinical data [98], it is clear that this approach is an effective prodrug strategy for delivering in vivo a nucleoside 5′-monophosphate into cells.
2′-C-Methyl nucleotide 3′,5′-cyclic phosphate prodrugs
The first reported example of a 3′,5′-cyclic phosphate prodrug construct for inhibiting HCV was reported in an attempt to develop a set of 2′-C-methyl ribonucleosides with modified purine bases where the purine base has a hydrazido sulfonamide at the C-6 position (Figure 15) [99]. The parent nucleosides
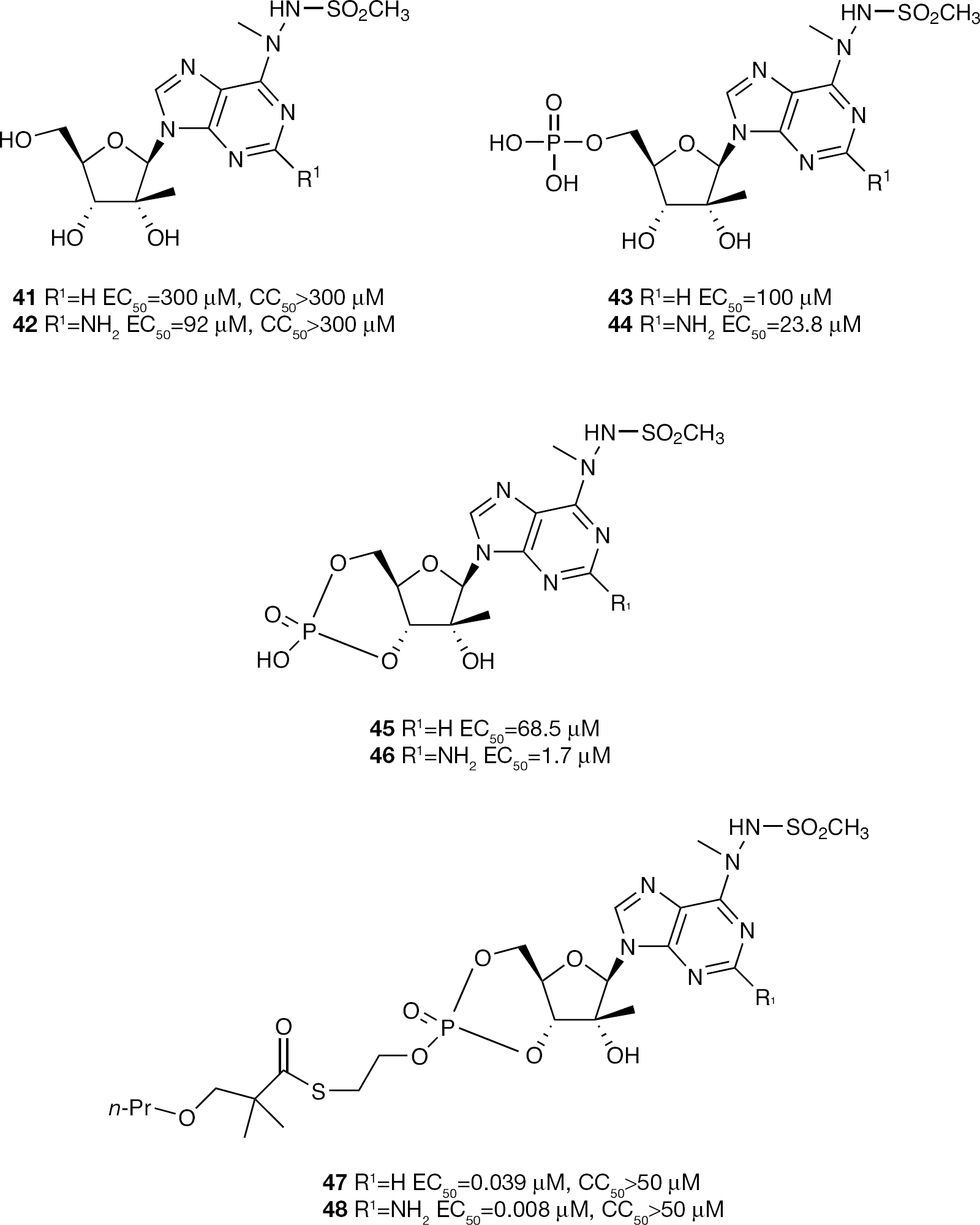
C-6-Hydrazido sulfonamide 2′-C-methyladenosine and 2′-C-methyl-2-amino-adensosine nucleoside and nucleotide cyclic phosphate prodrug inhibitors of HCV replication
Another application of the 3′,5′-cyclic phosphate prodrug approach focused on the study of 3′,5′-cyclic phosphoramidates in an attempted to improve the activity of 2′-C-methylcytidine (Figure 16) [100]. It was postulated that relative to the 5′-phosphoramidate prodrug, the 3′,5′-cyclic version would reduce the molecules rotational degrees of freedom and potentially improve entry into cells. In addition, use of a cyclic phosphate would remove the need for a phenolic substituent on the phosphoramidate moiety. Preparation of 2′-C-methycytidine 3′,5′-cyclic phosphoramidates like
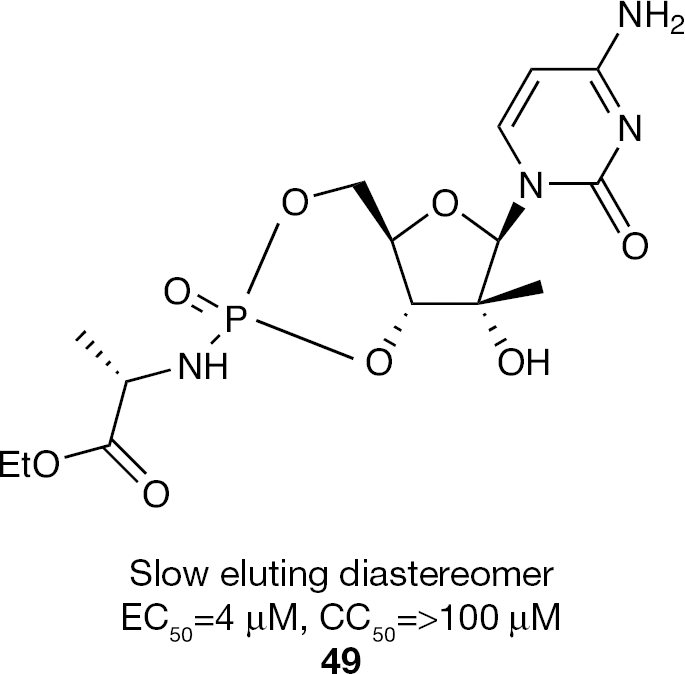
2′-C-Methylcytidine-3′,5′-cyclic phosphoramidate prodrug inhibitor of HCV replication
2′-α-F-2′-β-C-Methyl nucleotide 3′,5′-cyclic phosphate prodrugs
The 3′,5′-cyclic phosphate prodrug strategy was also applied to the 2′-α-F-2′-β-C-methyl class of nucleosides and ultimately resulted in the clinical development candidate PSI-352938 (
Nucleoside/nucleotide prodrugs: comparison of Phase I and Phase IIa human clinical trial results a
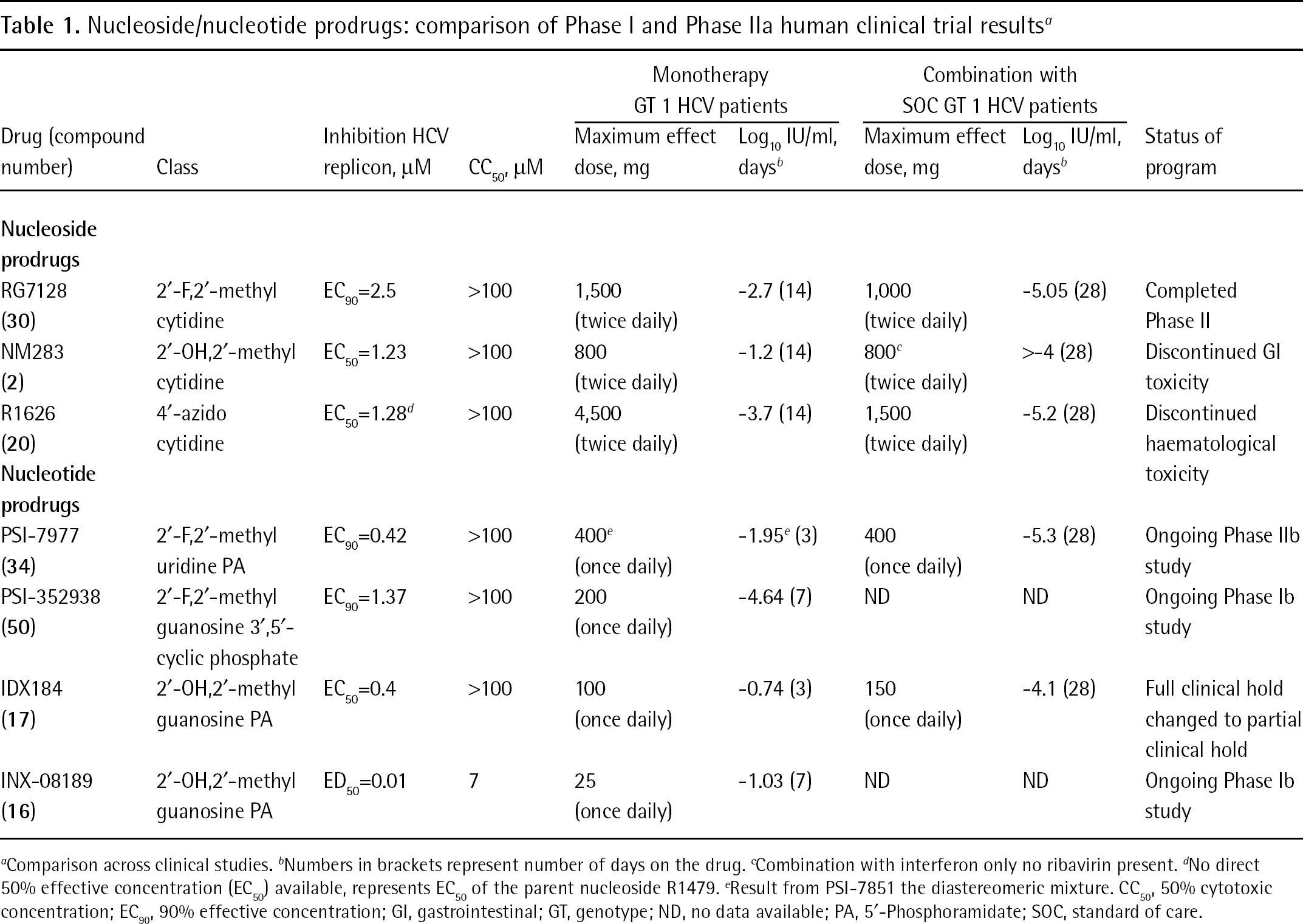
Comparison across clinical studies.
Numbers in brackets represent number of days on the drug.
Combination with interferon only no ribavirin present.
No direct 50% effective concentration (EC50) available, represents EC50 of the parent nucleoside R1479.
Result from PSI-7851 the diastereomeric mixture. CC50, 50% cytotoxic concentration; EC90, 90% effective concentration; GI, gastrointestinal; GT, genotype; ND, no data available; PA, 5′-Phosphoramidate; SOC, standard of care.
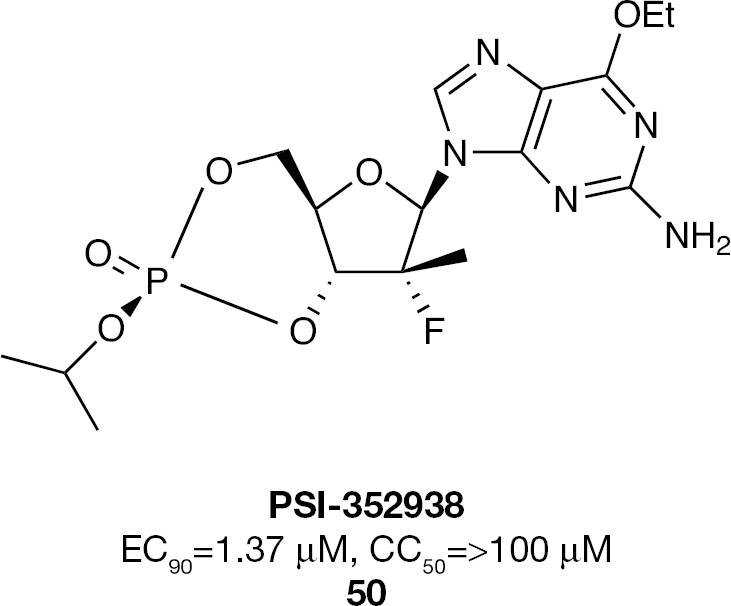
O-6-Ethyl-2′-α-F-2′-β-C-Methylguanosine-3′,5′-cyclic phosphate prodrug inhibitor (PSI-352938) of HCV replication
PSI-352938 (
Cyclic 1-aryl-1,3-propanyl nucleotide phosphate esters: HepDirect prodrugs
Preparation of a nucleoside 5′-monophosphate cyclic diester using a 1-aryl-1,3-propanyl prodrug moiety to accomplish liver targeting has been called HepDirect prodrug technology [33,105]. HepDirect technology was developed specifically to deliver drugs to the liver by taking advantage of liver-specific enzymes which would cleave the prodrug moiety thus revealing the active drug in hepatocytes. HepDirect prodrugs have been reported to be very stable in aqueous solutions, blood and non-hepatic tissues other than that of the gastrointestinal tract. For HepDirect technology, activation is achieved via a cytochrome P450 mediated oxidation (Figure 18). The described mechanism for nucleoside prodrug cleavage starts with hydroxylation of the C-4 tertiary carbon of the pro-moiety by CYP3A4 followed by rapid ring opening and then β-elimination to reveal the nucleoside monophosphate and an aryl vinyl ketone byproduct. Although release of aryl vinyl ketone as a byproduct of HepDirect cleavage raises toxicological concerns, it is hypothesized that this byproduct would be rapidly scavenged by high levels of glutathione in the liver, thus minimizing the associated risks. The HepDirect liver targeting prodrug approach was demonstrated in the clinic with pradefovir, a HepDirect prodrug version of adefovir [33].
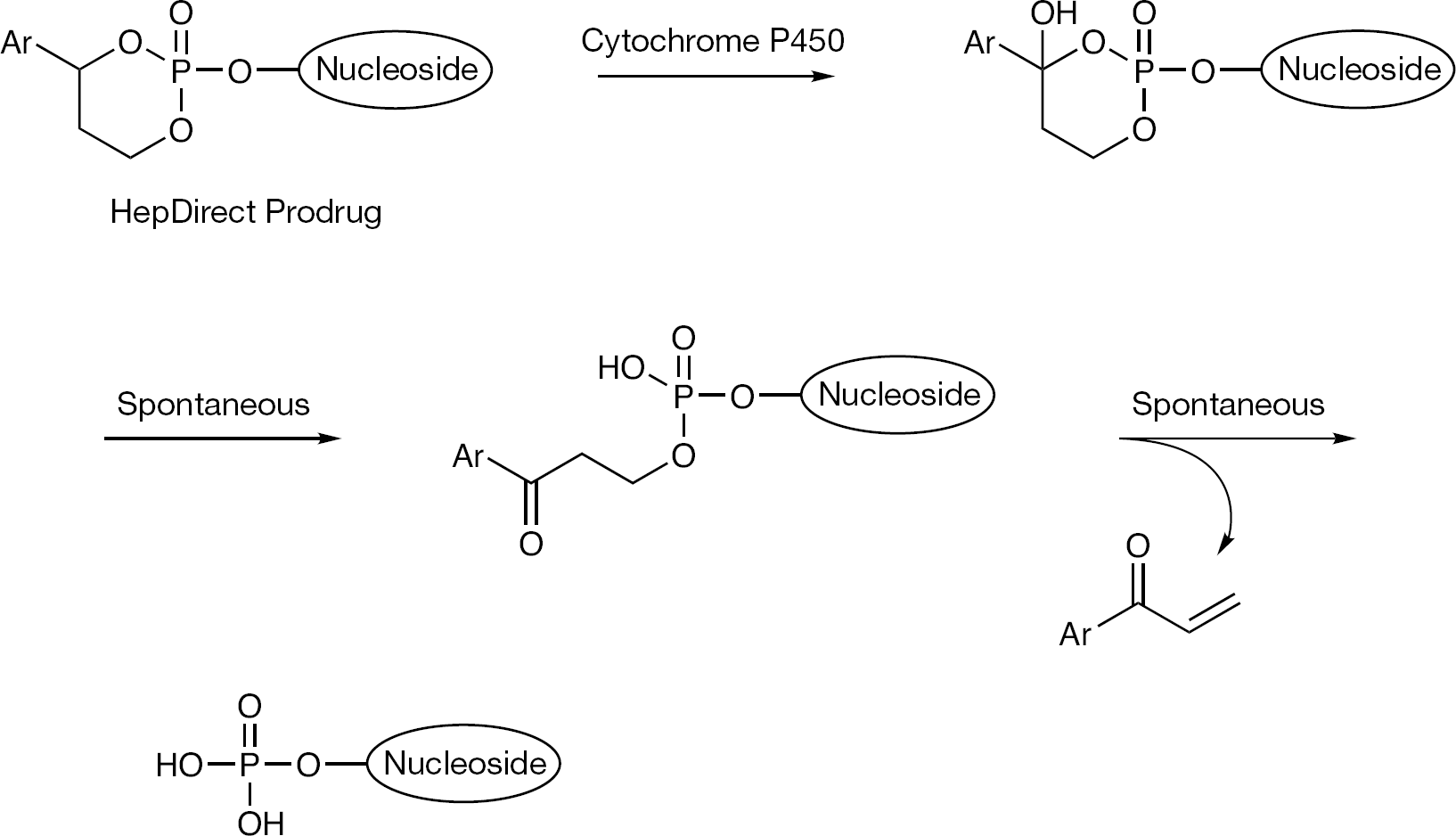
HepDirect prodrug technology and the prodrug decomposition pathway.
2′-C-Methyladenosine HepDirect prodrugs
2′-C-Methyladenosine (
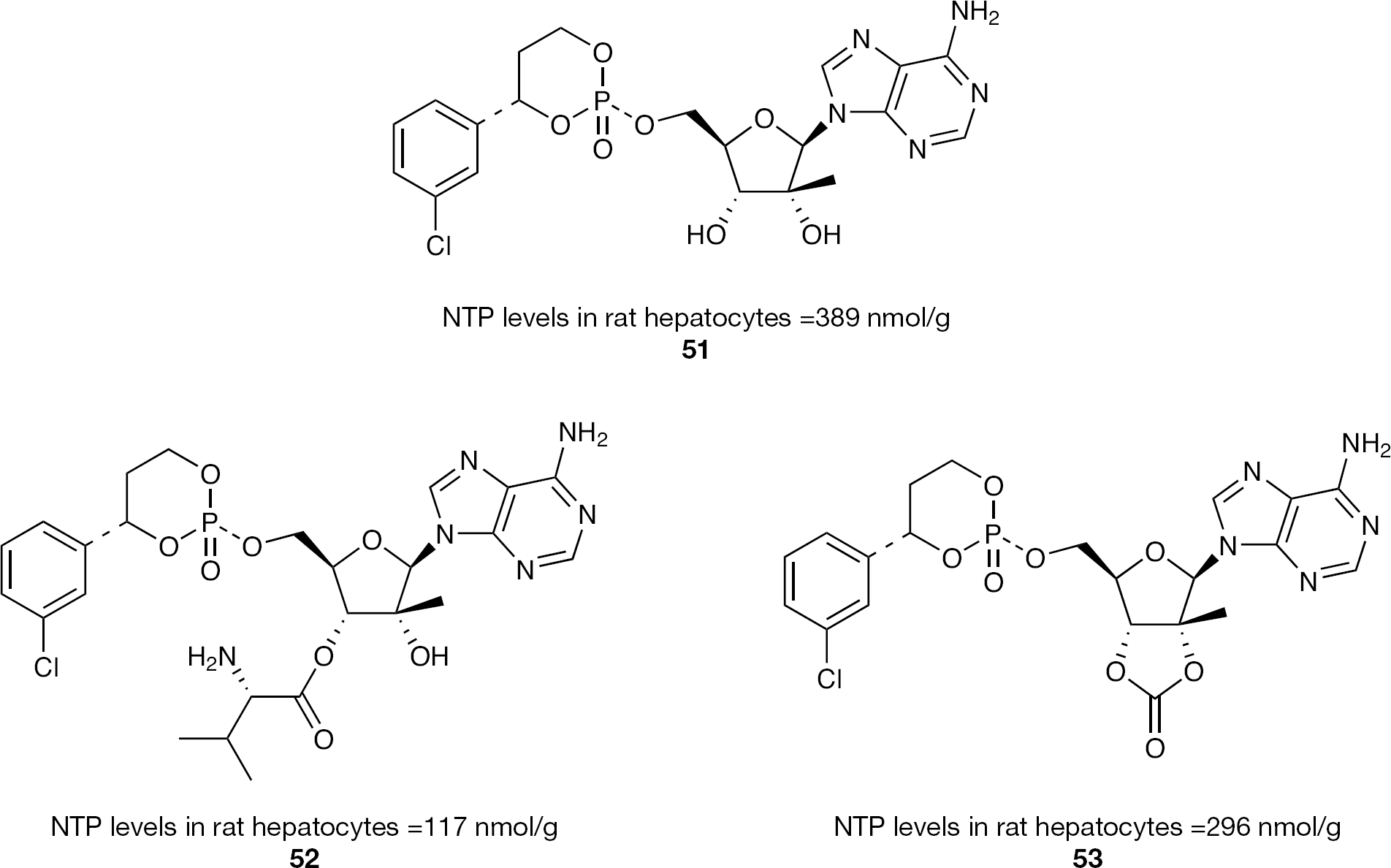
HepDirect prodrug analogues of 2′-C-methyladenosine nucleoside monophosphates
Bis(S-acyl-2-thioethyl) nucleotide phosphate prodrugs: SATE prodrugs
The bis(S-acyl-2-thioethyl) phosphate strategy (SATE) has been applied widely in the field of nucleoside 5′-phosphate prodrugs [33,34]. As described above in the case of IDX184 which contains a SATE monoester, an acid and an equivalent of episulfide is liberated as a result of each thioester cleavage (Figure 20). Episulfide is highly reactive and is a known mutagen, consequently it has been assumed that release of such an agent from a pronucleotide compound would be of some concern [109].
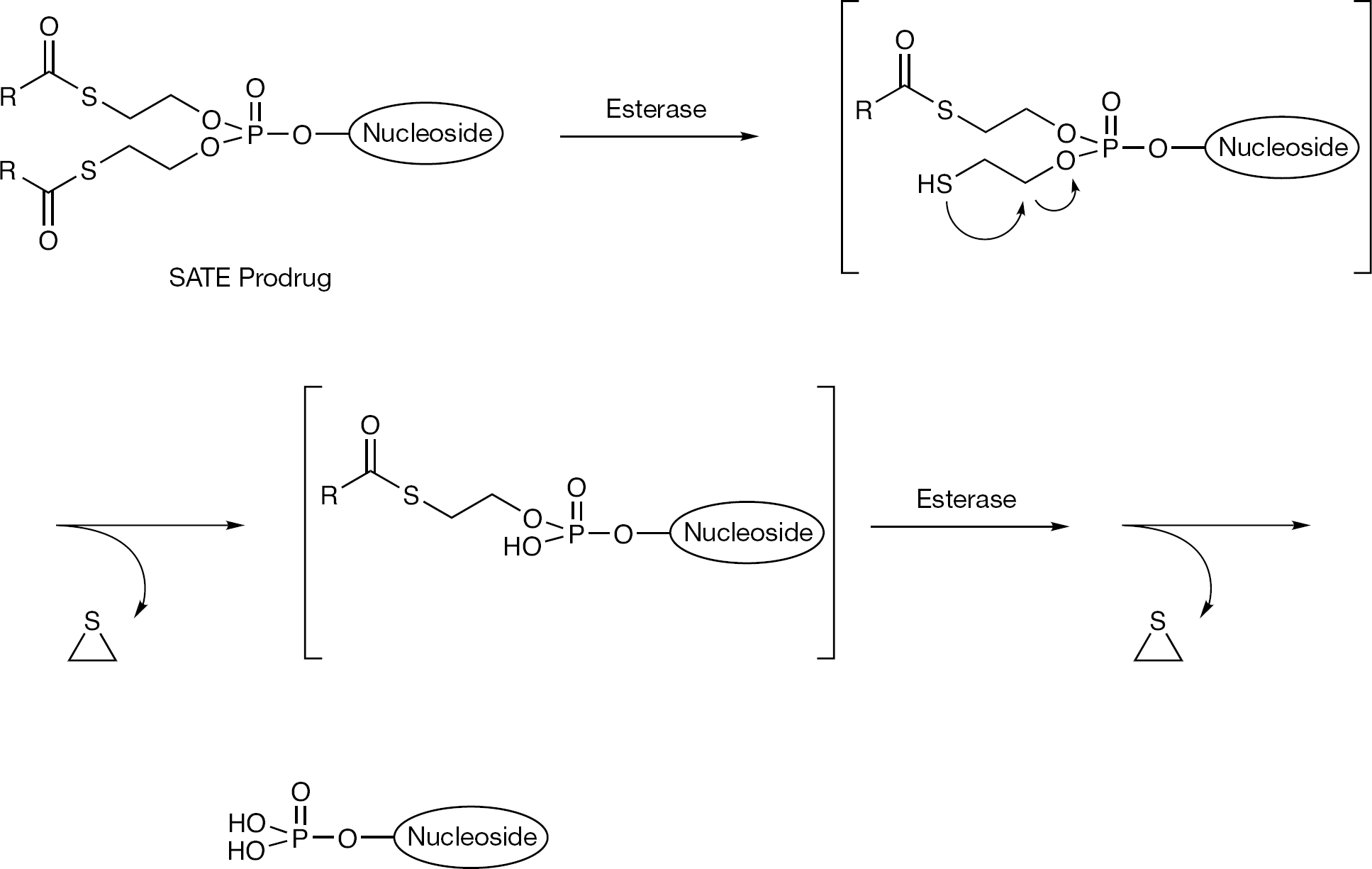
SATE prodrug decomposition pathway
The only reported application of the bis-SATE prodrug method to nucleotide HCV inhibitors was toward 2′-α-F-2′-β-C-methyl 7-ethynyl-7-deaza adenosine
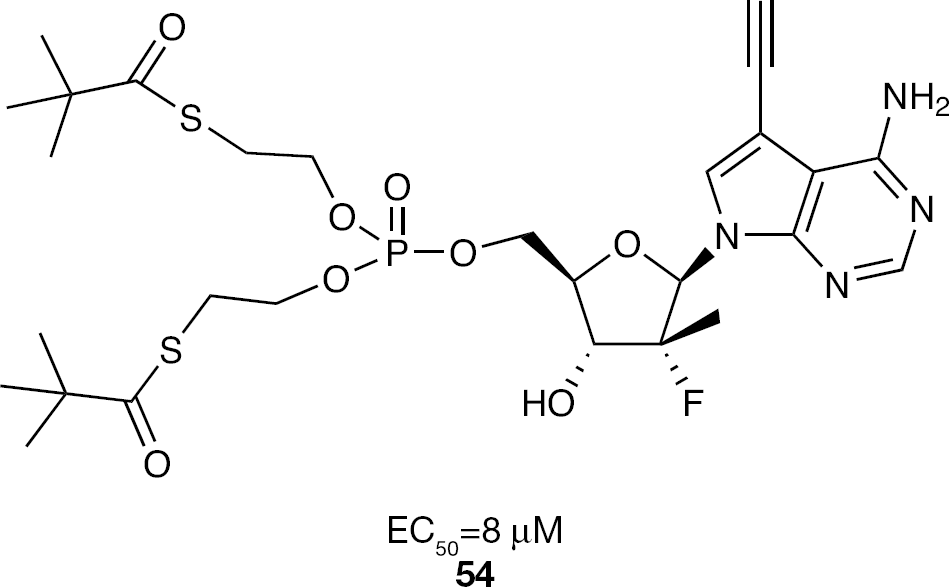
SATE prodrug of the 5′-monophosphate of 7-deaza-7-ethynyl-2′-α-F-2′-β-C-methyladenosine
Discussion
Never before has the use of phosphate prodrugs played such a significant role in the discovery and development of nucleoside-based drugs as has been the case in the area of inhibitors of HCV. The translation of multiple examples of phosphate prodrugs of nucleotides into the clinic and the demonstration of clinical proof of concept and beyond are equally unprecedented. HCV has offered a unique opportunity to apply phosphate prodrug technology. Since HCV is a disease of the liver, drug hunters are able to leverage the robust characteristics of liver metabolism and first pass metabolism as the means for achieving target organ drug levels and obviating the need for maintaining high circulating levels of a potentially unstable prodrug. However, because of liver targeting, decomposition of the phosphate prodrug to the desired nucleoside 5′-monophosphate does release prodrug byproducts into the liver at potentially high concentrations. This release of sometimes potentially reactive or toxic metabolites does pose a concern in developing prodrugs of nucleoside monophosphates. This concern is particularly relevant to HCV patients with advanced liver disease where liver function is compromised and therefore, may have a lower capacity to clear or process metabolites. Some of the prodrug strategies implemented for delivering nucleoside monophosphates such as the SATE or HepDirect strategies are known to release reactive metabolites that should be of concern in assessing their long term safety potential. Other prodrug strategies such as the phosphoramidate release different metabolites such as phenol or naphthol. Although phenol is readily cleared by the liver, the toxicity of substituted phenols may be of concern in deciding to progress a nucleotide prodrug forward into development [110,111]. Each of these liability concerns must be put into context of dose and PK. It is possible to circumvent any toxicity concerns if the dose of drug administered is sufficiently small to not warrant concern or if the PK profile of the metabolites limits exposure. Clearly the increased potency of these nucleotide prodrugs relative to the first generation nucleoside HCV inhibitors increases the therapeutic index and reduces the chances of observing undesired side effects. However, long term animal toxicity studies and clinical safety assessment over extended dosing periods will ultimately address the concerns over any potential human toxicity.
The nucleotide phosphate prodrugs which have entered clinical study have demonstrated superiority over the first generation nucleoside inhibitors developed for treating HCV (Table 1). The use of the nucleoside 5′-monophosphate prodrugs has demonstrated dramatic potency enhancements relative to first generation nucleoside inhibitors in the cellular subgenomic replicon assay, higher levels of triphosphate in whole cells and in livers of animals when dosed orally. To a large extent this improved in vitro and whole animal profile has translated into the clinical setting. In the clinic, dosing frequency has been reduced from twice daily to once daily, drug load has been greatly reduced and impressive viral load declines and RVR rates have been achieved. However, in vitro HCV replicon potency alone can not itself justify the robust initial clinical results observed for these phosphate prodrugs. The least potent inhibitor in the subgenomic replicon assay, PSI-352938 (
It has been suggested that nucleosides/nucleotides will eventually become the backbone of anti-HCV therapy, as they have for HIV HAART. Their broad genotype coverage and resistance profile put them in a unique position and make them the drug class of choice to combine with DAAs of different mechanisms of action. With the identification of both purine and pyrimidine nucleotide prodrugs with different resistance profiles and metabolic pathways, the possibility also exists for combining two nucleosides/nucleotides into one regimen as has been done in the HIV field. Studies to explore different combinations of DAAs including dual nucleotide combinations have begun. Results in a 14-day dual nucleotide proof of concept study with PSI-7977 (
Footnotes
MJS is an employee of Pharmasset, Inc. and receives salaried compensation in his role as Vice President of Chemistry.