
Abstract
Background:
We examined the anti-influenza virus activity of tricin, 4′,5,7-trihydroxy-3′,5′-dimethoxyflavone, against five viruses: A/Solomon islands/3/2006 (H1N1), A/Hiroshima/52/2005 (H3N2), A/California/07/2009 (H1N1pdm), A/Narita/1/2009 (H1N1pdm) and B/Malaysia/2506/2004 strains in vitro and against A/PR/8/34 virus in vivo.
Methods:
The effect of tricin was studied by an infectious virus yield reduction assay. The anti-influenza virus mechanism of the tricin was examined by western blot analysis, real-time reverse transcriptase PCR analysis, haemagglutination inhibition (HI) assay and neuraminidase (NA) inhibition assay. The anti-influenza virus efficacy of tricin was further examined in a murine influenza virus infection model.
Results:
Tricin of 3.3 to 30 μM significantly reduced seasonal A (H1N1), (H3N2) viruses, novel A (H1N1pdm) virus, as well as B virus in a dose-dependent manner. The 50% effective concentrations of tricin were 3.4 μM for seasonal A (H3N2) virus, 4.9 μM for B virus and 8.2 μM for A/Narita (H1N1pdm) virus. Tricin decreased the expression of haemagglutinin (HA) protein and matrix (M) protein, and messenger RNA expression of HA and M of influenza virus in the infected cells. Tricin exhibited little or no effects on influenza virus HI and NA activities. In the mouse infection model, tricin was significantly effective in reducing body weight loss, and also effective in prolonging survival times of infected mice.
Conclusions:
Tricin was indicated to possess anti-influenza virus activity and to ameliorate body weight loss and survival rate of influenza-A-virus-infected mice. Tricin is a novel compound with potential anti-influenza virus activity in vitro and in vivo.
Introduction
Influenza virus infection frequently causes severe acute respiratory infection and is sometimes accompanied by fever, muscle pain, pneumonia, otitis media, meningitis and encephalitis [1,2]. These clinical symptoms often become severe, especially in high-risk groups such as the elderly and infants [3,4]. Treatment with anti-influenza virus agents plays an important role in influenza virus control. Amantadine and its analogue, rimantadine, act as viral M2 channel blockers, and zanamivir and oseltamivir are viral neuraminidase (NA) inhibitors, and have been used for the treatment and prevention of influenza virus infection [5–7]. However, amantadine-resistant viruses have begun to appear at a high rate clinically, and the appearance of resistant viruses has also been reported in treatment with NA inhibitors [8–10]. Therefore, new or alternative anti-influenza virus agents that are effective against resistant viruses need to be developed.
Sasa albo-marginata is well known as Kumazasa in Japan. Extracts of Kumazasa have been used as traditional medicine and as wrapping materials for foods throughout Asia. The water-soluble fraction of this plant has a number of biological activities, including antiulcerogenic and anti-inflammatory properties [11–13]. With the development of drug resistance, which is a constant concern, the search for new antiviral agents from a variety of sources, including plants, has assumed more urgency than in the past [14]. A recent study in our laboratory revealed that tricin (4′,5,7-trihydroxy-3′,5′-dimethoxyflavone), which is derived from the hot water extract of Sasa albo-marginata (Figure 1), has anti-human cytomegalovirus activity [15]. In the present study, we show that tricin has anti-influenza virus properties in vitro and in vivo.
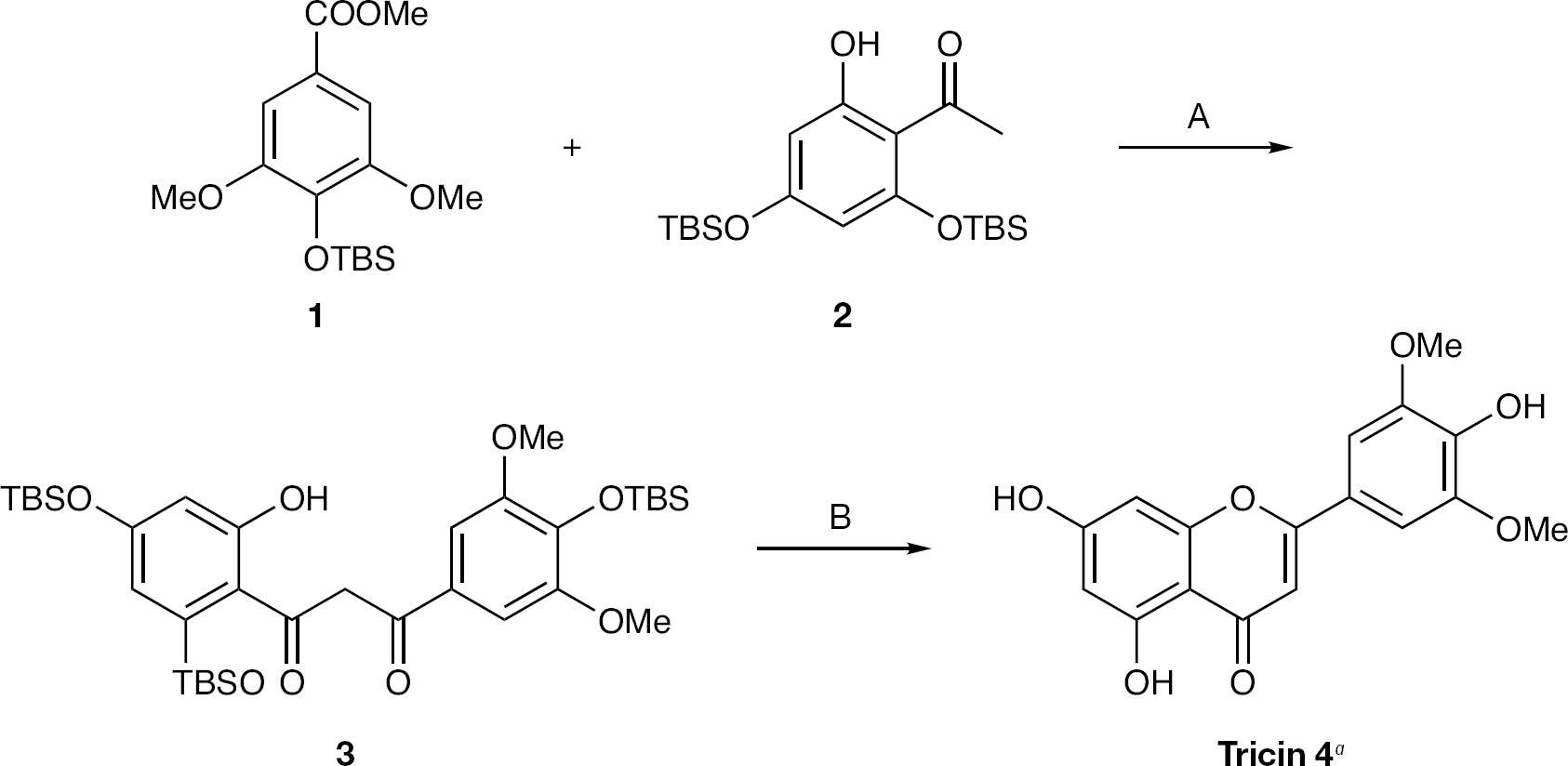
Synthesis of 4′,5,7-trihydroxy-3′,5′-dimethoxyflavone (tricin)
Methods
Cells and viruses
Madin–Darby canine kidney (MDCK) cells were purchased from the American Type Culture Collection, and MDCK cells were grown and maintained in Eagle's minimum essential medium (EMEM; Nissui Pharmaceutical Co. Ltd, Tokyo, Japan) supplemented with 5% fetal calf serum (Bocknek Ltd, Rexdale, ON, Canada), L-glutamine (0.3 mg/ml; Nacalai Tesque, Inc., Kyoto, Japan), streptomycin (100 μg/ml; Nacalai Tesque, Inc.) and penicillin (100 units/ml; Nacalai Tesque, Inc.). For the influenza virus infection medium, EMEM supplemented with 0.4% bovine serum albumin (BSA; Cohn Fraction V, pH 7.0; Wako Pure Chemical Industries, Ltd, Osaka, Japan), L-glutamine (0.15 mg/ml; Nacalai Tesque Inc.), vitamin solution (MEM Vitamin Solution 100X, GIBCO, Grand Island, NY, USA), streptomycin (50 μg/ml; Nacalai Tesque, Inc.) and penicillin (50 units/ml; Nacalai Tesque, Inc.), was used. Influenza A/Solomon islands/3/2006 (H1N1), A/New Caledonia/20/99 (H1N1), A/Hiroshima/52/2005 (H3N2), A/California/07/2009 (H1N1pdm) [16,17], A/Narita/1/2009 (H1N1pdm) [18] and B/Malaysia/2506/2004 viruses were propagated in MDCK cells. For the in vivo assay, a mouse-adapted influenza A/PR/8/34 (H1N1) virus was used [19].
Compound
Tricin, 4′,5,7-trihydroxy-3′,5′-dimethoxyflavone (Figure 1) was synthesized and suspended in dimethyl sulfoxide (DMSO). We prepared tricin through a condensation reaction of methyl benzoate (
Amantadine hydrochloride was purchased from Wako Pure Chemical Industries, Ltd, and dissolved in distilled water. Oseltamivir carboxylate was provided by F Hoffmann-La Roche Ltd (Basel, Switzerland), and dissolved in distilled water.
Animals
Female DBA/2 Cr mice (6 weeks old, 17–19 g) were purchased from Japan SLC Inc. (Hamamatsu, Japan). The mice were housed six or seven per cage with food and water ad libitum under a 12 h light/12 h dark diurnal cycle. The temperature in the room was kept at 24 ±2°C. The mice were acclimated for ≥3 days before use in the experimental procedures. Animal studies were performed in accordance with the animal experimentation guidelines of Kyusyu University of Health and Welfare, and in an approved biosafety level facility.
Infectious virus yield reduction assay
Tricin was examined for its anti-influenza virus activity by an infectious virus yield reduction assay. When MDCK cells in 6-well plastic plates (Falcon number 3046, Becton Dickinson, Franklin Lakes, NJ, USA) reached confluence, the cells were inoculated with each influenza virus at a multiplicity of infection (MOI) of 1.0, 0.1 or 0.01. After adsorption for 1 h, cells were incubated in 1 ml of EMEM containing 0.4% BSA, 5 μg/ml trypsin (acetylated trypsin, type V-S, from bovine pancreas; Sigma-Aldrich Japan, Tokyo, Japan) with various concentrations of tricin (0.37 to approximately 30 μM) or without tricin for 8 h after viral infection. Infectious virus production in the supernatant was titrated using plaque assay.
Western blot analysis
Proteins from influenza-virus-infected cells were separated by SDS-PAGE on 8% gel. Proteins were then transferred to a polyvinylidene difluoride membrane (Hybond-p; Amersham Pharmacia Biotech AB, Uppsala, Sweden) according to the manufacturer's instructions using 20 mM Tris and 150 mM glycine (pH 8.3) in 20% methanol as the blotting buffer. Membranes were incubated for 1 h at room temperature with blocking reagent (5% skim milk, Tris-buffered saline-0.5% Tween-20, pH 7.6 [TBS-T]), followed by 1 h at room temperature with a primary antibody (mouse monoclonal antibody, C102, specific for a haemagglutinin [HA] of influenza A virus; Gene Tex Inc., Irvine, CA, USA; GA2B, specific for a matrix [M] protein of influenza A virus; Funakoshi Corporation, Tokyo, Japan; and C4, specific for β-actin; Chemicon International Inc., Temecula, CA, USA) diluted 1:2,000 in TBS-T. β-actin was used to monitor actin levels as an internal control of protein induction. Membranes were washed three times in TBS-T and incubated with peroxidase-conjugated second antibody diluted 1:10,000 in TBS-T for 1 h at room temperature. After washing three times in TBS-T, immune complexes were detected using the ECL system (Amersham Pharmacia Biotech AB) according to the manufacturer's instructions.
Analysis of gene expression
Confluent monolayers of MDCK cells in 6-well plates (Falcon number 3046; Becton Dickinson) were infected with the influenza A/Solomon islands/3/2006 (H1N1) virus, at an MOI of 1.0. At 1 h after infection, the inoculum was removed, and cells were treated in 2 ml of DMEM with or without the indicated concentrations of tricin. RNA samples were collected at 4 and 5 h post-infection.
Total RNA was extracted from mock- or virus-infected cells treated with or without tricin using the chaotropic Trizol method, followed by Isogen-chloroform extraction and isopropanol precipitation [20]. Prior to the reverse transcriptase (RT) reaction, potentially contaminating residual genomic DNA was eliminated with DNase I (Takara Shuzo, Otsu, Japan). RNA accumulation was monitored by quantitative real-time RT-PCR (qRT-PCR). RT-PCR analysis of influenza HA and M gene expression was carried out using 0.2 μg of total RNA per reaction. The RT reaction was performed with random primers (5 min at 25°C, 30 min at 42°C and 5 min at 85°C) using the iScript cDNA Synthesis kit from Bio-Rad Laboratories, Inc. (Hercules, CA, USA), according to the manufacturer's instructions. cDNA products were amplified for HA, M and β-actin gene expression via qRT-PCR with specific primers using iQ SYBR Green Supermix (Bio-Rad Laboratories, Inc.) for 34 cycles (10 s at 95°C, 20 s at 55.5°C and 20 s at 72°C) by Mini Opticon realtime PCR with Gene Expression Macro software (Bio-Rad Laboratories, Inc.). PCR primers used were as follows: influenza HA primers (forward: 5′-GGT GTT CAT CAC CCG TCT AAC AT-3′, reverse: 5′TG TTT GAC ACT TCG CGT CAC AT-3′); influenza M primers (forward: 5′-AGA TGA GTC TTC TAA CCG AGG TCG-3′, reverse: 5′-TGC AAA AAC ATC TTC AAG TCT CTG-3′) [21–23]; and β-actin primers (forward: 5′-ATC ATG TTT GAG ACC TTC AAC-3′, reverse: 5′-CAG GAA GGA AGG CTG GAA GAG-3′) [14]. Results were normalized against β-actin RNA levels.
Haemagglutination inhibition assay
Haemagglutination inhibition assay was carried out as described by Suzuki et al. [24]. Briefly, virus suspension (4 HA units/20 μl) was incubated for 1 h at 4°C with a twofold serial dilution of 190 μM tricin (20 μl) with 0.01% gelatin-containing phosphate-buffered saline (PBS), in 96-well microtitre plates (Falcon number 3077, Becton Dickinson). After adding 0.5% (v/v) of chicken erythrocytes to each well, the plates were kept for 1 h at 4°C.
Neuraminidase inhibition assay
NA inhibition assay was carried out using an NA-StarTM influenza NA inhibitor resistance detection kit (Applied Biosystems, Foster City, CA, USA). The assay was performed according to the manufacturer's protocol using an LB940 plate reader (Berthold Technologies GmbH & Co.KG, Bad Wildbad, Germany). The influenza A/Solomon islands/3/2006 virus culture supernatant (cultured on MDCK cells) was initially serially diluted in phenol red-free EMEM to provide an appropriate NA titre to the assay (signal-to-noise ratio of 40:1). Final drug concentrations ranged from 0.0009 to 10 μM for tricin and from 0.03 to 320 nM for oseltamivir carboxylate.
Influenza virus infection in mice
Tricin suspended in 5% Arabic gum was examined in an intranasal influenza virus infection model in mice. DBA/2 Cr mice were intranasally infected or mock-infected with a mouse-adapted influenza A/PR/8/34 virus at 1,600 plaque forming units/25 μl or PBS, respectively, under anaesthesia [19,25]. Tricin suspension (20 or 100 μg/kg) of 0.2 ml was orally administered to the mice by gavage, once at 4 h prior to and twice after virus infection on day 0, and three times daily to day 5 after infection. A 5% Arabic gum solution was used for the control and mock-infected mice. Six mice from each group were weighed daily from day 0 to day 8 after infection, and the changes were calculated based on the body weight of each mouse on day 0. The number of surviving mice in a group was observed daily from day 0 to day 14.
Statistical analysis
Statistically significant differences between tricin-treated and untreated groups were evaluated using Tukey's test. Based on net body weight change data, the differences between the treated and untreated groups were analysed using repeated measures two-way ANOVA from day 0 to day 8 after infection.
Results
In vitro anti-influenza virus activity of tricin
We examined the anti-influenza virus activity of tricin against A/Solomon islands/3/2006 (H1N1), A/Hiroshima/52/2005 (H3N2), A/California/07/2009 (H1N1pdm), A/Narita/1/2009 (H1N1pdm) and B/Malaysia/2506/2004 strains using an infectious virus yield reduction assay. The numbers of virus titres were significantly reduced at 3.3 μM tricin against A/Solomon islands/3/2006 (H1N1) and A/Hiroshima/52/2005 (H3N2) strains and were obviously reduced at 3.3 to 30 μM tricin against all viral strains, indicating that tricin inhibits virus production in a dose-dependent manner (Figure 2A). The 50% effective concentration (EC50) of tricin ranged from 3.4 to 4.6 μM for influenza A viruses and 4.9 μM for influenza B virus (Table 1). The EC50s of tricin against novel influenza A viruses were 8.2 μM for A/Narita/1/2009 (H1N1pdm) and 10.2 μM for A/California/07/2009 (H1N1pdm) strains. However, viral replication of this novel influenza A virus was >95% inhibited by 30 μM tricin treatment (Figure 2A). The anti-influenza virus activity of tricin was compared with that of amantadine against A/Solomon islands/3/2006 (H1N1) strain. After infection to MDCK cells, the cells were incubated in the presence of various concentrations of either tricin or amantadine and cultured for 8 h. Tricin and amantadine significantly inhibited the replication of the influenza A virus at concentrations ranging from 1.1 to 30 μM and 0.21 to 5.7 μM, respectively (Figure 2B). Next, to examine the MOI dependence of antiviral activity of tricin, we determined the anti-viral activity of tricin against the influenza A/Solomon islands/3/2006 (H1N1) virus at various MOI (0.01, 0.1 or 1). An inhibitory effect of influenza A virus replication by tricin was observed in cells infected at the MOIs used here (Figure 2C). The size of plaques under this condition appeared to be slightly small by the treatment with tricin (KY et al., data not shown).
Comparison of anti-influenza virus activity of tricin against various influenza viruses
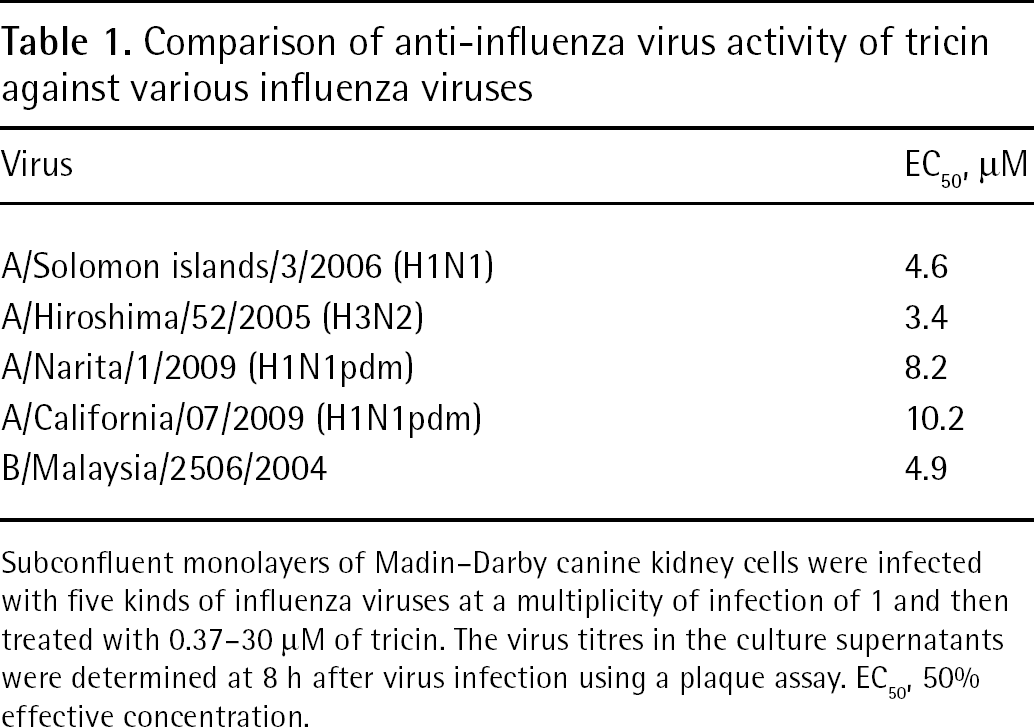
Subconfluent monolayers of Madin–Darby canine kidney cells were infected with five kinds of influenza viruses at a multiplicity of infection of 1 and then treated with 0.37–30 μM of tricin. The virus titres in the culture supernatants were determined at 8 h after virus infection using a plaque assay. EC50, 50% effective concentration.
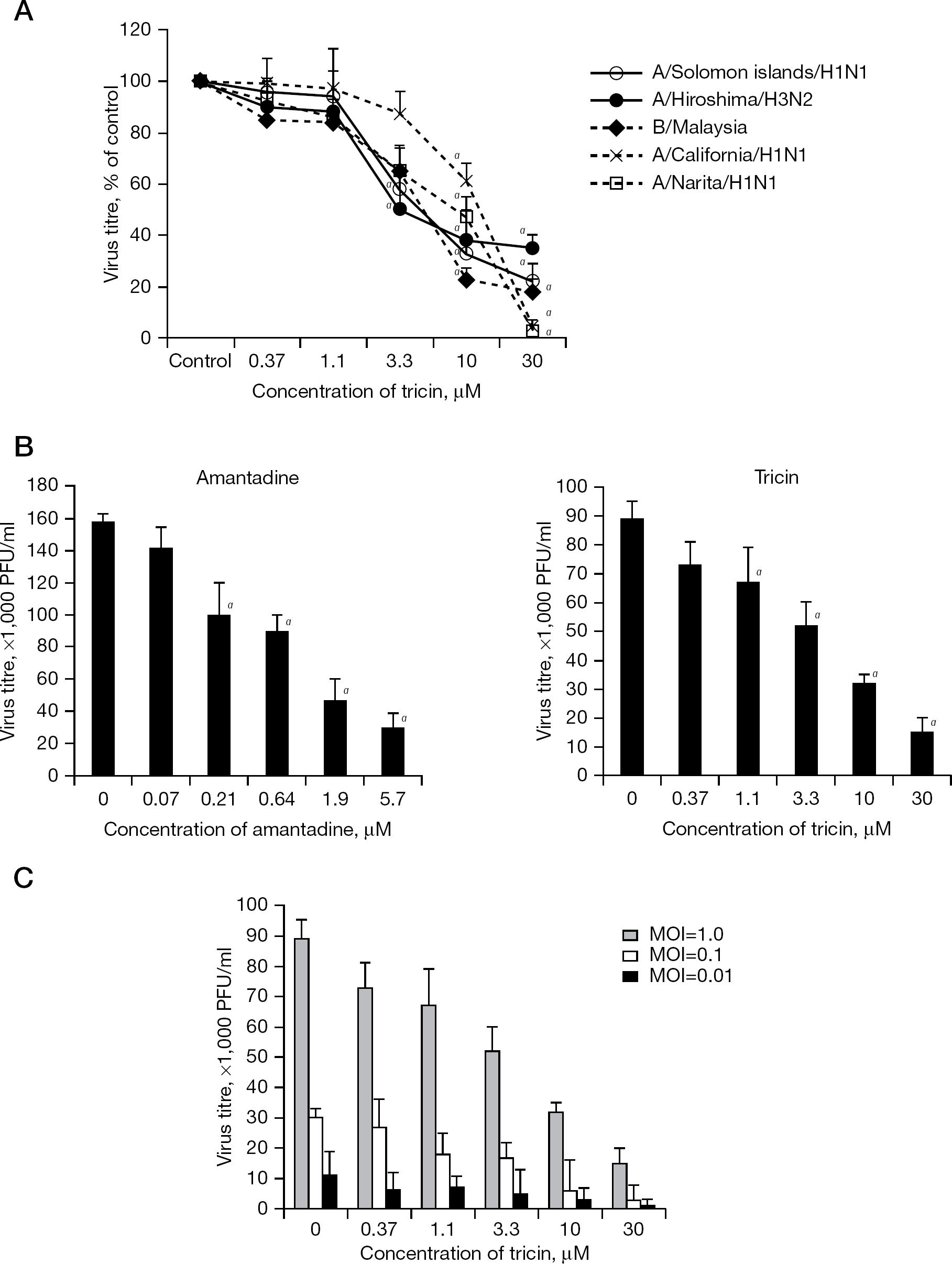
Inhibitory effects of tricin on influenza virus replication by an infectious virus yield reduction assay
In the next experiment, the virucidal effect of tricin was examined. Cell-free influenza A/Solomon islands/3/2006 (H1N1) virus was incubated with 30 μM tricin for 1 to 8 h. However, the infectivity of cell-free viruses was unchanged (Figure 3A). The cytotoxic effects of tricin were then examined after incubation (1, 24 and 72 h) of MDCK cells with concentrations of 0.37–30 μM tricin. However, no cytotoxic effects were observed, even at the highest tricin concentration (Figure 3B).
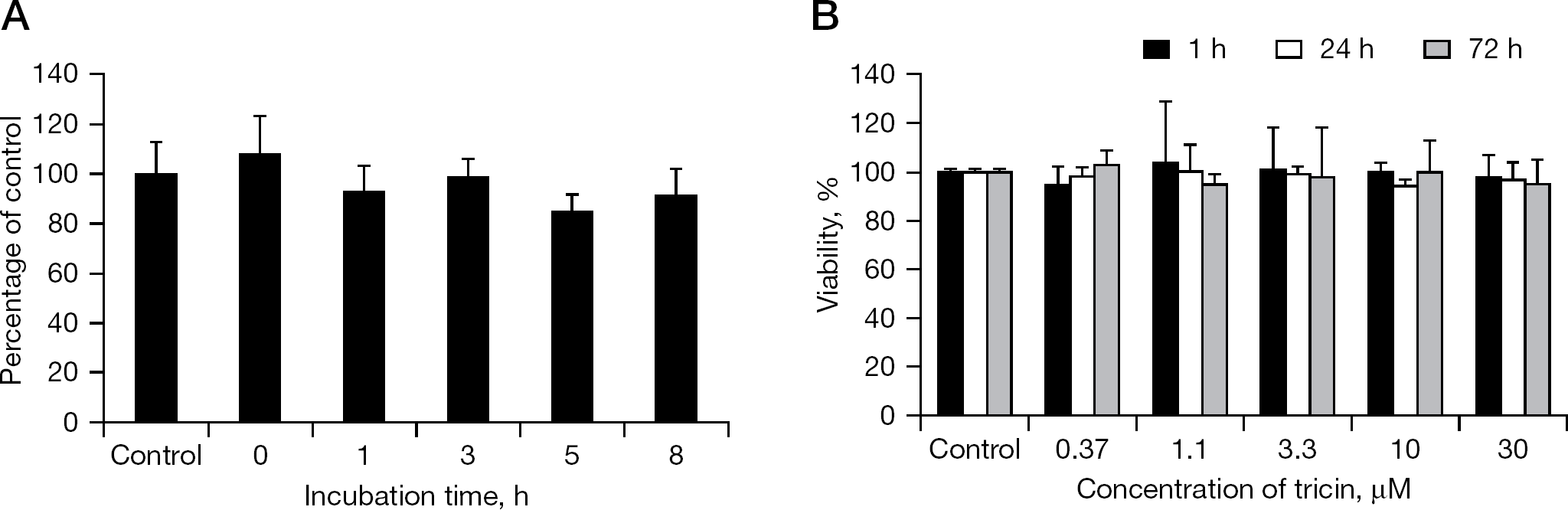
Toxic effects of tricin
Effect of tricin on influenza virus protein synthesis
To determine the molecular nature of influenza HA and M proteins expressed in the tricin-treated cells (3.3, 10 and 30 μM), we examined their synthesis in MDCK cells at the indicated time intervals by western blot analysis. As shown in Figure 4, influenza HA and M proteins were synthesized in virus-infected, tricin-untreated cells at 5 and 8 h post infection. By contrast, HA protein synthesis was strongly reduced by treatment of the influenza-virus-infected cells with tricin at 5 and 8 h post infection (Figure 4; lanes 2, 3 and 4). Moreover, the M protein synthesis was also inhibited by tricin treatment (Figure 4; lanes 7, 8 and 9).
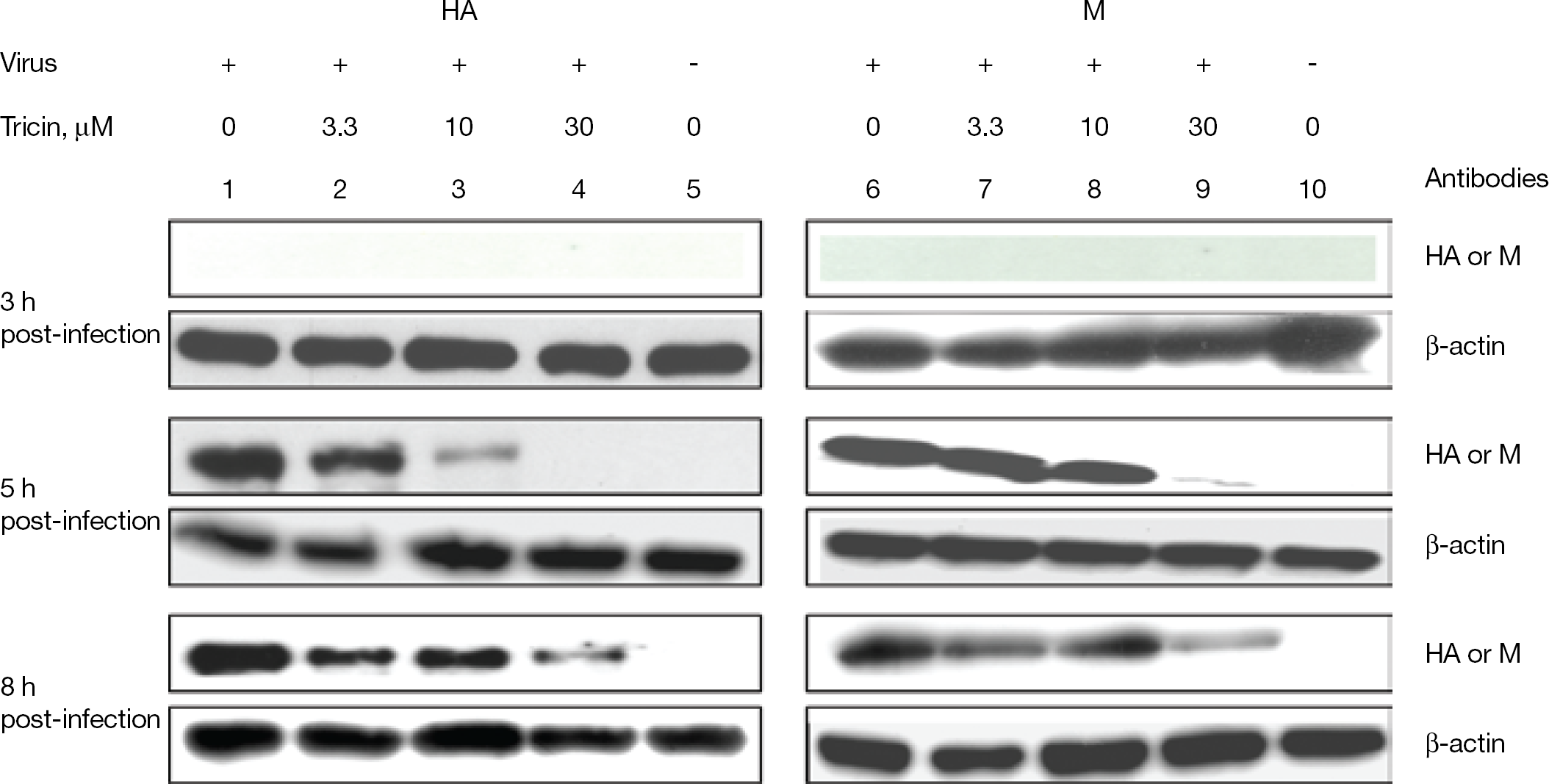
Regulation of protein synthesis by tricin in the influenza A/Solomon islands/3/2006 (H1N1) virus-infected cells
Detection of viral genes by RT-PCR analysis
To evaluate the effect of tricin on influenza virus gene expression, the MDCK cells infected with influenza A/Solomon islands/3/2006 (H1N1) virus were incubated in the presence or absence of tricin (3.3–30 μM). At 4 or 5 h post-infection, RNA was extracted from the infected cells, and viral RNA expression was determined by real-time RT-PCR analysis. This compound at 3.3 μM inhibited expression of HA messenger RNA (mRNA) by 41% at 4 h post-infection, whereas at 30 μM, expressions of both HA and M mRNA were inhibited by approximately 90% at 5 h post-infection (Figure 5). In addition, tricin inhibited the expression of HA mRNA in influenza-virus-infected cells at concentrations ranging from 3.3 to 30 μM. These results indicated that tricin suppresses expression of HA mRNA and M mRNA at 4 and 5 h post-infection in a dose-dependent manner.
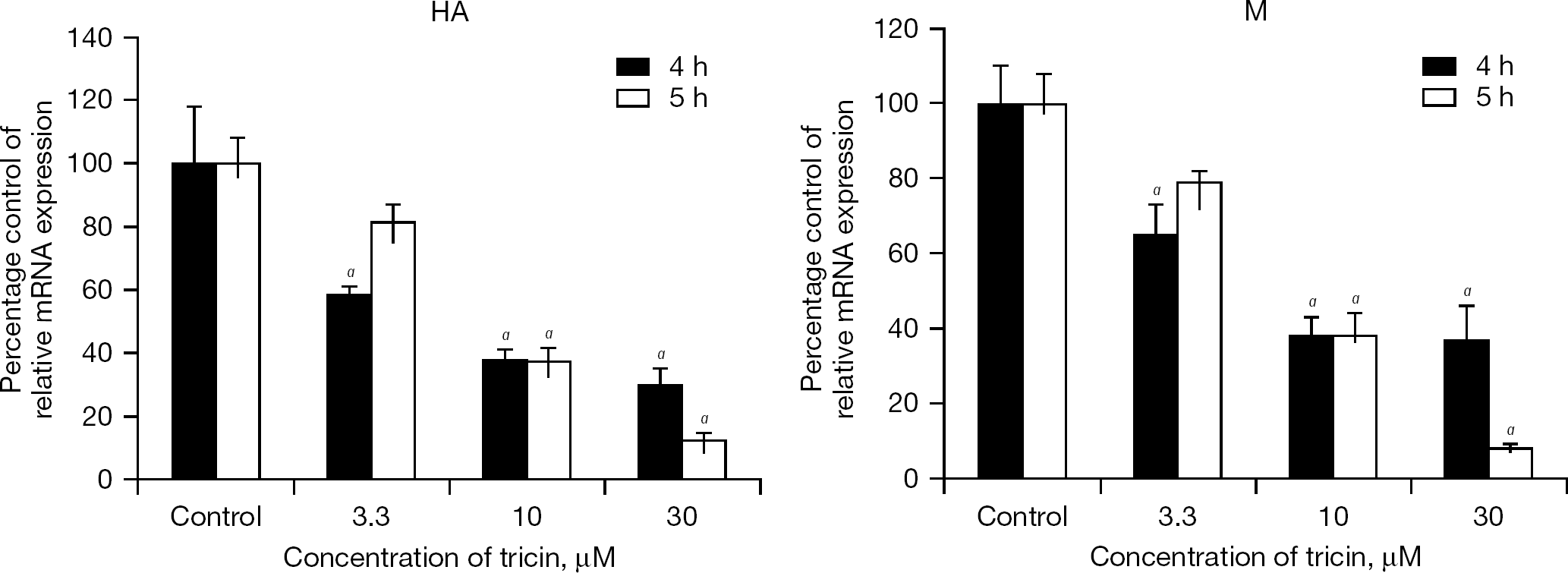
Effect of tricin on mRNA expression in the influenza A/Solomon islands/3/2006 (H1N1) virus-infected cells
Effect of tricin on the biological activities of influenza A virus
To investigate whether the effects of tricin on plaque inhibition are directly due to the biological activities in the virus replication cycle of influenza A viruses, we carried out HA inhibition and NA inhibition activity assays. First, we examined the effect of tricin on haemagglutination. Following incubation of erythrocytes with influenza A/Solomon islands/3/2006 (H1N1) and A/Hiroshima/52/2005 (H3N2) viruses, pretreated with 0.74 to approximately 190 μM of tricin, no inhibition of haemagglutination was observed (Figure 6A). The tricin-untreated control, however, demonstrated a reduction in haemagglutination. These results suggest that tricin does not interfere with viral attachment to host cell surfaces. Next, we examined NA inhibitor activity of tricin against influenza A/Solomon islands/3/2006 (H1N1) virus using an NA-StarTM influenza NA inhibitor resistance detection kit. When serially diluted agents (oseltamivir carboxylate, DMSO and tricin) were incubated with the virus, the values of light signal intensity were completely inhibited at >32 nM oseltamivir carboxylate, which was the positive control (Figure 6B). However, the light intensity values following treatment with tricin were unchanged, even at the highest concentration of tricin, and similar to those of the negative control (treatment with NA-Star assay buffer or DMSO; Figure 5B). Thus, tricin was found to exhibit a potential anti-influenza virus efficacy without NA inhibition. These results indicated that the tricin does not affect the biological activities of influenza A viruses, including HA and NA activities.
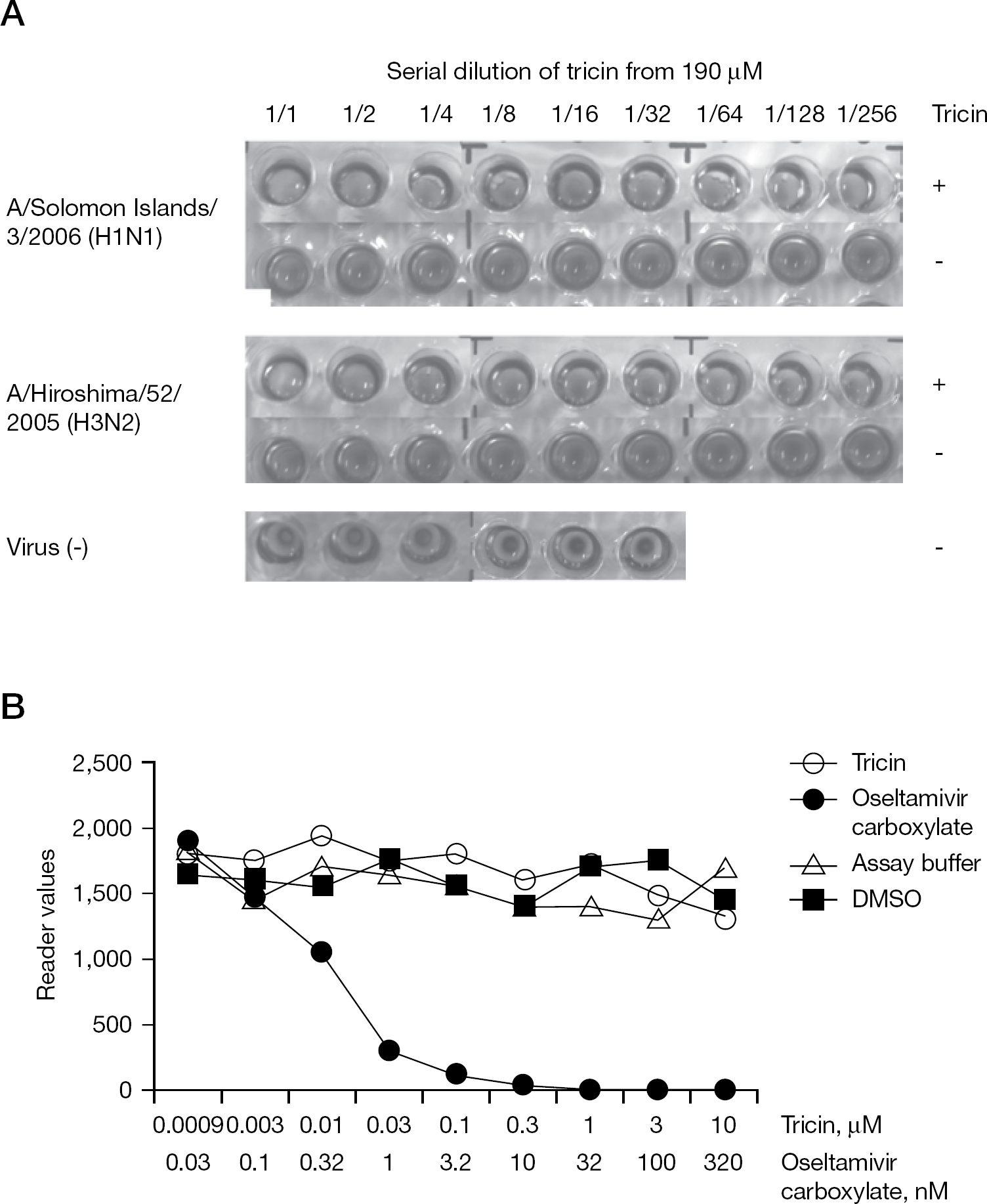
Effect of tricin on the biological activities of influenza A virus
Anti-influenza virus efficacy of tricin in vivo
Tricin efficacy was examined in a murine influenza virus infection model. In this model, weight changes of infected mice are a useful indicator of the morbidity of mice after a challenge with influenza virus [26,27]. We used 20 and 100 μg/kg of tricin as the doses for mice. Efficacies of the two doses were assessed by weight changes of the infected mice from day 0 to day 8 after infection. As shown in Figure 7A, the mean body weight of the infected mice administered 5% Arabic gum solution (control) decreased markedly from day 0 to day 8 after infection compared with that in the mock-infected mice. The administration of tricin significantly reduced the loss of mean body weight in the infected mice from day 0 to day 8 after infection (20 μg/kg, P=0.05; 100 μg/kg, P=0.03). Single oral administration of 2,000 mg/kg tricin did not produce body weight loss in the uninfected mice (MK et al., data not shown). Thus, tricin was found to exhibit potential anti-influenza virus efficacy in mice without toxicity.
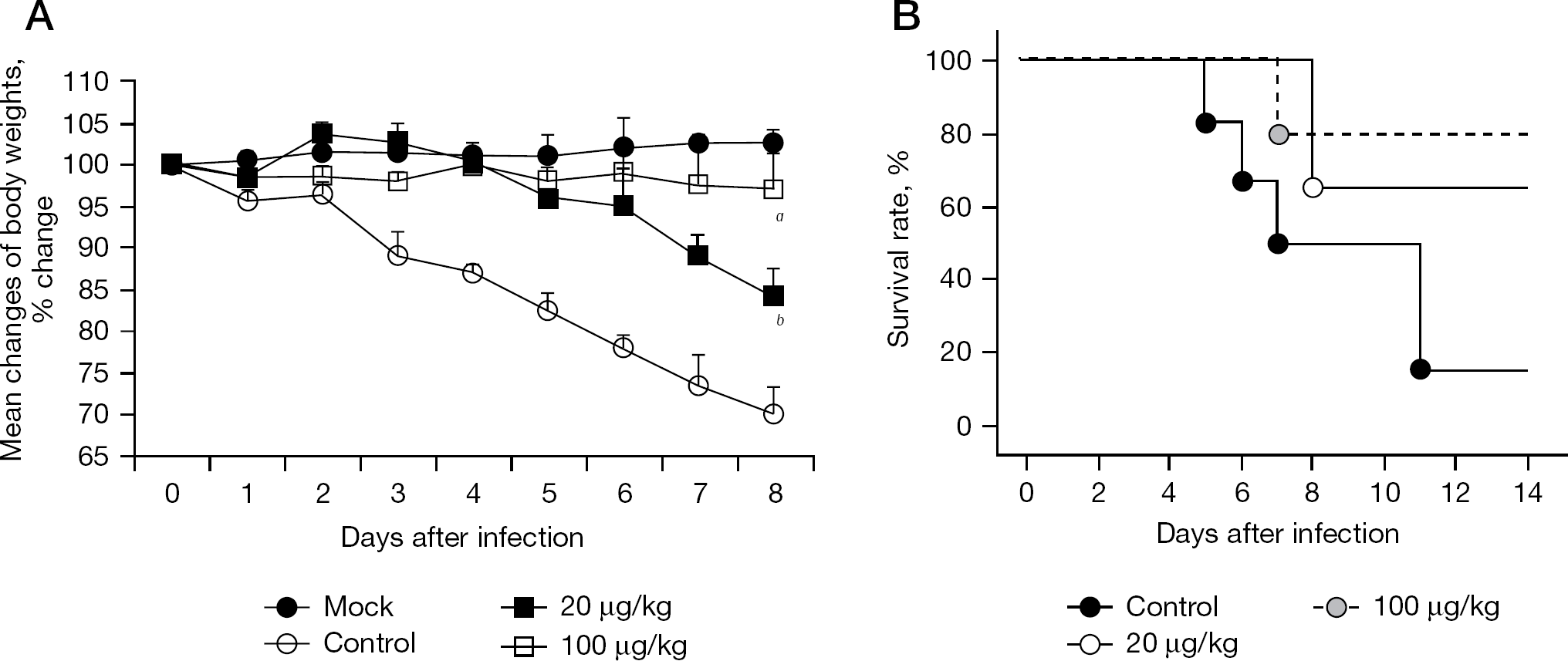
Anti-influenza efficacy of tricin in vivo
To confirm the potential anti-influenza virus efficacy of tricin, we examined the dose-dependent efficacy of tricin on the survival rate of the infected mice (Figure 7B). The infected mice were orally administered tricin at 20 and 100 μg/kg. Five infected mice that received 5% Arabic gum solution (control) died by day 14 after infection. However, 67% and 83% of the infected mice that received tricin at 20 and 100 μg/kg, respectively, were still alive on day 14 after infection. Thus, tricin was effective against flu virus infection in a dose-dependent manner and its anti-influenza virus efficacy in mice was confirmed.
Discussion
In this study, we found that tricin exhibited anti-influenza virus activity in vitro and in vivo. This is the first study demonstrating the anti-influenza virus activity of tricin. Tricin inhibits influenza virus production in a dose-dependent manner at doses of 3.3 to 30 μM (Figure 2A). The EC50 of tricin ranged from 3.4 to 10.2 μM for influenza A strains, 4.9 μM for influenza B virus, 8.2 μM for novel influenza A/Narita/1/2009 (H1N1pdm) and 10.2 μM for A/California/07/2009 (H1N1pdm) strains (Table 1). Moreover, viral replication of this novel influenza A virus was >95% inhibited by 30 μM tricin treatment (Figure 2A). This novel influenza A virus was also resistant to amantadine. Therefore, the anti-influenza virus mechanisms of tricin may differ from those of amantadine. Amantadine and its analogue, rimantadine, act as viral M2 channel blockers, and zanamivir and oseltamivir are viral sialidase inhibitors [5–7]. Amantadine and rimantadine are active against influenza A virus only, and produce rapid drug-resistant variants. Studies on the mechanisms of action are consistent with tricin mainly affecting the early processes of the viral replication cycle. The first step of the influenza virus replication cycle involves the binding of viral HA to sialic acid receptors on the host cell surface. Erythrocyte haemagglutination is mediated by the interaction of viral HA and sialic acid containing glycoconjugates on the surfaces of erythrocytes [28–30]. However, tricin had no inhibitory effect on haemagglutination and NA activity (Figure 6A and 6B). These results suggest that the decrease in progeny virus production is not due to the inhibition of viral HA and NA. Tricin was added to before or after virus-infected cells at various times; we found that tricin inhibited viral replication both before and after viral adsorption to the host cell (KY et al., data not shown). The data suggests that tricin might act at the early to middle stage of the viral replication cycle, presumably via blockage of viral protein production by host-mediated molecules. Indeed, we found that tricin may act as an immunomodulator (TM et al., unpublished observations). Tricin has been reported to have a chemical composition and pharmacological activity of an anti-human cytomegalovirus agent [15]. We also found that prostaglandin E2 accumulation and induction of cyclooxygenase-2 (COX-2) protein synthesis by human cytomegalovirus infection were inhibited by tricin treatment [31]. Therefore, tricin may also be a novel COX synthesis inhibitor. Lee et al. [32] and Li et al. [33] reported that influenza A virus has been shown to induce COX-2 accumulation. Furthermore, COX inhibitors substantially block influenza A virus-induced inflammatory responses. Although the effects of COX inhibitors on influenza disease in humans have not yet been tested, there are indications that this class of drugs will help control herpes simplex virus and influenza virus in an animal model; COX-2 inhibitors have been shown to suppress herpes simplex virus reactivation in a mouse model of latency [34] and exhibit anti-influenza virus activity in mice [35].
Tricin administered orally significantly reduced body weight loss of infected mice (Figure 7A) and prolonged survival times (Figure 7B). The dose of 20 μg/kg of tricin was effective in countering body weight loss and prolonging the survival time of infected mice, indicating that the efficacy of tricin against influenza virus was dose-dependent. In the in vitro study, tricin did not show cytotoxicity against MDCK cells (Figure 3B), and similar to results of the in vivo study, no body weight loss was observed in mice administered tricin at 2,000 mg/kg. Thus, tricin is likely adsorbed from the alimentary tract in mice thereby exhibiting anti-influenza virus action.
In conclusion, our results predict that tricin might be candidates as natural anti-influenza virus compounds and useful as therapeutic agents in influenza-virus-infected hosts. Further investigation into the mechanisms of its antiviral action, as well as the chemical modification of tricin, should be conducted to further develop these compounds for clinical use.
Footnotes
Acknowledgements
This work was supported in part by the Specific Research Fund of Hokuriku University.
The authors declare no competing interests.