
Abstract
Background:
Dengue fever, dengue haemorrhagic fever, and dengue shock syndrome are caused by infections with any of the four serotypes of the dengue virus (DENV), and are an increasing global health risk. The related West Nile virus (WNV) causes significant morbidity and mortality as well, and continues to be a threat in endemic areas. Currently no FDA-approved vaccines or therapeutics are available to prevent or treat any of these infections. Like the other members of Flaviviridae, DENV and WNV encode a protease (NS3) which is essential for viral replication and therefore is a promising target for developing therapies to treat dengue and West Nile infections.
Methods:
Flaviviral protease inhibitors were identified and biologically characterized for mechanism of inhibition and DENV antiviral activity.
Results:
A guanidinylated 2,5-dideoxystreptamine class of compounds was identified that competitively inhibited the NS3 protease from DENV(1–4) and WNV with 50% inhibitory concentration values in the 1–70 μM range. Cytotoxicity was low; however, antiviral activity versus DENV-2 on VERO cells was not detectable.
Conclusions:
This class of compounds is the first to demonstrate competitive pan-dengue and WNV NS3 protease inhibition and, given the sequence conservation among flavivirus NS3 proteins, suggests that developing a pan-dengue or possibly pan-flavivirus therapeutic is feasible.
Introduction
The four dengue virus (DENV) serotypes (DENV[1–4]) are among the most significant public health threats in tropical and subtropical regions, and dengue fever is the most rapidly spreading mosquito-borne viral disease. Up to 55% of the world's population is at risk for dengue infection. Due to the geographical expansion of the Aedes species mosquito vector, an estimated 50–100 million individuals in 124 countries currently suffer dengue infections annually [1]. Furthermore, infected travellers may enter non-endemic countries and become a source of domestic transmission [2]. Occurrences of local transmission in Hawaii, Texas, and most recently in Key West and Miami, Florida [3,4] demonstrate the growing threat of this disease in the US. Establishment of endemic dengue in the US would create a substantial disease and economic burden. For comparison, endemic dengue costs to the health sector and overall economy in eight affected countries approaches USD 2 billion annually [5].
The signs and symptoms of dengue infection may range from mild to severe discomfort and malaise (dengue fever) to life-threatening dengue haemorrhagic fever (DHF) and dengue shock syndrome (DSS). Even those with relatively uncomplicated dengue fever suffer significant morbidity. Over 2 million patients annually develop DHF or DSS and require hospitalization with intensive care. Without the currently accepted, but inadequate, supportive treatment, DHF mortality can exceed 20% [1,6]. Furthermore, infection with one serotype does not provide cross-protective immunity against the other serotypes and, in contrast, is believed to increase the probability of developing life-threatening DHF or DSS upon infection with a second serotype by mechanisms such as antibody dependent enhancement [7,8]. Despite decades of intensive research efforts, there are no approved vaccines or therapeutics available to prevent or treat DENV infections.
The related West Nile virus (WNV) is considered another significant disease-causing flavivirus, and is transmitted primarily by Culex mosquitoes. Morbidity and mortality associated with WNV infection is particularly concerning due to a significant incidence of long-term sequelae [9]. Those that develop the neuroinvasive form of West Nile disease experience increased mortality [10]. Cases of WNV infection in the US peaked in 2003 and have subsequently decreased steadily, although it still remains a threat in the US, Europe, and other developed countries [9,11,12]. Currently there is no licensed vaccine or specific antiviral therapeutic available to prevent or treat WNV infections.
The DENV along with the WNV, tick-borne encephalitis, yellow fever and Japanese encephalitis are members of the Flaviviridae and contain an enveloped, 11 kb positive-stranded RNA genome [13]. This genome encodes a single polyprotein containing three structural proteins and seven non-structural (NS) proteins involved in viral replication and particle maturation. Similar to other members of this family, the polyprotein requires proteolytic cleavage to release its individual functional proteins [13,14].
The DENV NS3 protease domain is located within the approximately 170 N-terminal amino acid residues of the 618 amino acid full-length NS3 protein, and requires the NS2b cofactor for expression in Escherichia coli of a soluble protein with optimal enzymatic activity [15–17]. The NS3 viral protease processes the majority of the required polyprotein cleavages and is essential for virus production, making it an ideal target for intervention [18,19]. In the closely related HCV, NS3 is a validated drug target and inhibitors of HCV NS3 protease are in clinical trials for the treatment of chronic HCV infections [20,21]. Clinical trials using a first generation inhibitor [22] were shown to be effective in reducing HCV viral load, but the early clinical candidate BILN-2061 was abandoned due to cardiotoxicity in animal studies [23]. Improved second generation compounds are showing more promise [24–26].
The NS3 proteases of all four DENV serotypes have significant amino acid sequence homology (65–74%) and shared substrate preference [27], suggesting that therapeutics targeting DENV NS3 protease have the potential for displaying pan-dengue antiviral activity. More importantly, the homology among the residues lining the DENV substrate binding sites is significantly higher and is essentially identical on the non-prime side of the site. Furthermore, the high homology with other flaviviruses suggests that a pan-flavivirus NS3 protease inhibitor may be feasible.
Here we report the results of inhibitor validation activities, occurring after a compound screening campaign, using the WNV and DENV NS3 proteases. In doing so, the majority of the initial hits were eliminated by identifying them as promiscuous non-specific inhibitors. However, the guanidinylated 2,5-dideoxystreptamine class of compounds demonstrated bona fide and potent NS3 protease inhibition against the DENV(1–4) and WNV NS3 proteases. These compounds have been previously demonstrated to inhibit furin and anthrax lethal factor proteases [28,29] and are shown here to be competitive inhibitors of WNV and DENV NS2b/NS3 proteases from all four serotypes.
Methods
Chemistry
Materials
Peptide substrates were custom synthesized to order by AnaSpec (Fremont, CA, USA). Codon optimized synthetic genes were obtained from DNA2.0 (Menlo Park, CA, USA) using NS2b and NS3 protease amino acid sequences from prototype viral strains (WNV Genbank number 1710261A; DENV-1 Singapore/S275/1990 Genbank accession number P33478; DENV-2 New Guinea C Genbank accession number AF038403; DENV-3 Philippines/H87/1956 Genbank accession number P27915; DENV-4 Dominica/814669/1981 Genbank accession number P09866). Chromatography media were obtained from GE Healthcare (Piscataway, NJ, USA). All other standard materials and supplies were obtained from Fisher Scientific (Pittsburgh, PA, USA), Sigma–Aldrich (St Louis, MO, USA), Invitrogen Corp. (Carlsbad, CA, USA) or as indicated.
Compound synthesis
The guanidinylated 2,5-dideoxystreptamine compounds were synthesized as previously described [28,29].
Production of DENV(1–4) and WNV NS2b/NS3 proteases
Synthetic genes, codon-optimized for expression in Escherichia coli, for the four DENV NS3 proteases encoding residues 1–184 together with the 48 amino acid hydrophilic domain of NS2b (residues 48–95) attached via a small flexible nine amino acid linker (Gly4SerGly4) were obtained from DNA2.0. The genes were inserted into the pQE30 expression vector (Qiagen, Valencia, CA, USA) using the BamHI and SalI sites of the plasmid. The N-terminally 6×His tagged protein was expressed in Escherichia coli XL1 Blue (Agilent, Santa Clara, CA, USA). Cells were grown at 37°C until an optical density of 0.6 was reached, then induced with 1 mM IPTG for 4 h at 37°C. The DENV NS3 protease from all serotypes was purified from cell lysate in a denatured state followed by an on-column refolding procedure. Therefore, harvested cells were lysed by sonication in denaturing lysis buffer (20 mM potassium phosphate pH 7.4, 300 mM NaCl, 20 mM imidazole and 6 M guanidine-HCl), cleared by centrifugation and the supernatant applied to a 5 ml IMAC column (HiTrap; GE Healthcare Life Sciences, Piscataway, NJ, USA) equilibrated in lysis buffer. The column was washed with 15 ml of lysis buffer and bound protein was refolded on the IMAC column by a reverse gradient to lysis buffer lacking the guanidine-HCl. Bound refolded DENV NS2b/NS3 was eluted with a linear gradient to 20 mM potassium phosphate pH 7.4, 300 mM NaCl, containing 500 mM imidazole. Eluted protein was collected, and then buffer exchanged into 20 mM Tris-HCl pH 8.0, and further purified using Q15 anion-exchange chromatography (Source-15Q; GE Healthcare Life Sciences). After loading protein and washing with 20 mM Tris-HCl pH 8.0, bound protease was eluted on a gradient up to 1 M NaCl in wash buffer, collected, buffer exchanged to remove salt, and concentrated to ≥2 mg/ml in 20 mM Tris-HCl pH 8.0/20% glycerol using an Amicon micro-concentrator (10,000 Da MWCO; Millipore, Billerica, MA, USA). Purified protease was stored at −80°C until needed for enzymatic assays.
Expression of a similar synthetic construct for the WNV NS3 protease under the same conditions resulted in a fully soluble protein extract. Therefore, harvested cells were routinely lysed by sonication in 20 mM potassium phosphate pH 7.4, 300 mM NaCl, 20 mM imidazole and the native protein purified by IMAC and Q15 anion-exchange chromatography similarly to the DENV NS3 protease purification protocol above.
Virology
NS2b/NS3 protease assays: FRET peptide cleavage assay
For compound screening and routine determination of the 50% inhibitory concentration (IC50), the DENV NS2b/NS3 proteolytic activity was measured in a continuous format using a fluorescence resonance energy transfer (FRET)-based assay with the FRET-3 substrate (QXL570-Phe-Ala-Ser-Gly-Lys-Arg-Ser-Gln-Ile-Gly-Leu-Lys(5-TAMRA)-NH2). The 100 ul assay conducted in a 96-well plate contained 50 mM Tris-HCl pH 8.8, 20% Glycerol, 0.050% BRIJ 35, 0.1 mg/ml BSA, 5 μM FRET-3, 25–200 nM NS2b/NS3 protease, 2% DMSO and varying concentrations of inhibitor. This level of DMSO does not adversely affect the activity of the NS3 proteases and DMSO only control reactions were performed for each NS3 protease and included in IC50 curve fitting for each inhibitor. Inhibitors were diluted into assay buffer from 10 mM stock solutions prepared in 100% DMSO. Assay reactions are started with the addition of enzyme and continuously monitored for 15 min at 25°C using fluorescence excitation and emission wavelengths of 550 and 585 nm, respectively. Initial rates of product formation (<10% peptide cleavage) are determined and fitted by non-linear least squares routines to standard equations to determine IC50 values using KaleidaGraph 4 (Synergy Software, Reading, PA, USA).
NS2b/NS3 protease assays: Colorimetric peptide cleavage assay
The colorimetric assay depends upon the proteolytic release of the C-terminal para-nitroaniline (pNA) P1-prime moiety from the substrate Phe-Ala-Ser-Gly-Lys-Arg-pNA. This pNA substrate is a mimic of the P6 to P1 residues at the NS3-NS4A natural cleavage site of the WNV polyprotein capped with a C-terminal pNA tag. Release of pNA due to enzyme catalyzed proteolysis results in an increase in absorbance at 405 nm. The pNA assay, due to its extensive dynamic linear range, and insensitivity to inner filter effects of inhibitor compounds or substrate itself, which limit the utility of FRET-based assays, was used for the mechanism of inhibition studies. The concentration of the pNA peptide substrate was varied and cleavage monitored by ΔA405 nm under the same assay conditions as described for the FRET assay. A molar extinction coefficient of 10,500 M−1cm-1 was used to calculate rates of peptide cleavage. For mechanism studies, rate data was fitted by global analysis to competitive and mixed inhibition equations using GraFit 5.0 (Erithacus Software, Surrey, UK).
Compound library screening
The FRET 96-well plate based assay was used to screen our approximately 12,000 member corporate compound library for WNV NS2b/NS3 inhibition. Assay conditions were as described above except that the concentration of the tested compounds was fixed at 25 μM. Compounds that inhibited the proteolytic activity >40% were identified as hits and chosen for further characterization regarding IC50 determination against both the WNV and the four DENV NS2b/NS3 proteases, identification of potential promiscuous behaviour and mechanism of inhibition.
Antiviral testing: Virus growth assay
Compounds were evaluated for inhibition of DENV-2 growth on Vero cells. For this, viral growth was measured by ELISA detection of virally produced envelope protein as described below. Vero cell cytotoxicity was also assessed for each compound using an MTT-based viability assay with the same format.
For the assays, 100 μl EMEM (Fisher Scientific) supplemented with L-glutamine, NEAA and heat-inactivated fetal bovine serum (Invitrogen Corp.) containing 2×104 Vero cells were seeded into each well on 96-well plates and allowed to grow at 35°C, 5% CO2 for 3 days to reach approximately 80% confluence. Test compounds (10 mM) dissolved in 100% DMSO were diluted in culture medium and added to each well to achieve final concentrations from 0.78 μM to 100 μM in a twofold dilution series in triplicate. After 30 min of pre-incubation of cells with test compound, 20 plaqueforming units of DENV-2 were added to each well and incubated for 4 days at 35°C, 5% CO2. Culture medium was removed and cells were fixed by adding 300 μl of ice cold 1:1 methanol-ethanol solution and incubating for 60 min at −20°C. Wells were then washed three times with phosphate-buffered saline (PBS) to remove the fixative. For immunochemical detection of DENV-2 envelope protein, the fixed cells were probed with the dengue-E reactive monoclonal antibody D1-4G2-4-15 (produced from hybridoma line HB-112; ATCC, Manassas, VA, USA) in 100 μl blocking diluent (0.5% BSA, 0.5% goat serum in water), covered and incubated at 35°C for 2 h. After washing three times with PBS, 100 μl/well of goat-anti-mouse-HRP conjugate in blocking diluent was added and plates further incubated for 1 h at 35°C. After another three PBS washes, TMB substrate solution was added and plates incubated for 10–20 min at room temperature. Stop solution was then added and the optical density determined at 650 nm. The absorption value for each test well was corrected for background and compared to values obtained from infected, untreated wells to determine relative level of reduction in virus growth.
Results
Production of DENV(1–4) and WNV NS2b/NS3 proteases
DENV NS3 proteases from all 4 serotypes were found to be largely insoluble after Escherichia coli cell lysis in non-denaturing IMAC buffer. These results are consistent with a report by Leung et al. [16], where it was observed that the expressed DENV NS2b/NS3 protein soluble fraction, was accompanied by a significant insoluble fraction. Since the catalytic parameters determined using our non-acetylated pNA peptide substrate for refolded protein, and for the protein purified from the soluble fraction were similar to each other and to those reported by Leung et al. [16], we chose to refold all expressed material to improve yields. The Escherichia coli expressed WNV NS3 protease was fully soluble and was therefore purified in its native state.
NS3 protease inhibitor identification and characterization
More than 140 hits, encompassing many unique and diverse structures, were identified with >40% inhibition against WNV NS2b/NS3. These were repurchased and in most cases, a dose-dependent behaviour was demonstrated and IC50 values calculated. We had a collection of >40 compounds demonstrating IC50 values <10 μM, and many of these compounds also inhibited the DENV-2 NS3 protease.
Prior to conducting cell-based antiviral testing with a subset of the hits, we attempted to verify that they functioned as competitive inhibitors. However, initial experiments on the most promising compounds gave unexpected results, indicating that these compounds were not displaying competitive inhibition. Furthermore, the observed inhibition levels were found to be variable, suggesting that some of these compounds could be binding in an unexpected manner or were perhaps false positives. We investigated the possibility that our compound set was populated with compounds that exhibited promiscuous inhibition due to insolubility or aggregation. These properties may lead to adsorption of the protein on the surface of the solid and in effect remove it from participation in the desired enzymatic reaction; a result which artifactually appears as enzyme inhibition [30]. Subsequent microscopic observation of assay wells containing test compounds revealed precipitated compound in the majority of cases even in the presence of BRIJ-35 detergent.
In an effort to follow-up on this initial finding, potential inhibitors were again tested after removing insoluble material in assays by centrifugation prior to adding enzyme. As hypothesized, the previously observed inhibition of many of these compounds was completely abolished. Although the majority of the hits were now considered false positives, some of the compounds prevailed and were investigated in more detail. Based on their ultraviolet/visible absorbance spectrum, some of these compounds would have the potential to cause inner filter effects [31] in our FRET assay. In order to test for this potential cause of artifactual inhibition, these compounds were tested again using the colorimetric pNA peptide cleavage assay. Since inner filter effects do not affect colorimetric assays, true inhibition of enzyme-catalyzed peptide cleavage could now be revealed. Unfortunately, the majority of the remaining hits did not show any level of inhibition using this substrate, again, suggesting non-specific inhibition. One caveat to using the pNA substrate is that due to the small pNA moiety and the lack of additional C-terminal amino acids in the peptide substrate, it does not appreciably occupy the prime side of the catalytic site but occupies primarily the non-prime portion of the substrate binding site that is N-terminal to the scissile bond. This lack of occupancy would possibly preclude detecting enzyme inhibition with compounds that bound primarily to the prime side of the substrate binding pocket. As a result, the possibility remained that the lack of observed inhibition was due to inhibitor binding selectively to the prime side. To address this hypothesis, the remaining active compounds were evaluated using the original FRET-3 substrate which spans both the prime and non-prime substrate binding regions of the active site. For this, an HPLC-based method was used to detect substrate cleavage and product formation rather than fluorescence to avoid confounding inner filter effects. With the exception of one class of compounds, all of the hits had now been eliminated from further consideration as true WNV or DENV NS2b/NS3 protease inhibitors. We concluded that the majority of our hits exhibited promiscuous, non-specific inhibition similar to what others have observed with flavivirus NS3 proteases and other screening campaigns [30,32–34]. The one remaining compound class which passed all of the above tests possesses several positively charged groups at physiological pH, perhaps acting as surrogates for the basic P1-P2 residues found in preferred peptide substrates. The activity of this surviving class of compounds, the guanidinylated 2,5-dideoxystreptamine derivatives, was investigated in further detail.
Since the set of guanidinylated 2,5-dideoxystreptamine compounds inhibited DENV-2 as well as WNV NS2b/NS3 protease, the representative compounds were tested versus the protease from the other three DENV serotypes. Figure 1 presents data for all compounds of this class tested for activity against the NS3 proteases of all four DENV serotypes and WNV. Aprotinin was used as a comparison and its observed IC50 values are in agreement with published values for WNV and DENV-2 NS3 protease inhibition [35]. The IC50 values obtained with this set of guanidinylated 2,5-dideoxystreptamine compounds are similar for each enzyme suggesting a common mechanism and binding site.
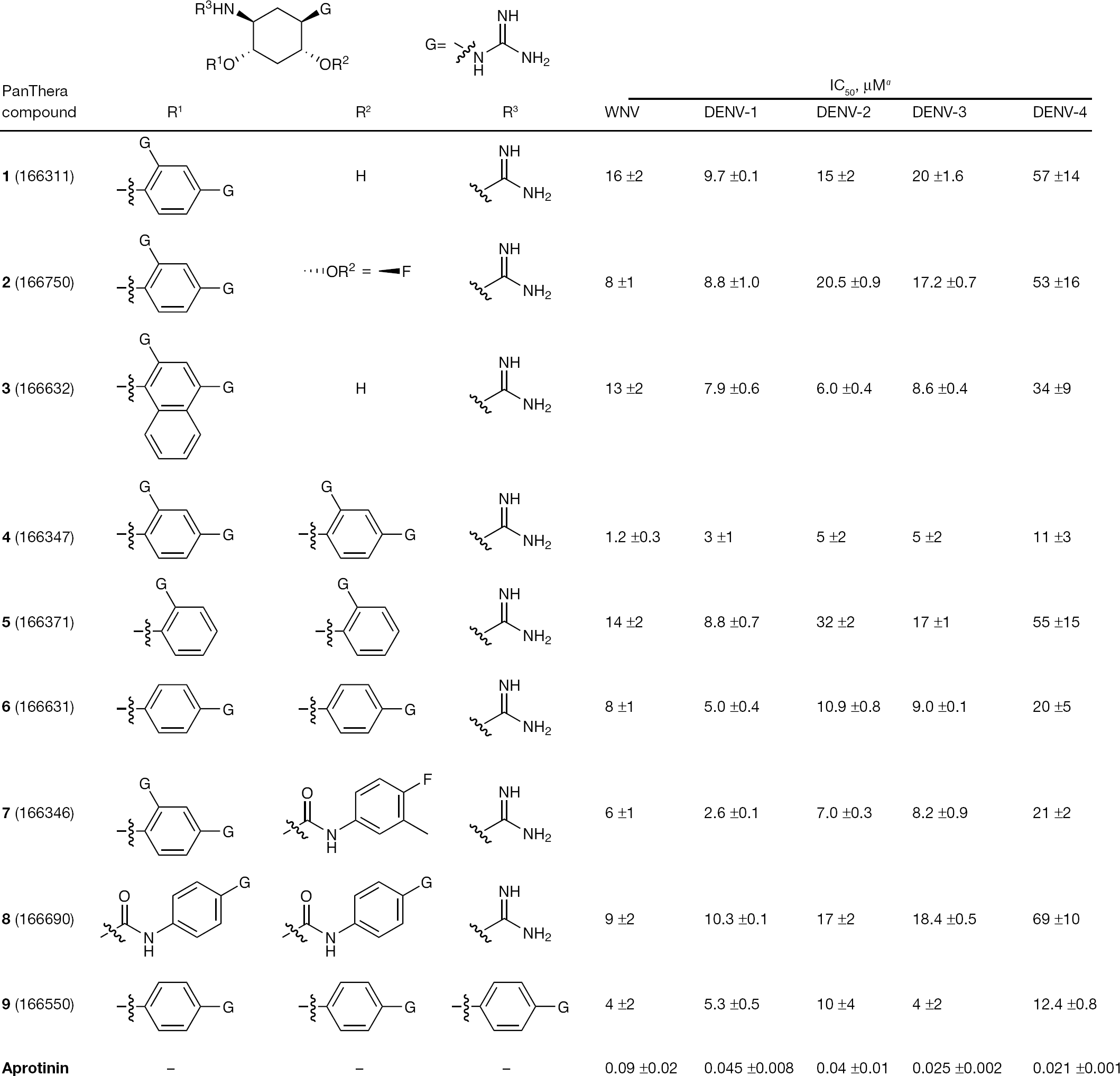
Pan-dengue and WNV NS2b/NS3 protease inhibition
Structure-activity relationships
As shown in Figure 1, the guanidinylated 2,5-dideoxystreptamine class of inhibitors is capable of blocking NS3 protease activity for all four DENV and WNV. In general, inhibition of DENV-4 NS2b/NS3 protease activity is lower by two- to fivefold relative to the remaining serotypes. The presence of a single guanidine group (G-group) at the para-position of a phenyl ring (R1 and R2) appears to be preferred by twofold relative to the ortho-position (confer
Mechanism of inhibition
The colorimetric pNA assay was used to determine the mechanism of protease inhibition to avoid complications due to inner filter effects associated with our more sensitive FRET-based assay. Substrate was varied from 0.05 to 1 mM, while the inhibitor 166347 was varied from 0 to 50 μM. Initial rates of product formation were globally fitted to the competitive and mixed inhibition models. The fitted parameter Vmax was converted to kcat by the equation kcat =Vmax/Et, and thus kcat =2.5 ±0.2 min−1, Km =810 ±112 μM, and Ki=9.8 ±0.8 μM for 166347 versus DENV-2 NS2b/NS3 protease. The double reciprocal plot for the competitive fit between the DENV-2 enzyme and 166347 is shown in Figure 2. Data was also fitted to the mixed inhibition equation but this did not result in an improvement of fit parameters and indicates no evidence of mixed competitive behaviour, since the resulting fitted noncompetitive Ki prime >> Ki or negative and irrelevant. The inhibitor 166347 was tested against West Nile NS2b/NS3 protease as well and demonstrated competitive behaviour (LC-H et al., graphical data not shown) with resulting fitted parameters of kcat =5.4 ±0.2 min−1, Km =71 ±12 μM, and Ki=0.7 ±0.1 μM and no evidence of mixed inhibition. This analysis was extended to an additional member of this class, 166550, which also demonstrated competitive behaviour for DENV-2 and WNV NS2b/NS3 proteases. Fitted parameters for 166550 and DENV-2 protease are kcat =2.1 ±0.2 min−1, Km =633 ±75 μM, Ki=7.1 ±0.5 μM and for WNV protease are kcat =4.0 ±0.1 min−1, Km =58 ±5 μM and Ki=3.6 ±0.3 μM (LC-H et al., graphical data not shown).
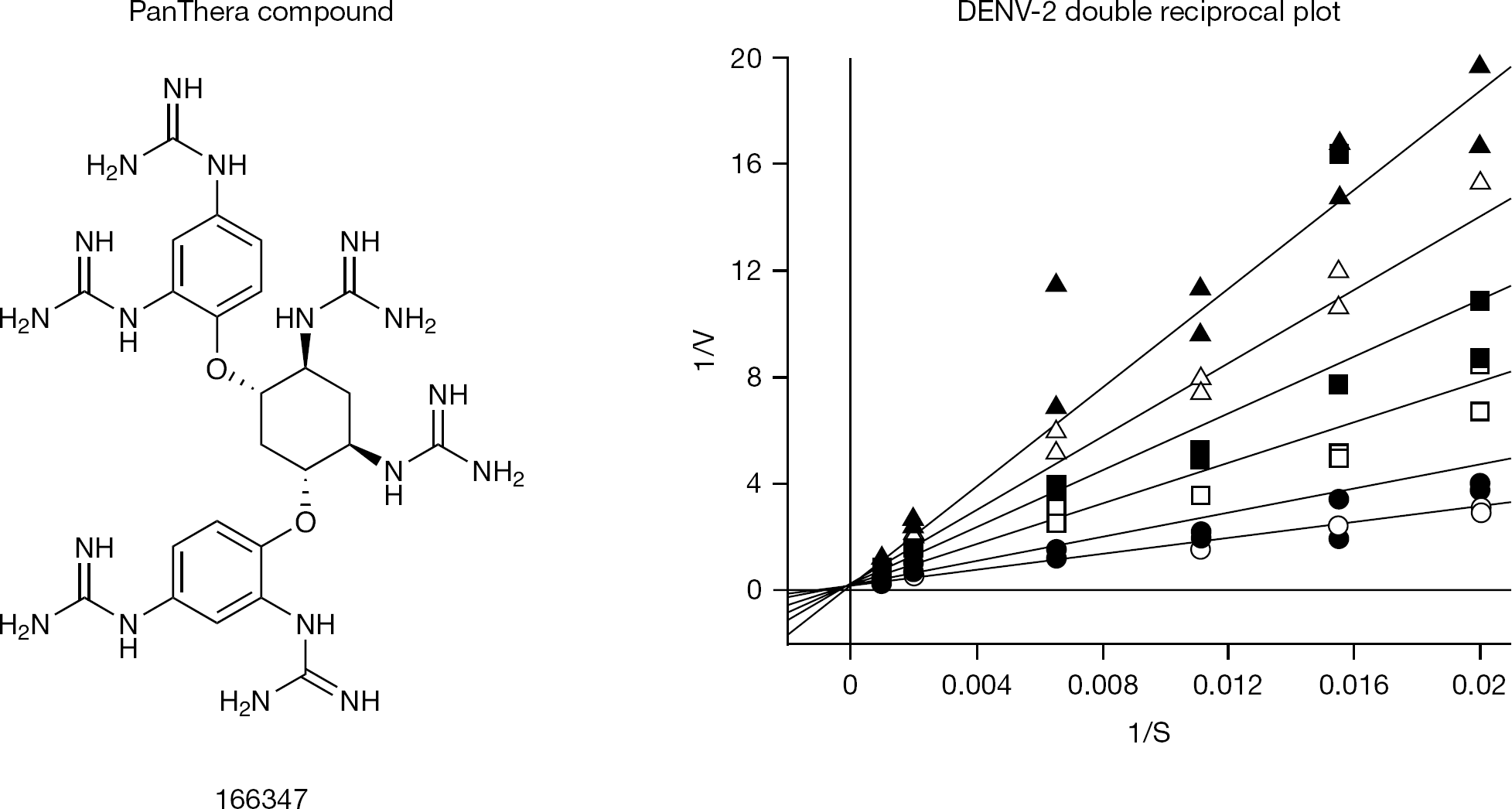
Compound 166347 demonstrates competitive inhibition with DENV-2 NS3 protease
Antiviral testing
Inhibitors of the essential NS3 protease have potential antiviral activity. Accordingly, antiviral activity was measured using a viral growth assay. Due to their highly cationic nature, we predicted that the cell based activity of these compounds would be poor. For this reason, the most lipophilic (cLogP=-0.06) compound from this class (166550) was chosen for DENV-2 antiviral testing. This compound, tested at a maximum concentration of 100 μM, showed neither signs of cytotoxicity nor a measurable reduction in virus growth of DENV-2 in Vero cells and are unremarkable (LC-H et al., data not shown). Presumably the highly charged nature of the compound blocked cellular entry or possibly the compound was bound to serum proteins in the culture medium and sequestered.
Discussion
Discovery of new inhibitors of flaviviral enzymes essential for viral growth, such as the DENV and WNV NS3 protease, is critical for future development of safe and effective flavivirus therapeutics. In this report we identify a new class of DENV and WNV NS3 protease inhibitors, the guanidinylated 2,5-dideoxystreptamines. The highly positively charged nature of the compounds suggests that they act as substrate mimetics and interact with the highly negatively charged non-prime portion of the substrate binding pocket. Inhibition kinetics show competitive behaviour with a substrate that only interacts with the non-prime side and catalytic machinery within the active site, suggesting that the compounds bind the non-prime side of the active site exclusively. The multiple guanidine groups are presumably an isosteric replacement for the non-prime side arginine and lysine residues required for optimal substrate cleavage [27] and are adequately arrayed during binding. This class of compounds has previously been identified as inhibitors of the anthrax lethal factor metalloprotease and the serine protease Furin [28,29], both of which have contiguous basic amino acids as consensus sequences for substrate preference. However, similar to the findings reported here, significant intracellular cell-based activity is lacking. Therefore these compounds may have a better chance being developed for therapeutic activity versus extracellular targets rather than intracellular, or used as leads in a pseudo-peptidomimetic approach for identifying flaviviral NS3 protease inhibitors. However, a directed medicinal chemistry approach to introduce lipophilicity may enhance antiviral activity dependent upon intracellular mechanisms.
We envision that the path toward flavivirus therapeutics, at least for the NS3 protease, may need to be similar to that taken in successful HCV NS3 protease drug discovery programmes. The HCV protease active site topology presented significant challenges due to its solvent exposed and relatively flat surface. This featureless topology and challenge is similarly shared by the DENV and WNV NS3 protease. Extensive use of high throughput screening approaches were applied to the HCV NS3 protease and proved unsuccessful in identifying promising lead compounds [24,37]. Early inhibition studies started with short peptide carboxylates modelled after the N-terminal cleavage products of peptide substrates [38–40]. Research subsequently relied on classical peptidomimetic approaches using various warheads (serinetraps) such as aldehydes, keto-amides and boronic acids [41,42], combined with macrocyclic fragments, in an effort to move away from initial peptide-based inhibitors [24,43–45]. Ultimately, the use of α-keto-amides replacing the scissile-bond of the peptide substrate and the availability of catalytically relevant HCV NS3 protease crystal structures in complex with the properly positioned NS4a cofactor were critical in formulating the new approaches that provided a path to success [46].
Significant efforts have been dedicated to identifying promising inhibitors of DENV and WNV NS3 proteases. Recently, a number of compounds have been reported to have DENV NS3 inhibitory activity [47–49], however, few have progressed beyond demonstrating in vitro antiviral activity. The possibility that some of the reported compounds may owe their activity to artifactual non-specific or promiscuous inhibition [30,33,50] has been recognized recently and various methods have been published to minimize their effect on hit identification [34,51,52]. Similar experiences occurred within HCV NS3 protease drug development programmes where compounds identified through high throughput screening efforts were not useful as leads for medicinal chemistry efforts [24]. Building on these early results, subsequent crystal structures of HCV NS3/NS4a ternary complexes with inhibitors were vital to early successful drug discovery efforts [43,53]. We believe that the discovery of DENV NS3 protease inhibitors should follow a similar path and therefore relevant crystal structures are vitally important to success.
DENV drug discovery progress has been significantly hampered by the lack of sufficient DENV NS2b/NS3 cocrystal structures showing the NS2b cofactor appropriately interacting with the active site. In all available DENV NS3 structures [17,54–56] the NS2b C-terminal domain deviates significantly from the WNV ligand-bound structures [57] and does not form the complete non-prime portion of the substrate binding pocket. Obtaining this structural information would represent a major advance in the DENV and flavivirus NS3 protease drug discovery field and therefore in future work we will pursue the cocrystal structures with DENV NS2b/NS3 protease and this new class of flavivirus NS3 protease inhibitors. This information will guide medicinal chemistry efforts on the best approach to modify these compounds by reducing their charged cationic nature and possibly incorporating a functional group that interacts with the active site nucleophilic and catalytic serine residue.
Footnotes
Acknowledgements
We would like to thank Erica Koslov-Davino for assistance in purification of the four DENV NS2b/NS3 proteins. We would also like to thank the National Institutes of Health (NIH) for their support of this work with grants R43 AI079966 and R43 AI069584. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or the NIH.
The authors declare no competing interests.