
Abstract
Background:
Several published studies indicate that the acyclic guanine nucleoside analogues possessing bis(1,2-hydroxymethyl) substituted cyclopropane rings mimicking the sugar moiety are potent inhibitors of replication of several herpes viruses.
Methods:
Established synthetic methods and antiviral and cytostatic activity assays were used for the evaluation of new 1,2,4-triazole and purine acyclic nucleoside analogues.
Results:
The synthesis of new types of acyclic nucleoside analogues which incorporate 1,2,4-triazole or purine moiety bound via flexible methylenic spacer to the bis(1,2-hydroxymethyl) cyclopropane ring. None of the new compounds showed pronounced antiviral activities at subtoxic concentrations on a broad panel of DNA and RNA viruses.
Evaluation of their affinity for herpes simplex type 1 (HSV-1) and varicella-zoster virus-encoded thymidine kinases (VZV TK) also showed that none of the compounds was able to significantly inhibit 1 μM deoxythymidine phosphorylation by HSV-1 and VZV TK at 500 μM concentrations. The in vitro cytostatic activity evaluation results indicated a weak antiproliferative activity for all tested compounds. Only 6-pyrrolylpurine derivative bearing a carboxylic group substituted cyclopropane ring produced a rather slight inhibitory effect at higher micromolar concentrations on a breast carcinoma cell line (MCF-7) and no cytotoxic effect on human normal fibroblasts (WI 38).
Conclusions:
The lack of antiherpetic activity may be due to poor, if any, recognition of the compounds by virus-induced nucleoside kinases as an alternative substrate to become metabolically activated.
Introduction
The discovery of the acyclic guanosine analogue acyclovir (ACV; Figure 1) as a selective anti-herpes simplex virus (HSV) agent [1] initiated investigation of new acyclic nucleoside analogues with better antiviral activity properties than ACV. These efforts resulted in the discovery of new broad-spectrum antiviral agents: ganciclovir (GCV; Figure 1) [2–4] and penciclovir (PCV; Figure 1) [5,6]. In addition, the crystal structure of HSV-1-encoded thymidine kinase (TK), a key enzyme for activation of antiherpetic nucleosides, in complex with GCV, revealed the importance of the two hydroxyl groups of GCV mimicking the 3′- and 5′-hydroxyl groups of the 2′-deoxyribose moiety in natural nucleosides for substrate recognition [7].
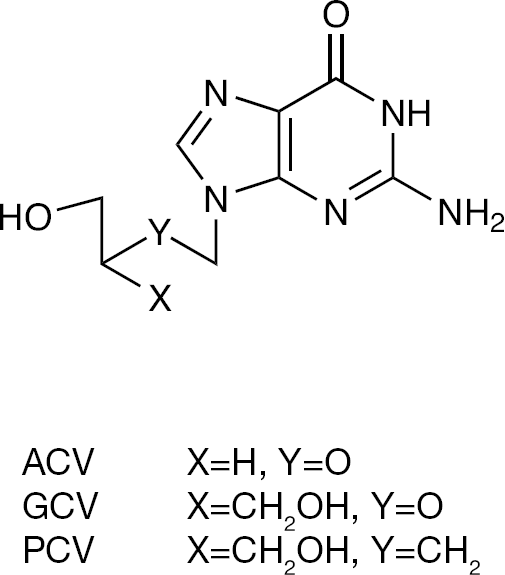
The antiviral agents ACV, GCV and PCV
Based on these findings, (1′S, 2′R)-9-{[1′,2′-bis(hydroxymethyl)cycloprop-1-yl]methyl}guanine (A-5021; Figure 2) was developed as a nucleoside analogue that potently inhibits the replication of several herpes viruses including HSV type-1 (HSV-1) and HSV type-2 (HSV-2), varicella-zoster virus (VZV), Epstein–Barr virus (EBV) and human herpesvirus type 6 (HHV-6). Though the sugar moiety of A-5021 has a cyclopropane ring, its overall feature is considered to be acyclic due to the flexible methylenic spacer. The antiviral activity of A-5021 is superior over ACV against HSV-1, HSV-2, VZV and HHV-6 [8–10]. The racemic 9-{[1′,2′-bis(hydroxymethyl)cycloprop-1-yl] methyl}guanine and especially its 1′S, 2′R enantiomer (A-5021) appears to be the most prominent in terms of antiviral activity among the guanine cyclopropane analogues possessing a methylene spacer between the base and the carbocyclic ring [8]. Preliminary tests indicated that the 1′S, 2′R-enantiomer of A-5021 displays 40-fold, and the racemic A-5021 nearly 20-fold, more potent activity against HSV-1 in vitro than ACV [11]. The 1′R, 2′S enantiomer of A-5021 was considerably less active [11].
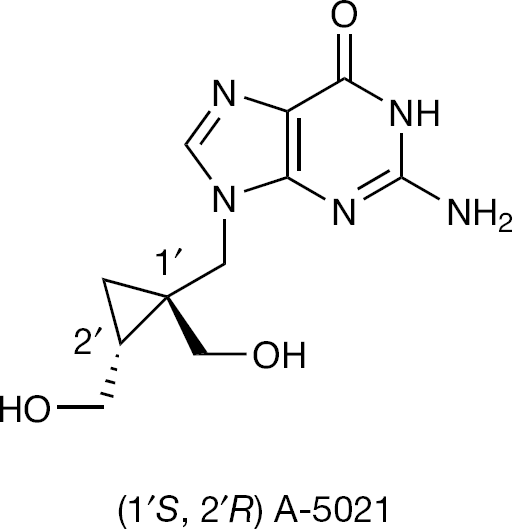
The guanine cyclopropane nucleoside analogue (1′S, 2′R) A-5021
According to subsequent determinations, A-5021 was 17- to 150-fold more active than ACV and approximately 60-fold more active than PCV against HSV-1, two- to eightfold more active than ACV and 10- to 12-fold more active than PCV against HSV-2, and five- to sevenfold more active than ACV and 2- to 18-fold more active than PCV against VZV, depending on the viral strain and type of cells used [9,12].
The selectivity index of A-5021 is superior to those of ACV and PCV against HSV-1 and against HSV-2 and VZV [11]. Antiviral effectiveness of A-5021 depends on the intracellular phosphorylation by the virus-encoded TK and is associated with a considerable intracellular stability of its triphosphate [13]. A-5021 has proved equipotent to ACV as an inhibitor of EBV [10]. The data reported in the literature point to the efficacy of A-5021 in HSV-infected mice [10,14]. Among all tested acyclic and carbocyclic guanine nucleosides, A-5021 has been identified as the most potent and selective cytostatic agent against HSV-1 TK gene-transfected human osteosarcoma cells [15]. A-5021 has also demonstrated cytostatic activity, equal to that of GCV, in HSV TK gene-transducted human cancer cells [10]. As compared with GCV, A-5021 is less toxic to the bone marrow cells, which makes it possible to use A-5021 as an alternative to GCV in the combined gene therapy/chemotherapy of cancer [16].
Among the compounds with the base moieties other than guanine, for example, adenine, 2,6-diaminopurine, all with the cis-1′,2′-bis-(hydroxymethyl) moiety, are moderately active. The activity of the latter two compounds may be due to the intracellular conversion to A-5021. In contrast to the strong activity against HSV-1, A-5021 was inactive against HIV. Among the compounds tested on HIV-1, the cytosine derivative was moderately active, and some thymine, hypoxantine, and 2,6-diaminopurine derivatives showed weak activity [8].
Ribavirin's 1,2,4-triazole-3-carboxyamide moiety mimics the native nucleosides adenosine or guanosine. For this reason, when ribavirin is incorporated into RNA, as a base analogue of either adenine or guanine, it pairs equally well with either uracil or cytosine, inducing mutations during the RNA-dependent replication of RNA viruses. Such hypermutation can be lethal to RNA viruses and is called ‘error catastrophy’ [17].
The previously described findings led us to synthesize a series of new 1,2,4-triazole and purine acyclic cyclopropane nucleoside analogues (Figure 3) with the primary aim to evaluate their potential antiviral and cytostatic potency.
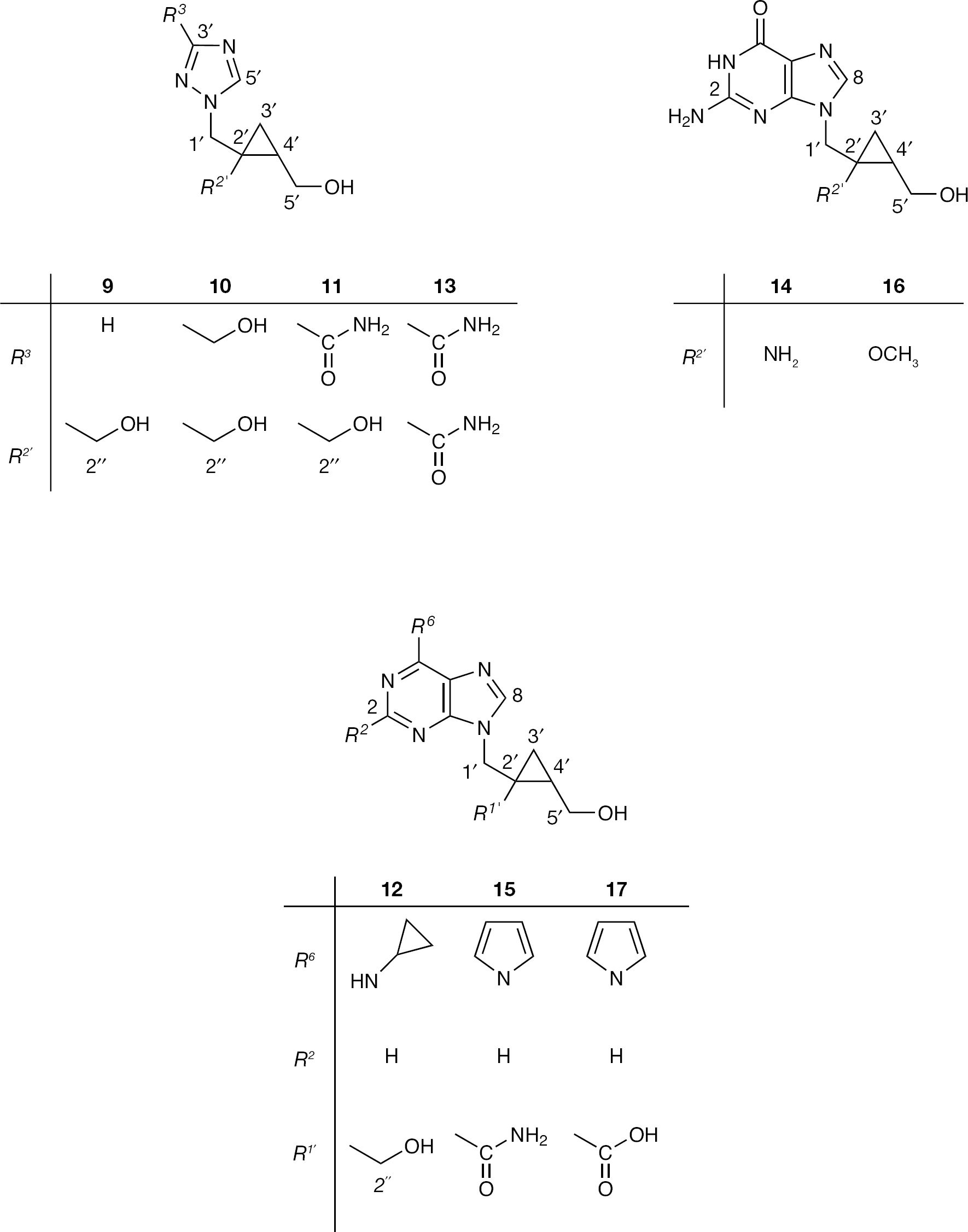
The new 1,2,4-triazole,
Methods
Chemistry: General methods
Melting points (uncorrected) were determined with Kofler micro hot-stage (Reichert, Vienna, Austria). Precoated silica gel 60F-254 plates (Sigma–Aldrich, Steinheim, Germany) were used for thin layer chromatography (TLC) and the spots were detected under UV light (254 nm). Column chromatography (CLC) was performed using silica gel (0.063–0.2 mm) Fluka; the glass column was slurry-packed under gravity. Mass spectra were recorded on an Agilent 6410 instrument (Agilent Technologies, Wilmington, NC, USA) equipped with electrospray interface and triple quadrupole analyzer (LC/MS/MS). High performance liquid chromatography was performed on an Agilent 1100 series system (Agilent Technologies) with UV detection (photodiode array detector) using Zorbax C18 reverse-phase analytical column (2.1×30 mm, 3.5 μm; Aligent Technologies). 1H and 13C NMR spectra were acquired on Bruker 300 MHz NMR spectrometer (Bruker, Rheinstetten, Germany). All data were recorded in DMSO-d6 at 298 K. Chemical shifts were referenced to the residual solvent signal of DMSO at δ 2.50 ppm for 1H and δ 39.50 ppm for 13C. Individual resonances were assigned on the basis of their chemical shifts, signal intensities, multiplicity of resonances and H-H coupling constants.
Procedures for the preparation of compounds
General procedure for the preparation of triazole (2–4) and purine (5−8) derivatives
To a stirred solution containing purine or triazole nucleoside bases and NaH (1.5 equivalents) in dimethylformamide (DMF; 15 ml) the compound 1-chloromethyl-2-oxo-3-oxabicyclo[3.1.0]hexane (
Preparation of compounds 2–8
Compound
1H NMR (DMSO; for enumeration of atoms cf. Figure 1) δ/ppm: 8.50 (s, 1H, H-5-triazole), 7.96 (s, 1H, H-3-triazole), 4.73 (d, 1H, J = 14.8, H-5), 4.36 (d, 1H, J = 14.8, H-5), 4.22 (d, 1H, J = 9.24, H-1), 4.13 (d, 1H, J = 9.24, H-1), 2.49 (m, 1H, H-4a), 1.46 (t, 1H, J1 = 4.7, J2=, H-4), 1.08 (t, 1H, J1= 4.7, J2= 4.7, H-4).
13C NMR (DMSO) δ/ppm: 175.97 (C-3), 151.98 (C-5-triazole), 144.80 (C-3-triazole), 68.63 (C-1), 47.69 (C-5), 27.99 (C-3a), 22.75 (C-4a), 16.99 (C-4); MS m/z 180.1 [MH]+.
Compound
1H NMR (DMSO) δ/ppm: 8.69 (s, 1H, H-5-triazole), 4.74 (d, 1H, J = 14.7, H-5), 4.55 (d, 1H, J = 14.7, H-5), 4.22 (d, 1H, J = 9.24, H-1), 4.13 (d, 1H, J = 9.24, H-1), 3.85 (s, 3H, -OCH3), 2.49 (m, 1H, H-4a), 1.46 (t, 1H, J1 = 4.7, J 2 =, H-4), 1.08 (t, 1H, J1= 4.7, J2= 4.7, H-4).
13C NMR (DMSO) δ/ppm: 175.88 (C-3), 160.35 (CO-triazole), 145.60 (C-3-triazole), 68.70 (C-1), 52.62 (OCH3-triazole), 49.27 (C-5), 27.78 (C-3a), 22.53 (C-4a), 17.18 (C-4); MS m/z 238.1 [MH]+.
1-[Methyl-1,2,4-triazol-(3-carboxylate)-1-yl]methyl-2-oxo-3-oxa-bicyclo[3.1.0]hexane (
1H NMR (DMSO) δ/ppm: 8.16 (br, 1H, NH2-triazole), 8.07 (s, 1H, H-5-triazole), 7.94 (br, 1H, NH2-triazole), 4.74 (d, 1H, J = 14.7, H-5), 4.55 (d, 1H, J = 14.7, H-5), 4.22 (d, 1H, J = 9.24, H-1), 4.13 (d, 1H, J = 9.24, H-1), 2.49 (m, 1H, H-4a), 1.46 (t, 1H, J1 = 4.7, J2 =4.6, H-4), 1.08 (t, 1H, J1= 4.7, J2= 4.7, H-4).
13C NMR (DMSO) δ/ppm: 175.91 (C-3), 159.32 (CO-triazole), 150.25 (C-5-triazole), 147.17 (C-3-triazole), 68.53 (C-1), 48.80 (C-5), 27.83 (C-3a), 22.46 (C-4a), 17.33 (C-4); MS m/z 223.0 [MH]+.
Compound
1H NMR (DMSO) δ/ppm: 8.75 (s, 1H, H-8-purine), 8.61 (s, 1H, H-2-purine), 8.29 (s, 2H, pyrrolyl), 8.45 (s, 2H, pyrrolyl), 4.74 (d, 1H, J = 14.7, H-5), 4.55 (d, 1H, J = 14.7, H-5), 4.22 (d, 1H, J = 9.24, H-1), 4.13 (d, 1H, J = 9.24, H-1), 2.49 (m, 1H, H-4a), 1.46 (t, 1H, J1 = 4.7, J2 =, H-4), 1.08 (t, 1H, J1 = 4.7, J2 = 4.7, H-4).
13C NMR (DMSO) δ/ppm: 175.72 (C-3), 153.29 (C-6-purine), 151.76 (C-2-purine), 146.44 (C-8-purine), 145.81 (C-5-purine), 121.07 (C-4), 120.06 and 112.43 (CH2-pyrrolyl) 68.23 (C-1), 42.26 (C-5), 27.63 (C-3a), 23.01 (C-4a), 16.79 (C-4); MS m/z 296.1 [MH]+.
Compound
1H NMR (DMSO) δ/ppm: 8.25 (s, 1H, H-8-purine), 8.11 (s, 1H, H-2-purine), 7.90 (br, 1H, NH-purine), 4.74 (d, 1H, J = 14.7, H-5), 4.55 (d, 1H, J = 14.7, H-5), 4.22 (d, 1H, J = 9.24, H-1), 4.13 (d, 1H, J = 9.24, H-1), 2.49 (m, 1H, H-4a), 1.54 (t, 1H, J1 = 4.7, J 2=, H-4), 1.08 (t, 1H, J1 = 4.7, J2 = 4.7, H-4), 0.73 (d, 2H, J = 6.7,cyclopropyl), 0.61 (d, 2H, J = 6.7, cyclopropyl).
13C NMR (DMSO) δ/ppm: 176.34 (C-3), 156.00 (C-6-purine), 152.91 (C-2-purine), 141.23 (C-8-purine), 119.34 (C-5-purine), 68.71 (C-1), 42.12 (C-5), 28.39 (C-3a), 23.45 (C-4a), 17.05 (C-4), 7.05 (CH2-cyclopropyl); MS m/z 286.20 [MH]+.
Compound
Compound
General procedure for the preparation of triazole (9–11) and purine (12) acyclic cyclopropane nucleoside analogues
Compounds (
Preparation of compounds 9–12
Compound
11H NMR (DMSO) δ/ppm: 0.44 (CH2–3′, AB, t, 1H, J = 4.76 Hz), 0.83 (CH2–3′, AB, dd, 1H, J = 8.46, 4.86 Hz), 1.24 (H-4′, quintet, 1H), 3.25 and 3.43 (CH2–2″, AB, dd, 2H, J = 110.52, 11.88 Hz), 3.34–3.38 (CH2–5′, m, 1H), 3.61 (CH2–5′, AB, dd, 1H, J = 11.22, 6.6 Hz), 4.00 and 4.32 (CH2–1′, dd, 2H, AB, J = 193.86, 14.11 Hz), 4.58 (OH-2″, t, 1H, J = 4.32 Hz), 4.64 (OH-5′, t, 1H, J = 4.56 Hz), 7.94 (H-3, s, 1H), 8.53 (H-5, s, 1H).
13C NMR (DMSO) δ/ppm: 151.49 (C-5-triazole), 144.50 (C-3-triazole), 61.07 (C-2′a), 60.17 (C-1′a), 54.17 (C-1′b), 27.23 (C-1′), 25.03 (C-2′), 14.40 (C-3′); MS m/z 184.1 [MH]+.
Compound
11H NMR (DMSO) δ/ppm: 0.43 (CH2–3′,AB, t, 1H, J = 5.16 Hz), 0.82 (CH2–3′, AB, dd, 1H, J = 8.52, 4.86 Hz), 1.23 (H-4′, quintet, 1H), 3.38 and 3.25 (CH2–2″, AB, dd, 2H, J = 130.53, 11.76 Hz), 3.60–3.62 (CH2–5′, m, 1H), 3.35–3.38 (CH2–5′, m, 1H), 4.30 and 3.92 (CH2–1′, AB, dd, 2H, J = 227.34, 14.11 Hz), 4.41 (CH2–3-triazole, d, 2H, J = 5.16 Hz), 4.61 (OH-2″, t, 1H, J = 11.82 Hz), 4.66 (OH-5′, t, 1H, J = 13.08 Hz), 5.17 (OH-3-triazole, t, 1H, J = 5.58 Hz), 8.44 (H-5, s, 1H).
13C NMR (DMSO) δ/ppm: 163.93 (C-5-triazole), 145.21 (C-3-triazole), 64.34 (CH2-triazole) 61.42 (C-2′a), 60.17 (C-1′a), 54.41 (C-1′b), 27.49 (C-1′), 25.31 (C-2′), 14.73 (C-3′); MS m/z 214.20 [MH]+.
Compound
11H NMR (DMSO) δ/ppm: 0.38 (CH2–3′, AB, t, 1H, J = 5.10 Hz), 0.80 (CH2–3′, AB, dd, 1H, J = 9.48, 17.04 Hz), 1.21 (H-4′, quintet, 1H), 3.20–3.39 and 3.50–3.62 (CH2–2″ and CH2–5′, 2× m, 2× 2H), 4.81 and 4.44 (CH2–1′, AB, dd, 2H, J = 223.53, 13.80 Hz), 4.54–4.83 (OH-2″ and OH-5′, m, 2H), 8.06 (H-5, s, 1H), 8.19 and 7.95 (CO-NH2-triazole, 2× s, 2× 1H).
13C NMR (DMSO) δ/ppm: 159.19 (CO-triazole), 149.51 (C-5-triazole), 146.69 (C-3-triazole), 61.32 (C-2′a), 60.64 (C-1′a), 54.01 (C-1′b), 27.56 (C-1′), 24.64 (C-2′), 13.71 (C-3′); MS m/z 227.38 [MH]+.
Compound
11H NMR (DMSO) δ/ppm: 0.62 (CH2–3′, AB, t, 1H, J = 2.70 Hz), 0.74–0.70 and 0.63–0.60 (CH2-cyclopropylamine, 2× m, 2×1H), 0.94 (CH2–3′, AB, dd, 1H, J = 4.74, 8.46 Hz), 1.33 (H-4′, quintet, 1H), 3.33 (CH-cyclopropylamine, m, 1H), 3.27–3.36 (CH2–2″, AB, m, 2H), and 3.41–3.47 and 3.58–3.61 (CH2–5′, 2× m, 2× 1H), 4.05 and 4.21 (CH2–1′, AB, dd, 2H, J = 98.52, 14.22 Hz), 4.53 (OH-5′, t, 1H, J = 5.16 Hz), 4.71 (OH-2″, t, 1H, J = 5.16 Hz), 7.81 (NH-cyclopropylamine, s, 1H), 8.16 (H-2, s, 1H), 8.24 (H-8, s, 1H).
13C NMR (DMSO) δ/ppm:155.48 (C-4-purine), 152.25 (C-2-purine), 140.79 (C-8-purine), 118.83 (C-6-purine), 60.59 (C-2′a), 55.99 (C-1′a), 47.96 (C-1′b), 26.86 (C-1′), 24.66 (C-2′), 18.50 (CH-cyclopropylamine), 14.07 (C-3′), 5.01 (CH2-cyclopropylamine); MS m/z 290.1 [MH]+.
General procedure for the preparation of triazole (13) and purine (14 and 15) acyclic cyclopropane nucleoside analogues
Compounds
Preparation of compounds 13–17
Compound
11H NMR (DMSO) δ/ppm: 0.96 (CH2–3′, AB, dd, 1H, J = 8.67, 4.86 Hz), 1.13 (CH2–3′, AB, t, 1H, J = 5.34 Hz), 1.39 (H-4′, quintet, 1H), 3.33–3.35 (CH2–5′, m, 2H), 4.39 and 5.31 (CH2–1′, AB, dd, 2H, J = 552.15, 28.74 Hz), 4.48 (OH-5′, t, 1H, J = 10.20 Hz), 7.06 and 7.43 (CO-NH2–2″, 2× s, 2× 1H), 8.07 (H-5, s, 1H), 8.26 and 8.01 (CO-NH2–3-triazole, 2× s, 2× 1H).
13C NMR (DMSO) δ/ppm:171.48 (CO-C-1′a), 159.71 (CO-triazole), 150.14 (C-5-triazole), 146.94 (C-3-triazole), 60.21 (C-2′a), 54.54 (C-1′b), 31.02 (C-1′), 27.57 (C-2′), 15.09 (C-3′); MS m/z 240.1 [MH]+.
Compound
11H NMR (DMSO) δ/ppm: 0.86 (CH2–3′, t, 1H, J = 5.70 Hz), 1.16 (CH2–3′, AB, dd, 1H, J = 8.04, 5.25 Hz), 1.39 (H-4′, quintet, 1H), 3.30–3.35 (CH2–5′, m, 1H), 4.13 (CH2–5′, AB, dd, 1H, J = 9.69, 4.89 Hz), 3.60 and 4.65 (CH2–1′, AB, dd, 2H, J = 314.28, 14.49 Hz), 4.58 (OH-5′, t, 1H, J = 5.07 Hz), 6.57 (NH2-guanine, s, 2H), 7.45 and 7.14 (CO-NH2–2″, 2× s, 2× 1H), 7.59 (H-8, s, 1H), 10.53 (NH-guanine, br, 1H).
13C NMR (DMSO) δ/ppm: 171.85 (C-1′a), 157.19 (C-6-purine), 154.07 (C-2-purine), 151.70 (C-4-purine), 60.51 (C-2′a), 54.55 (C-1′b), 27.56 (C-1′), 24.64 (C-2′), 13.71 (C-3′); MS m/z 2793 [MH]+.
Compound
11H NMR (DMSO) δ/ppm: 1.18 (CH2–3′, t, 1H, J = 5.55 Hz), 1.26 (CH2–3′, AB, dd, 1H, J = 8.61, 3.21 Hz), 1.57 (H-4′, quintet, 1H), 3.36–3.40 (CH2–5′, m, 2H), 4.05 and 4.93 (CH2–1′, AB, dd, 2H, J = 263.73, 14.49 Hz), 4.60 (OH-5′, t, 1H, J = 5.07 Hz), 7.15 and 7.42 (CO-NH2–2″, 2× s, 2× 1H), 8.29 and 6.44 (s, 2H), 8.54 (H-2, s, 1H), 8.74 (H-8, s, 1H).
13C NMR (DMSO) δ/ppm: 171.70 (C-1′a), 153.88 (C-4-purine), 152.17 (C-2-purine), 146.86 (C-6-purine), 120.55 and 112.93 (CH2-pyrrolyl), 60.51 (C-2′a), 54.55 (C-1′b), 27.56 (C-1′), 24.64 (C-2′), 13.71 (C-3′); MS m/z 313.3 [MH]+.
Compound
11H NMR (DMSO) δ/ppm: 1.04 (CH2–3′, m, 1H), 1.12 (CH2–3′, m, 1H), 1.40 (H-4′, quintet, 1H), 3.57–3.62 and 3.34–3.38 (CH2–5′, 2× m, 2× 1H), 3.87 and 4.38 (CH2–1′, AB, dd, 2H, J = 150.60, 12.30 Hz), 3.95 (CO-OCH3–2″, s, 3H), 4.58 (OH-5′, t, 1H, J = 5.07 Hz), 6.33 (NH2-guanine, s, 2H), 8.09 (H-8, s, 1H), 10.53 (NH-guanine, br, 1H).
13C NMR (DMSO) δ/ppm: 175.23 (C-1′a), 160.55 (C-6-purine), 160.50 (C-2-purine), 155. 7 (C-4-purine), 60.51 (C-2′a), 54.55 (C-1′b), 49.11 (-OCH3) 29.13 (C-1′), 28.50 (C-2′), 16.47 (C-3′); MS m/z 294.20 [MH]+.
Compound
11H NMR (DMSO) δ/ppm: 1.23 (CH2–3′, AB, dd, 1H, J = 6.69, 4.32 Hz), 1.32 (CH2–3′, AB, dd, 1H, J = 8.76, 3.90 Hz), 1.73 (H-4′, quintet, 1H), 3.53 and 3.70 (CH2–5′, 2× dd, 2× 1H, J = 11.19, 11.28 Hz), 4.31 and 4.52 (CH2–1′, AB, dd, 2H, J = 121.89, 14.22 Hz), 4.61 (OH-5′, t, 1H, J = 11.82 Hz), 8.30 and 6.44 (2× s, 2× 1H), 8.73 (H-2, s, 1H), 8.74 (H-8, s, 1H).
13C NMR (DMSO) δ/ppm: 173.45 (C-1′a), 153.55 (C-4-purine), 151.50 (C-8-purine), 146.27 (C-6-purine), 120.25 and 112.30 (CH2-pyrrolyl), 60.51 (C-2′a), 54.55 (C-1′b), 29.13 (C-1′), 28.50 (C-2′), 17.71 (C-3′); MS m/z 114.3 [MH]+.
Virology: Antiviral activity assays
The antiviral assays, other than the anti-HIV assays, were based on inhibition of virus-induced cytopathicity in human embryonic lung fibroblast cell line (HEL; HSV-1 [KOS], HSV-2 [G], vaccinia virus and vesicular stomatitis virus), Vero (parainfluenza-3, reovirus-1, Sindbis, Coxsackie B4 and Punta Toro virus), HeLa (vesicular stomatitis virus, Coxsackie virus B4 and respiratory syncytial virus) or Madin Darby canine kidney (influenza A [H1N1; H3N2] and influenza B) cell cultures. Confluent cell cultures (or nearly confluent for Madin Darby canine kidney cells) in microtitre 96-well plates were inoculated with 100 CCID50 of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures) in the presence of varying concentrations (200, 40, 8… μM) of the test compounds. Viral cytopathicity was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. The methodology of the anti-HIV assays was as follows: human T-lymphocyte cells (CEM; ∼3×105 cells/ml) were infected with 100 CCID50 of HIV(IIIB) or HIV-2(ROD)/ml and seeded in 200 μl wells of a microtitre plate containing appropriate dilutions of the test compounds. After 4 days of incubation at 37°C, HIV-induced giant cell formation was examined microscopically.
Cytotoxicity assays
Cytotoxicity measurements were based on the inhibition of HEL cell growth. HEL cells were seeded at a rate of 5×103 cells/well into 96-well microtitre plates and allowed to proliferate for 24 h. Then, medium containing different concentrations of the test compounds was added. After 3 days of incubation at 37°C, the cell number was determined by a Coulter counter. The cytostatic concentration was calculated as the compound concentration required to reduce cell growth by 50% relative to the number of cells in the untreated controls (CC50). CC50 values were estimated from graphic plots of the number of cells (percentage of control) as a function of the concentration of the test compounds. Cytotoxicity was expressed as minimum cytotoxic concentration (MCC) or the compound concentration that causes a microscopically detectable alteration of cell morphology.
Inhibitory activity of test compounds against HSV-1 and VZV TK-catalysed phosphorylation of dThd
The 50% inhibitory concentrations (IC50) of the test compounds against purified recombinant HSV-1 and VZV TK were assayed in a 50 μl reaction mixture containing 50 mM Tris/HCl, pH 8.0, 2.5 mM MgCl2, 10 mM dithiothreitol, 0.5 mM CHAPS, 3 mg/ml bovine serum albumin, 2.5 mM ATP, 1 μM [methyl-3H] deoxythymidine (dThd), and recombinant purified enzyme (4 nM). The samples were incubated at 37°C for 30 min in the presence or absence of different concentrations (fivefold dilutions) of the test compounds. At this time point, the enzyme reaction still proceeded linearly and did not convert more than 15% of the natural substrate under all experimental conditions. Aliquots of 45 μl of the reaction mixtures were spotted on Whatman DE-81 filter paper disks (Whatman, Clifton, NJ). The filters were washed three times for 5 min each in 1 mM ammonium formate, once for 1 min in water, and once for 5 min in ethanol. The radioactivity was determined by scintillation counting.
Cytostatic activity assays
Cell culturing
The cell lines HeLa (cervical carcinoma), SW620 (colorectal adenocarcinoma, metastatic), MiaPaCa-2 (pancreatic carcinoma), MCF-7 (breast epithelial adenocarcinoma, metastatic), HepG2 (hepatocellular carcinoma) and WI38 (normal diploid human fibroblasts) were cultured as monolayers and maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin in a humidified atmosphere with 5% CO2 at 37°C.
Proliferation assays
The panel cell lines were inoculated onto a series of standard 96-well microtitre plates on day 0, at 3,000 cells to 6,000 cells per well according to the doubling times of specific cell line. Test agents were then added in five, 10-fold dilutions (1×10–8 to 1×10–4 M) and incubated for another 72 h. Working dilutions were freshly prepared on the day of testing in the growth medium. The solvent (DMSO) was also tested for eventual inhibitory activity by adjusting its concentration to be the same as in the working concentrations (DMSO concentration never exceeded 0.1%). After 72 h of incubation, the cell growth rate was evaluated by performing the MTT assay: experimentally determined absorbance values were transformed into a cell percentage growth (PG) using the formulas proposed by NIH and described previously [18]. This method directly relies on control cells behaving normally at the day of assay because it compares the growth of treated cells with the growth of untreated cells in control wells on the same plate - the results are therefore a percentile difference from the calculated expected value.
The IC50 values for each compound were calculated from dose-response curves using linear regression analysis by fitting the mean test concentrations that give PG values above and below the reference value. If, however, all of the tested concentrations produce PGs exceeding the respective reference level of effect (for example, PG value of 50) for a given cell line, the highest tested concentration is assigned as the default value (in the screening data report that default value is preceded by a ‘>’ sign). Each test point was performed in quadruplicate in three individual experiments. The results were statistically analysed (ANOVA, Tukey post hoc test at P<0.05). Finally, the effects of the tested substances were evaluated by plotting the mean percentage growth for each cell type in comparison to control on dose-response graphs.
Results
Chemistry
The syntheses of new structurally diverse 1,2,4-triazole (
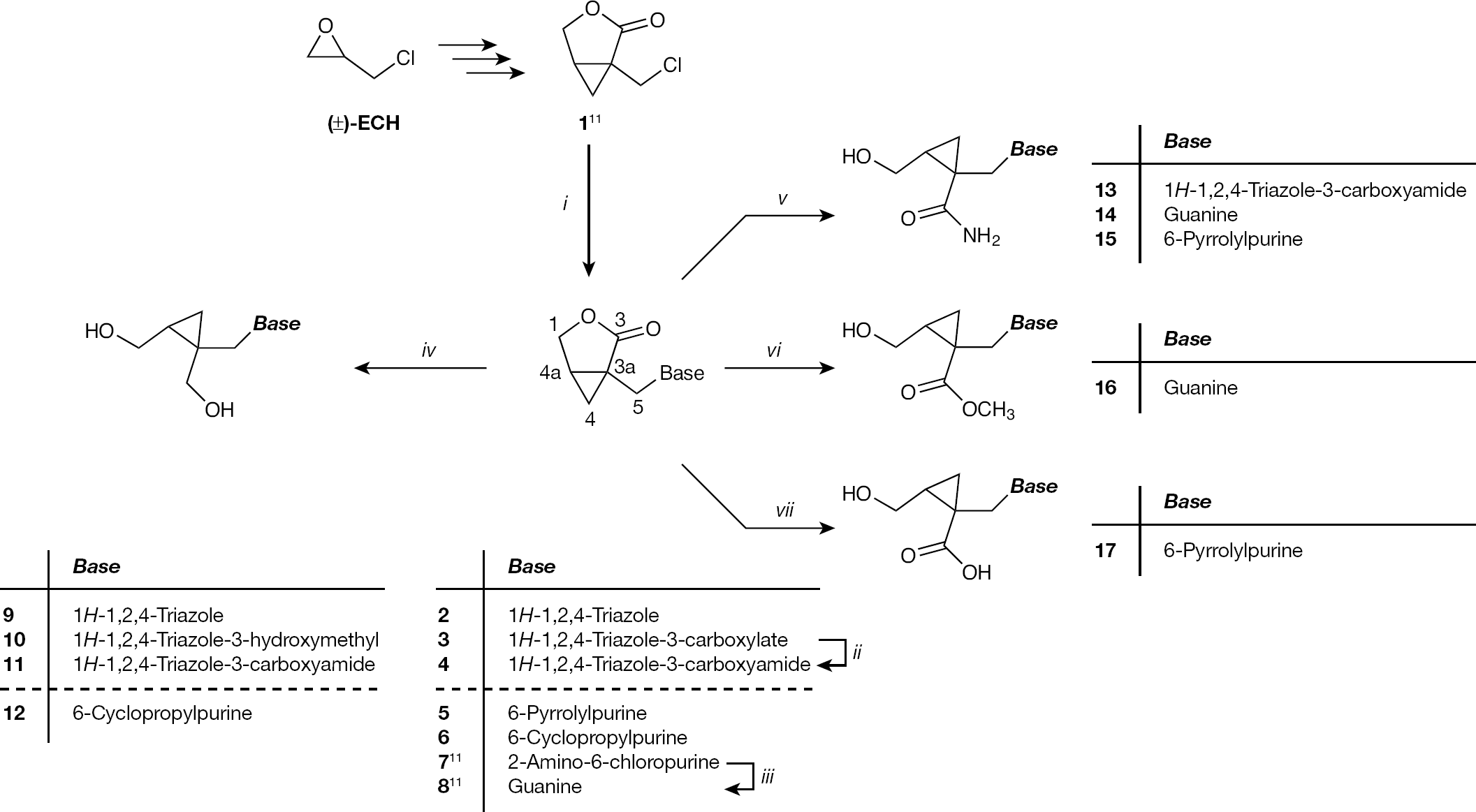
Synthesis of the new 1,2,4-triazole,
1H and 13C NMR spectra
Structures of newly synthesized compounds were deduced on the basis of analysis of their 1H and 13C NMR as well as their mass spectra. The assignment of 1H NMR spectra was performed from their chemical shifts, substituent induced chemical shifts, and signal intensities, magnitude and multiplicity of H-H coupling constants. DMSO-d6 was used as a solvent for all compounds; chemical shifts are referred to tetramethylsilane. 1H and 13C NMR spectra data are in accordance with proposed structures and those of structurally related cyclopropane derivatives [8].
Antiviral activity
Compounds
Cytostatic activity
The compounds were evaluated for their antiproliferative effect against a panel of malignant tumour cell lines: cervical carcinoma (HeLa), colorectal adenocarcinoma metastatic (SW620), pancreatic carcinoma (MiaPaCa-2), breast epithelial adenocarcinoma metastatic (MCF-7), hepatocellular carcinoma (HepG2), murine leukaemia cells (L1210), CEM cells and human cervix carcinoma cells (HeLa), and compared with their effects on the growth of normal human fibroblasts (WI 38; Additional file 1).
The results of the in vitro cytostatic activity evaluations revealed that majority of the 1,2,4-triazole (
Discussion
This study displays the synthesis of the new types of acyclic nucleoside analogues consisting of purine or 1,2,4-triazole moiety bound via flexible methylenic spacer to the bis(1,2-hydroxymethyl) substituted cyclopropane ring. The 1,2,4-triazole-3-carboxamide heterocyclic moiety in these nucleoside analogues is used to mimic the natural guanine base, whereas the two hydroxymethyl groups on the carbocyclic moiety serves for mimicking the 3′- and 5′-hydroxyl groups of the 2′-deoxyribose moiety in natural nucleosides. Systematic synthetic modification of either heterocyclic or carbocyclic moiety in these molecules gave an array of structurally diverse acyclic nucleoside analogues which were subjected to antiviral activity in vitro assays on a broad spectrum of DNA and RNA viruses, including HSV-1 and VZV-encoded TK.
Although the specificity requirements of the viral TKs are much less stringent then those for cellular nucleoside (thymidine) kinases, the in vitro testing showed that none of the evaluated compounds were able to significantly inhibit 1 μM dThd phosphorylation by HSV-1 and VZV TK at 500 μM concentrations, pointing to their poor, if any, substrate affinity for these enzymes. This can explain the lack of anti-herpes virus activity of the tested compounds. While these compounds exerted no antiviral activity potency at subtoxic concentrations, the in vitro cytostatic activity studies revealed that among all evaluated compounds, the 6-pyrrolylpurine derivative containing substituted carboxylic group in position 1′ of the cyclopropane ring (
In conclusion, a series of a new type of the acyclic cyclopropane nucleoside analogues composed of 1,2,4-triazole (
Footnotes
Acknowledgements
Support for this study was provided by the Ministry of Science, Education and Sports of the Republic of Croatia (Projects number 125-0982464-2922, number 335-0982464-2393 and number 335-0000000-3532) and by the ‘Geconcerteerde Onderzoeksacties’ of the Katholieke Universiteit Leuven (GOA project number 10/014). We thank Lizette van Berckelaer, Leentje Persoons, Frieda De Meyer, Lies Van den Heurck, Steven Carmans and Anita Camps for excellent technical assistance.
The authors declare no competing interests.