
Abstract
Highly active antiretroviral therapy (HAART) significantly decreases plasma viral load, increases CD4+ T-cell counts in HIV-1-infected patients and has reduced progression to AIDS in developed countries. However, adverse side effects, and emergence of drug resistance, mean there is still a demand for new anti-HIV agents. The HIV integrase (IN) is a target that has been the focus of rational drug design over the past decade. In 2007, raltegravir was the first IN inhibitor approved by the US Food and Drug Administration for antiretroviral combination therapy, while another IN inhibitor, elvitegravir, is currently in Phase III clinical trials. This article reviews the development and resistance profiling of small molecule HIV-1 IN inhibitors.
Introduction
Highly active antiretroviral therapy (HAART) refers to the standard combination therapy regimen for HTV-1-infected patients that effectively suppresses HIV-1 replication, resulting in sustained clinical efficacy in most patients. HAART usually comprises two nucleoside reverse transcriptase inhibitors (NRTIs) and one protease inhibitor (PI), but other classes of antiretroviral agents are also available, for example, non-nucle-oside reverse transcriptase inhibitors (NNRTIs) and entry inhibitors. Some patients do not tolerate current HAART regimens, resulting in poor adherence that facilitates emergence of drug-resistant variants.
Therefore, additional therapeutic agents that inhibit new targets required for HIV-1 replication are warranted. Since the mid-1990s, the retroviral, integrase (IN) enzyme, has been a focus of attention [1–3] and several IN inhibitors have undergone clinical trials.
Function of HIV-1 integrase
HIV-1 IN facilitates the importation of the viral preintegration complex through nuclear pores and catalyzes integration of the viral complementary DNA into the host chromosome, as depicted in Figure 1. Integration is a complex process comprised of four main steps, nuclear import, 3′-end processing and strand transfer steps that are intrinsic functions of IN, and DNA end repair which is performed by cellular DNA repair mechanisms (Figure 2). During 3′-end processing, the IN cleaves a dinucleotide from each viral DNA terminus (that is, long terminal repeat [LTR]), producing reactive CpA 3-hydroxyl ends. These reactive hydroxyl groups are utilized in a nucleophilic attack into the target host DNA, by a one-step transesterification reaction that leads to full-site integration. At the same time, most of the unintegrated DNA is subjected to degradation by cellular enzymes. For a comprehensive review considering the role of unintegrated DNA see Sloan and Wainberg [4].
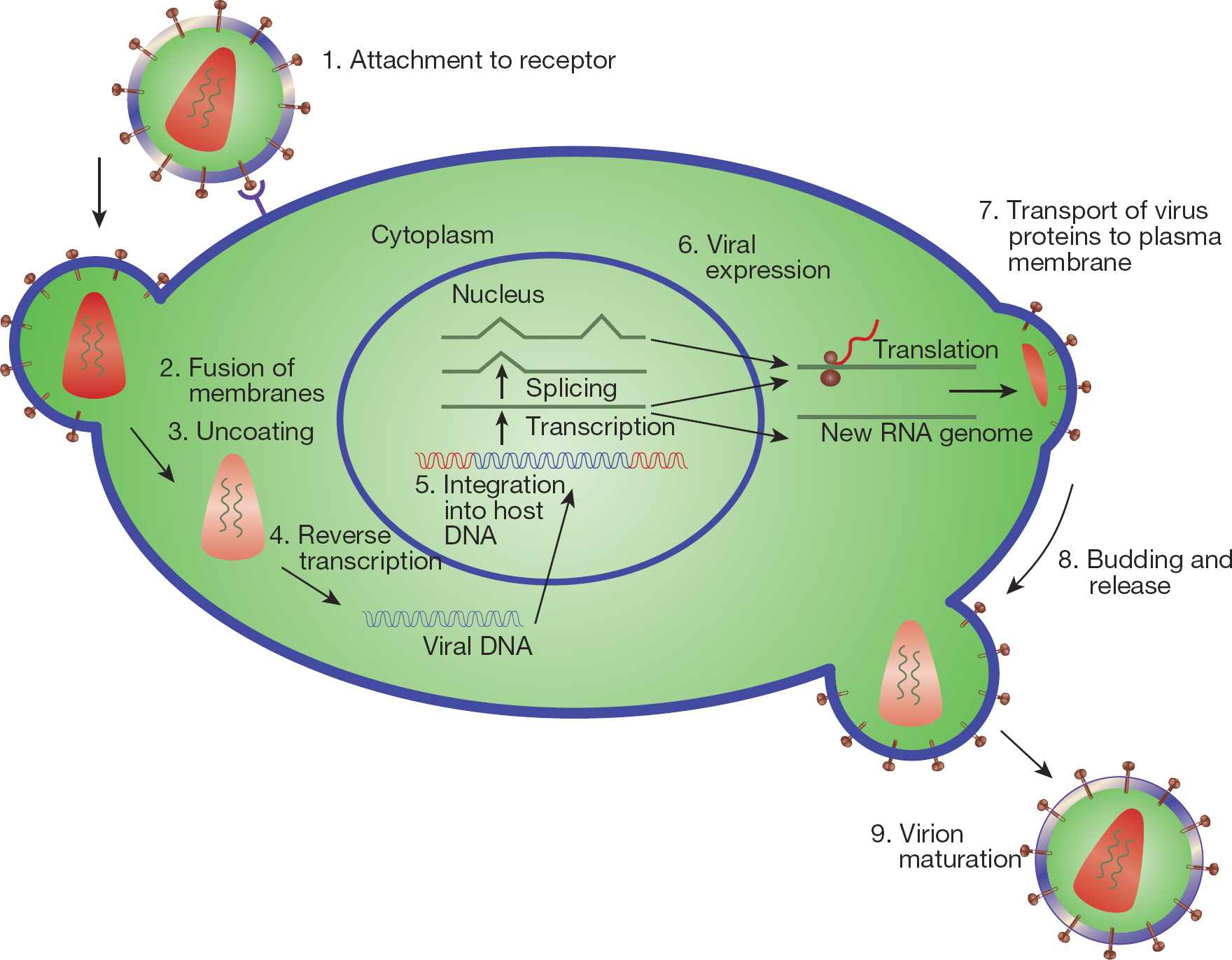
Replicative cycle of HIV type-1 and drug targets
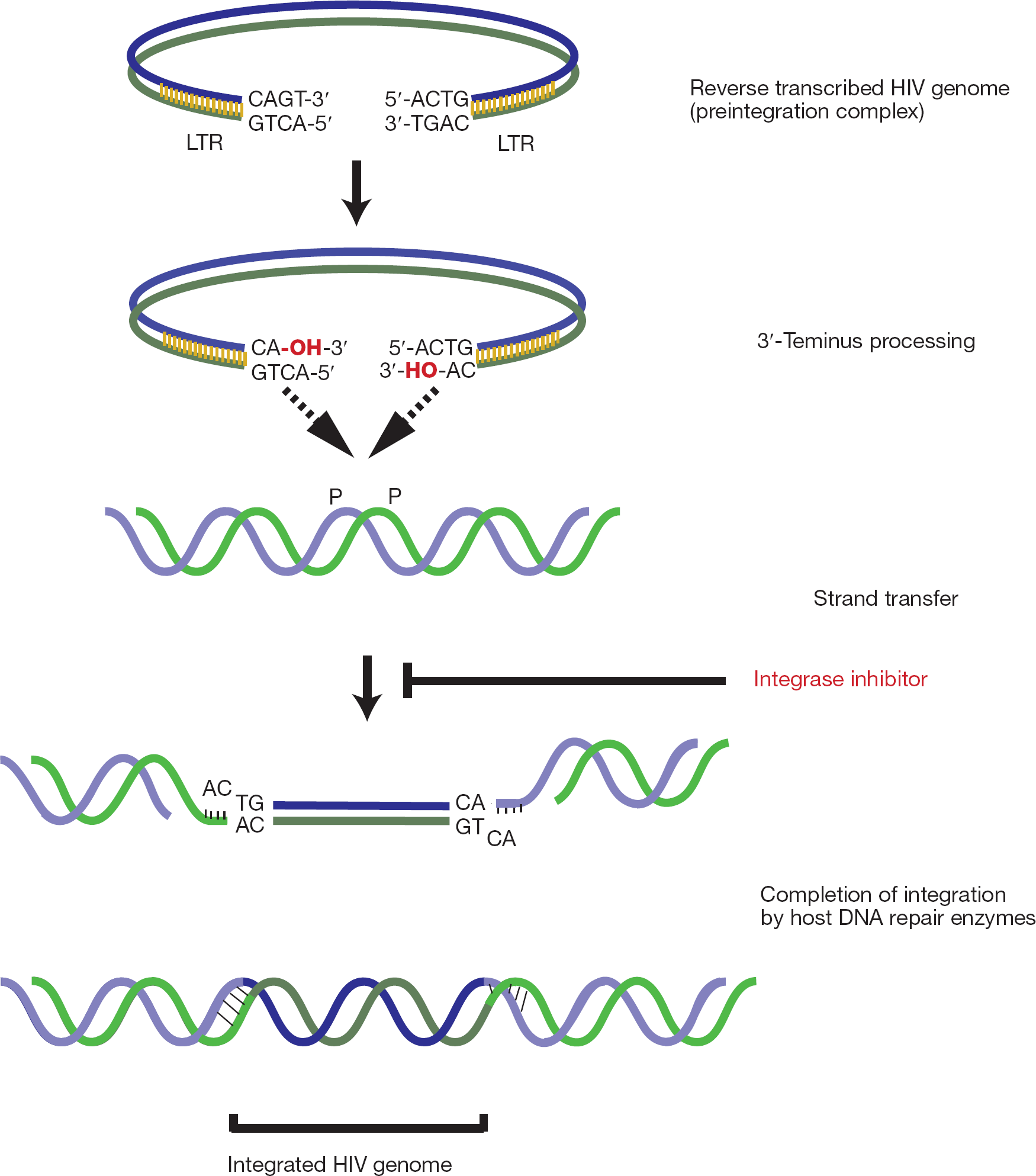
Outline of the integration process
HIV-1 IN is a 32 kDa enzyme comprising 288 amino acid residues, and it can be divided into three functional domains. Recently the crystal structures of all three individual IN domains: catalytic core domain [5,6], N-ter-minal core domain [7] and C-terminal core domain have been resolved. HIV-1 IN domains are delineated on the basis of the susceptibility of their linker regions to proteolysis, functional and/or the structural studies. A predicted conformational loop encompassing residues 141–148, hinged by Gly140 and Gly149, plays an essential role in the enzymatic functions [8]. However, the flexibility of this loop makes its structure difficult to determine, such that it was either missing or found in multiple different conformations. Furthermore, the temperature factors were typically high for the loop residues in these X-ray structures. Therefore, the structure of the loop appears to be inherently flexible and may depend on the presence of substrates and/or inhibitors [9].
The catalytic core domain contains the characteristic triad of acidic residues, the DDE motif, comprising residues Asp64, Asp116 and Glu152 as shown in Figure 3A. Mutagenesis of these residues and their counterparts in related retroviral INs abolishes or severely diminishes all catalytic activities [10,11]. It has been demonstrated that coordination of divalent metal ions like Mg2+ and Mn2+ to these residues plays a key role in catalysis. The N-terminal domain and DDE motif in the catalytic core are highly conserved among retroviridae, while the C-terminal domain is less conserved.
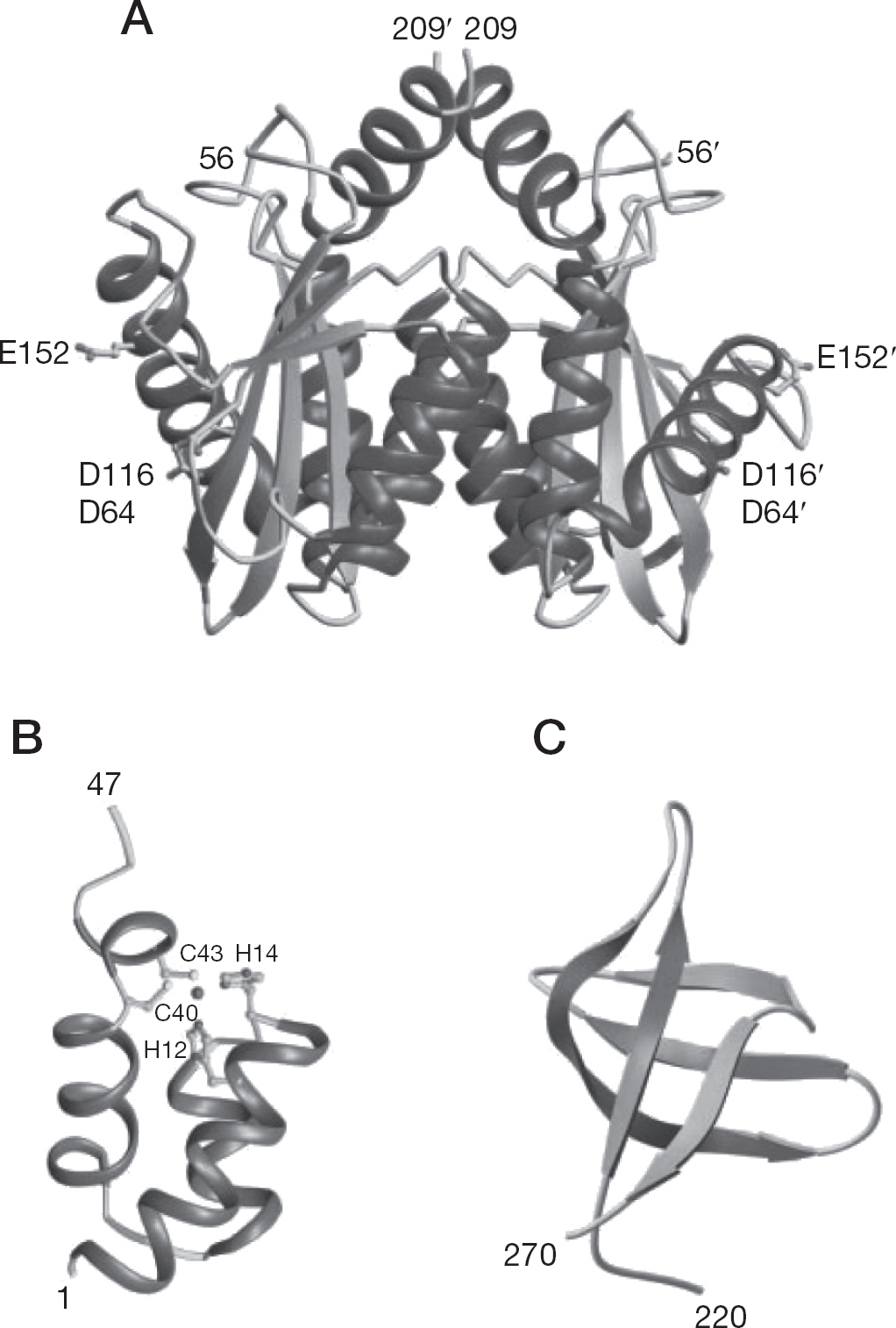
Structures of the three domains of HIV type-1 integrase shown as ribbon diagrams
IN catalytic core is dimeric in solution and in the crystal structures (Figure 3A) [12]. The spacing between the active sites in the nearly spherical dimer is not enough to grip two strands of target DNA. The sites of insertion on each strand of target DNA are separated by five base pairs. The functional unit of IN might therefore be expected to have a pair of active sites separated by a similar spacing. However, in the crystal structures (Figure 3A), the active sites in the dimer are separated by more than 30 Å, measured as a straight line through the protein and by an even greater distance measured around the circumference of the dimer. Assuming that the dimer interface is maintained in the functional IN complex, at least a tetramer of IN is required for the integration reaction. The N-terminal domain involved in multimerization contains two His and two Cys residues involved in zinc coordination by this domain [13,14]. However, its structure (Figure 3B) is dissimilar to that of the typical zinc finger proteins [15–17]. Detailed structure/functional understanding of the C-terminal domain (Figure 3C) involved in DNA binding remains to be elucidated and is controversial. It is believed that some cellular nuclear protein(s) seem(s) to be co-involved in integration as well as IN [18].
DKA class of HIV-1 integrase inhibitors
The first selective inhibitor of HIV-1 integrase, 5-CITEP (1-(5-chloroindol-3-yl)-3-hydroxy-3-(2H-tetrazol-5-yl)-propenone; Figure 4) was reported by Goldgur et al. [19] in 1999. Further studies carried out by Hazuda et al. [20] reported L-731,988 (Figure 4) in 2000. In this case, the tetrazole ring of 5-CITEP is replaced isosterically with carboxylic acid; and the keto enol is tautomeric with the diketo functionality. Another compound, L-708,906 (Figure 4), inhibited the replication of HIV-1 (50% effective concentration [EC50]=0.2 μM at 3′-end processing). It also inhibits DNA strand transfer at a 50% inhibitory concentration (IC50) of 3.5 μM.
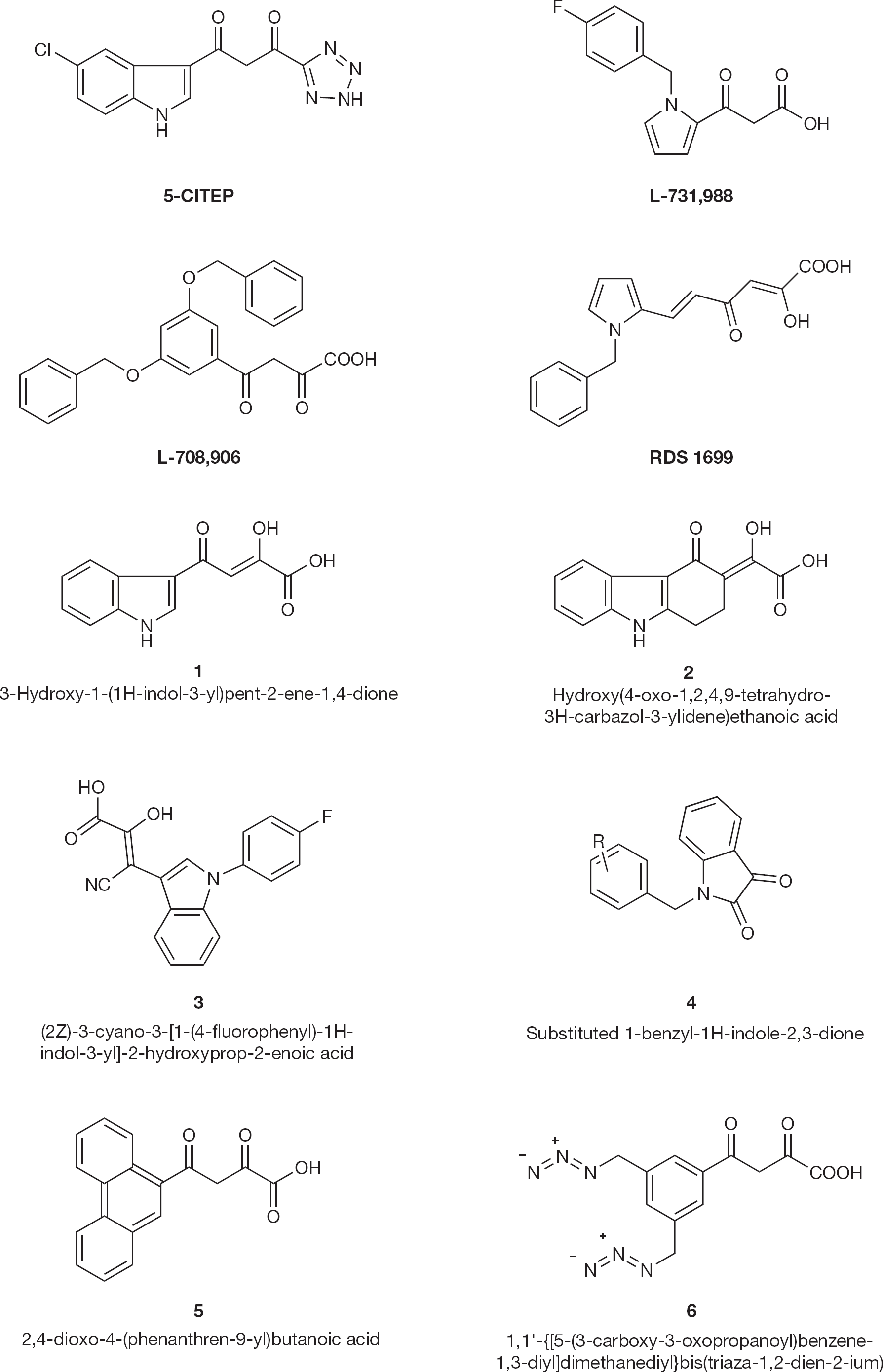
HIV type-1 integrase inhibitors 1–6
Li and Vince [21] described modification studies on the key α, γ-diketo acid (DKA) pharmacophore substituting the carboxylate moiety to its bioisosteres or other electron-pair bearing heterocycles. They further studied the conformation and geometry of the central DKA moiety and developed a series of carbazolone-containing α, γ-DKAs. As anticipated, these compounds inhibit IN activity in the low micromolar concentrations, indicating that the geometry of the DKA moiety is crucial for potency. Modification of the compound (1; Figure 4) to the conformationally restrained and 4bT-substituted carbazol-4-one DKA (
Di Santo et al. [22] designed some aryl diketo hexanoic acid derivatives as HIV-1 IN inhibitors and designed RDS1699 that showed anti-HIV activity in cell-based assays (EC50 =1.5 μM), again indicating that DKA structure seems a key moiety for IN inhibition.
Some dioxybutanoic acid derivatives have been reported by Wai et al. [23] as potent and specific inhibitors of HIV-1 multiplication targeting the integration process. The anti-IN activity was also retained when the terminus carboxylic function was masked by a tetrazole ring and the dioxobutanoic group was shortened into an oxopro-panoic moiety. Attempts have also been made to check the effect of lengthening of the dioxobutanoic group, for example, a series of 6-aryl-2,4-dioxo-5-hexanoic acids were synthesized (
Sechi et al. [24] have done a 3D computational search on the NCI database to identify the possible bioisosters of DKAs. Several structural platforms with potential as HIV-1 IN inhibitors were selected and their pharmacophoric fragments were incorporated into aromatic or heteroaromatic frameworks to give eight general structures (
In their search for new integrase inhibitors, Patil et al. [25] discovered a series of new substituted phenan-thridinones (
Certain other azido-containing β-DKA derivatives (
Catechol and poly phenol class of HIV-1 integrase inhibitors
A substituted catechol moiety is common in HIV-1 IN inhibitors, such as bis-catechols [27–29], caffeic acid phenethyl ester (CAPE; Figure 5) [30], flavones and flavonoids [31,,–38].
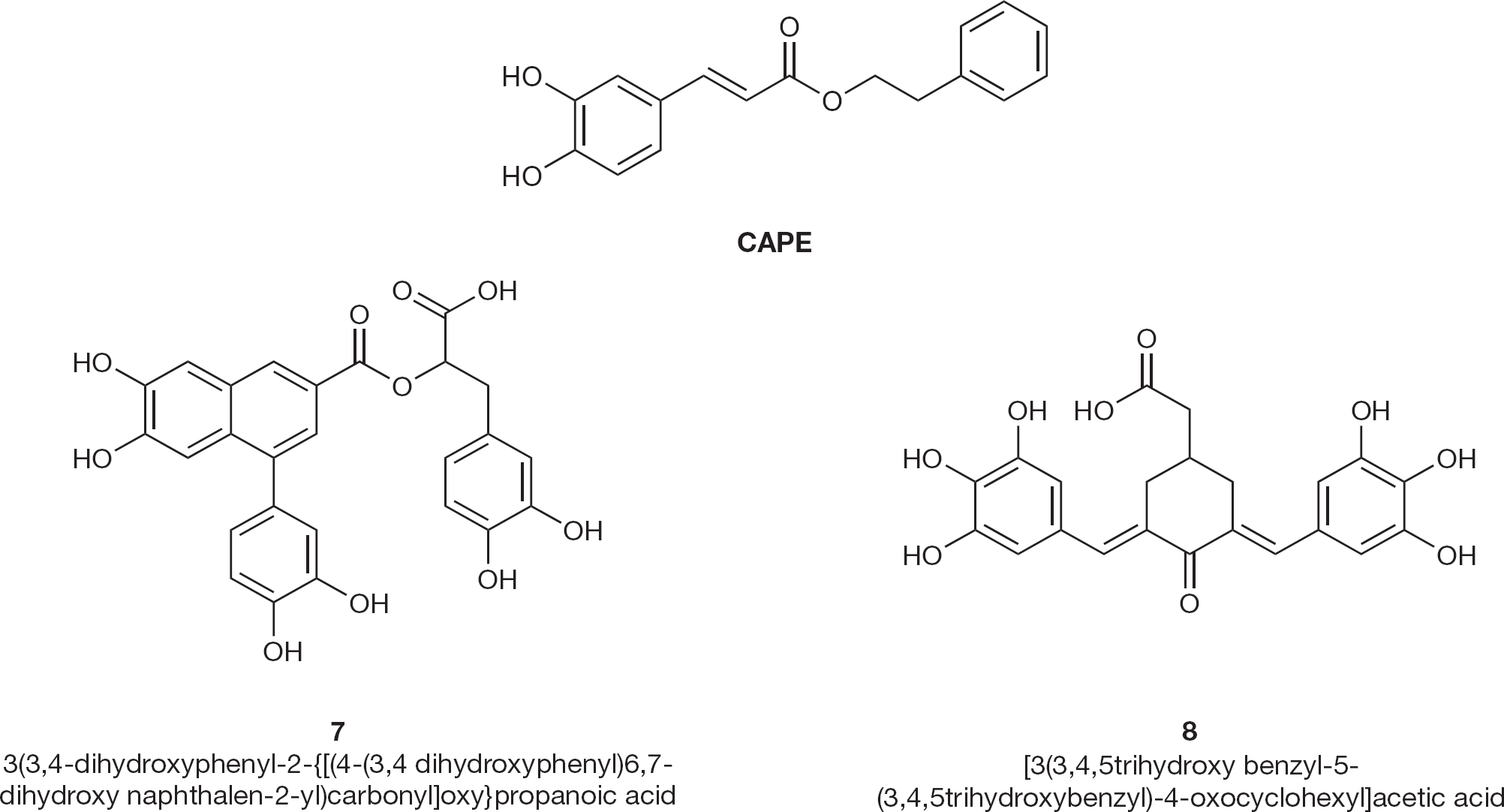
HIV type-1 integrase inhibitors 7 and 8
High throughput screening of natural product inhibitors of HIV-1 IN by Ovenden et al. [39] found that the methanol extract from the buds of a Eucalyptus globoidea was active. Bioassay guided fractionation resulted in the purification and structural elucidation of the lignan globoidnan A (
A number of 2,6-bisbenzylidenecyclohexane-1-one derivatives have been synthesized and tested by Costi et al. [40]. Compound 8 (Figure 5) resulted in one of the most potent and selective derivatives in acutely infected MT-4 cells (EC50 and CC50 values of 2 and 40 μM, respectively).
Development of IN inhibitors either lacking the catechol moiety or modifications which overcome its toxic properties, led to the development of dicaffeoyl quinic acids (DCQAs) and related compounds, L-chicoric acid (7-dicaffeoyl tartaric acid) [41,42]. The representative compounds (9; Figure 6) inhibit HIV-1 replication in culture and inhibit integration in biochemical assays. During the process of evolution of resistant species from HIV-1, the L-chicoric acid derivatives showed cross resistance to other IN inhibitor resistant variants in enzymatic assay, however, the derivatives were indeed proved as inhibitors in a cell-based assay. Probably, chicoric acid derivatives interact with gp120 through hydroxyl groups.
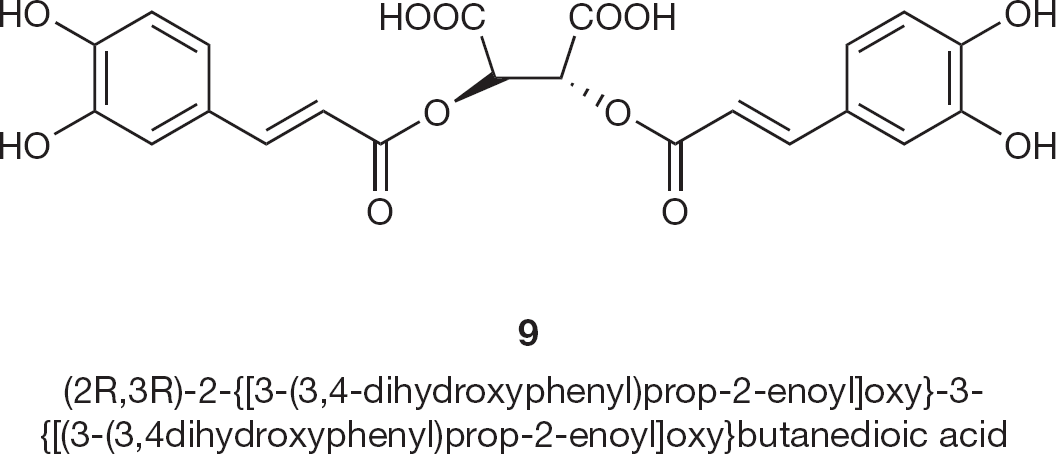
HIV type-1 integrase inhibitor 9
Quinoline class of HIV-1 integrase inhibitors
Bénard et al. [43] synthesized a new series of HIV-1 IN inhibitors featuring a quinoline subunit and ancillary aromatic ring linked by functionalized spacers such as amide, hydrazide, urea and 1-hydroxyprop-1-en-3-one moieties (10 and 11; Figure 7) and tested them in in vitro and ex vivo assays.
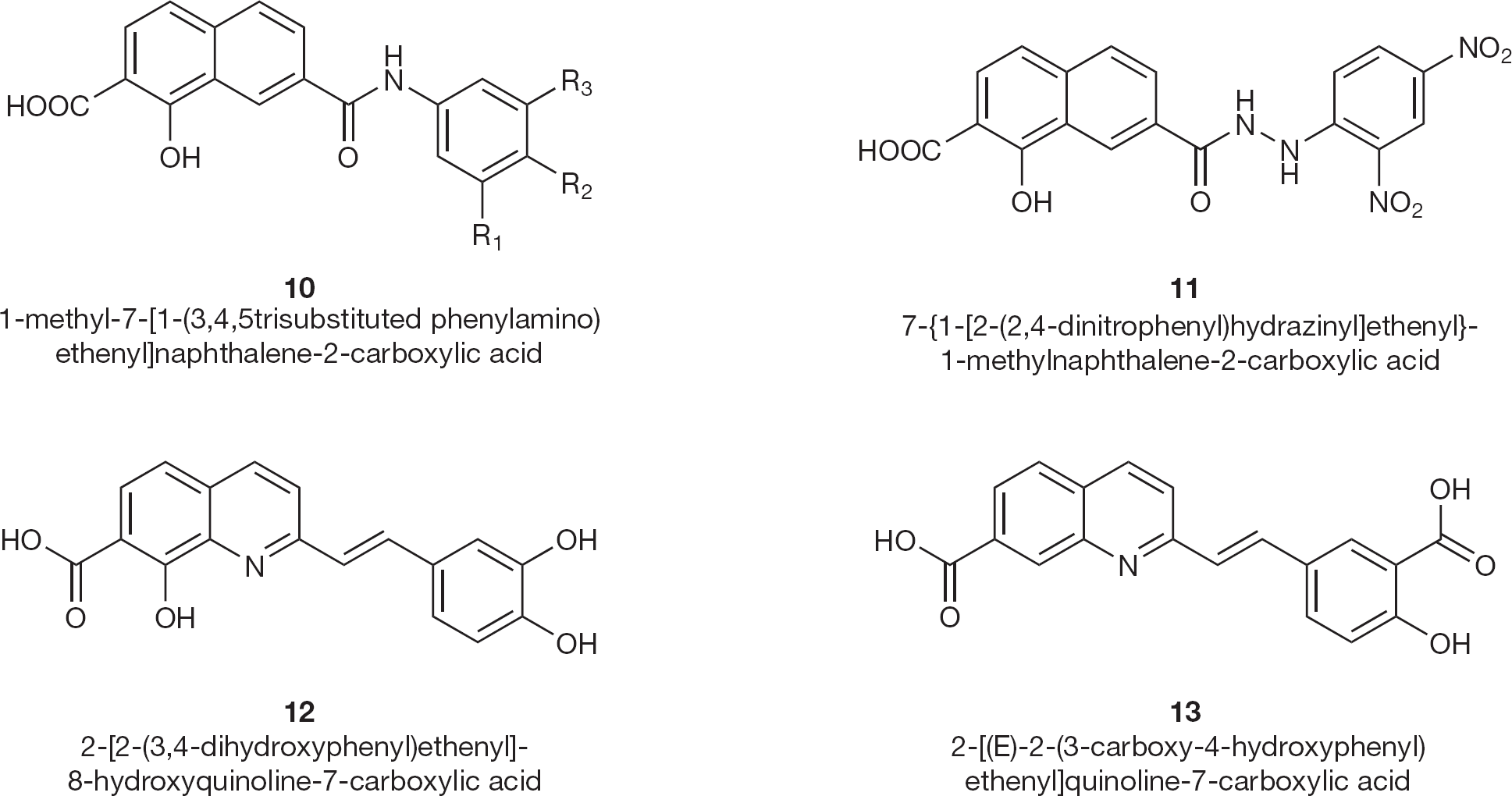
HIV type-1 integrase inhibitors 10–13
Certain styrylquinoline compounds (12 and 13; Figure 7) have been shown to be potent HIV-1 IN inhibitors in vitro [44]. Several polynucleotidyl transferases, related to HIV IN, contain two divalent metal cations separated by 4 Å in their active site. Potential IN inhibitors based upon the quinoline substructure linked to an aryl nucleus possessing various hydroxy substitution patterns, can easily interact at this site. These styrylquinoline derivatives target the IN core domain since they inhibited the disintegration assay carried out by the active site containing deletion mutation.
Sato et al. [45] described the modification of a qui-nolone antibiotic to produce a novel integrase inhibitor, JTK-303 (GS 9137/elvitegravir; Figure 8). It shows an IC50 of 7.2 nM in the strand transfer assay and an EC50 of 0.9 nM in an acute HIV-1 infection assay.
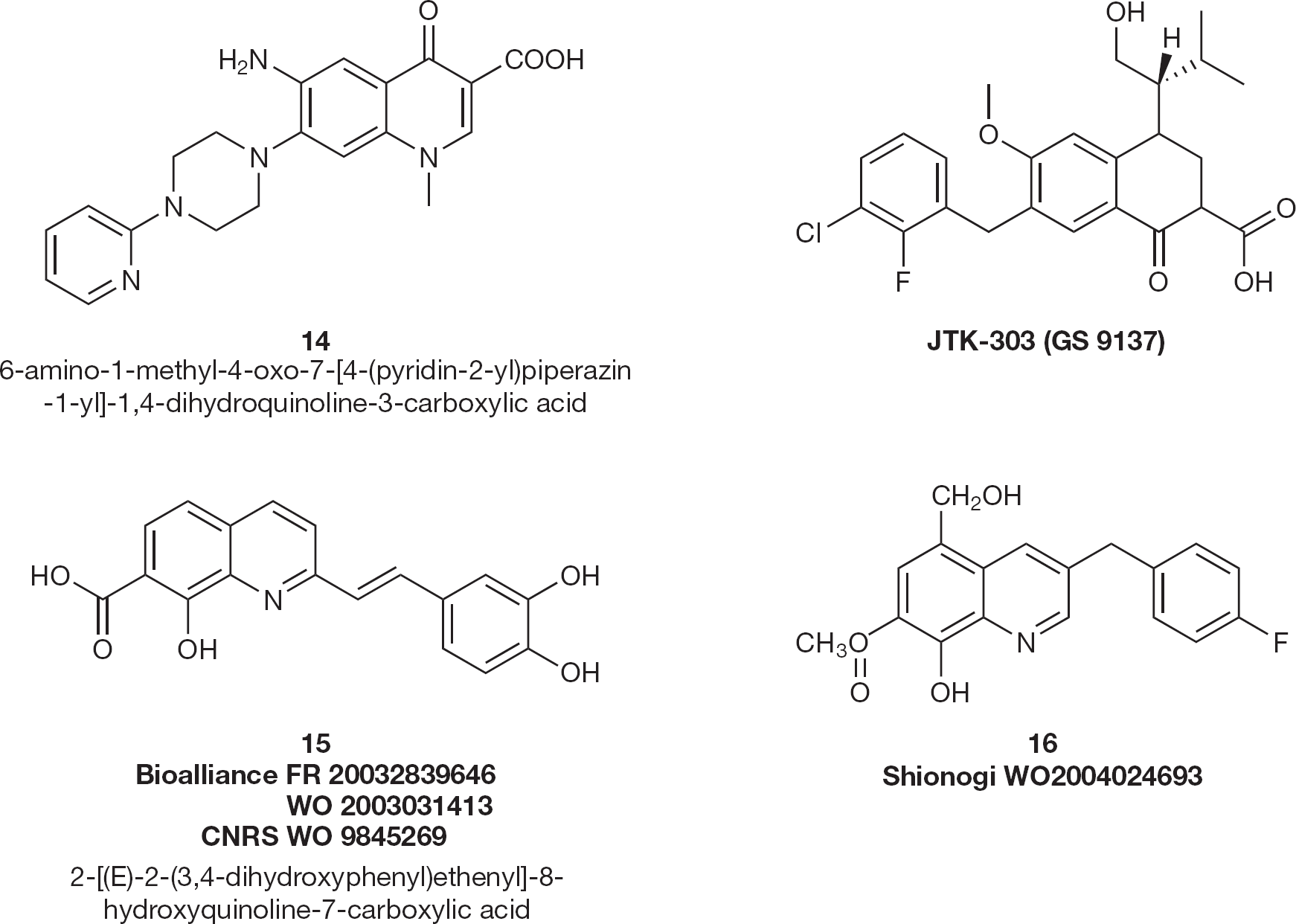
HIV type-1 integrase inhibitors 14–16
A series of 6-aminoquinolone compounds were evaluated by Cecchetti et al. [46] for their in vitro activity against HIV-1. Compound 14 (EC50 =0.1 μM; Figure 8) was the most active in inhibiting HIV-1 replication on de novo infected C8166 human lympho-blastoid cell lines.
D'Angelo's group [47] was the first to include 8-hydroxyquinoline-7-carboxylic acid (15; Figure 8) into the structure of new HIV-1 IN inhibitors with IC50 in the range of 0.3–4.0 μM. A substitution on the right side of the molecule (mainly by hydroxyl groups) has led to the absence of selectivity towards the two steps catalyzed by IN [48].
In vivo, styrylquinolines were found to act prior to integration by reducing the amount of the late cDNA, suggesting for the first time that IN-targeting molecules may affect the accumulation of DNA during reverse transcription [49]. More recently, Shionogi patented 8-hydroxyquinoline-7-carboxylic acid methyl esters substituted on position 3 by a substituted benzyl group (
Zhuang et al. [51] synthesized a series of 8-hydroxy-[1,6]-naphthyridine compounds that inhibited the strand transfer reaction with an IC50 of 10 μM in vitro. One of the most potent compounds in this series is L-870,810 (Figure 9) which inhibits HIV-1 replication in cell culture with an IC95 value of 100 nM.
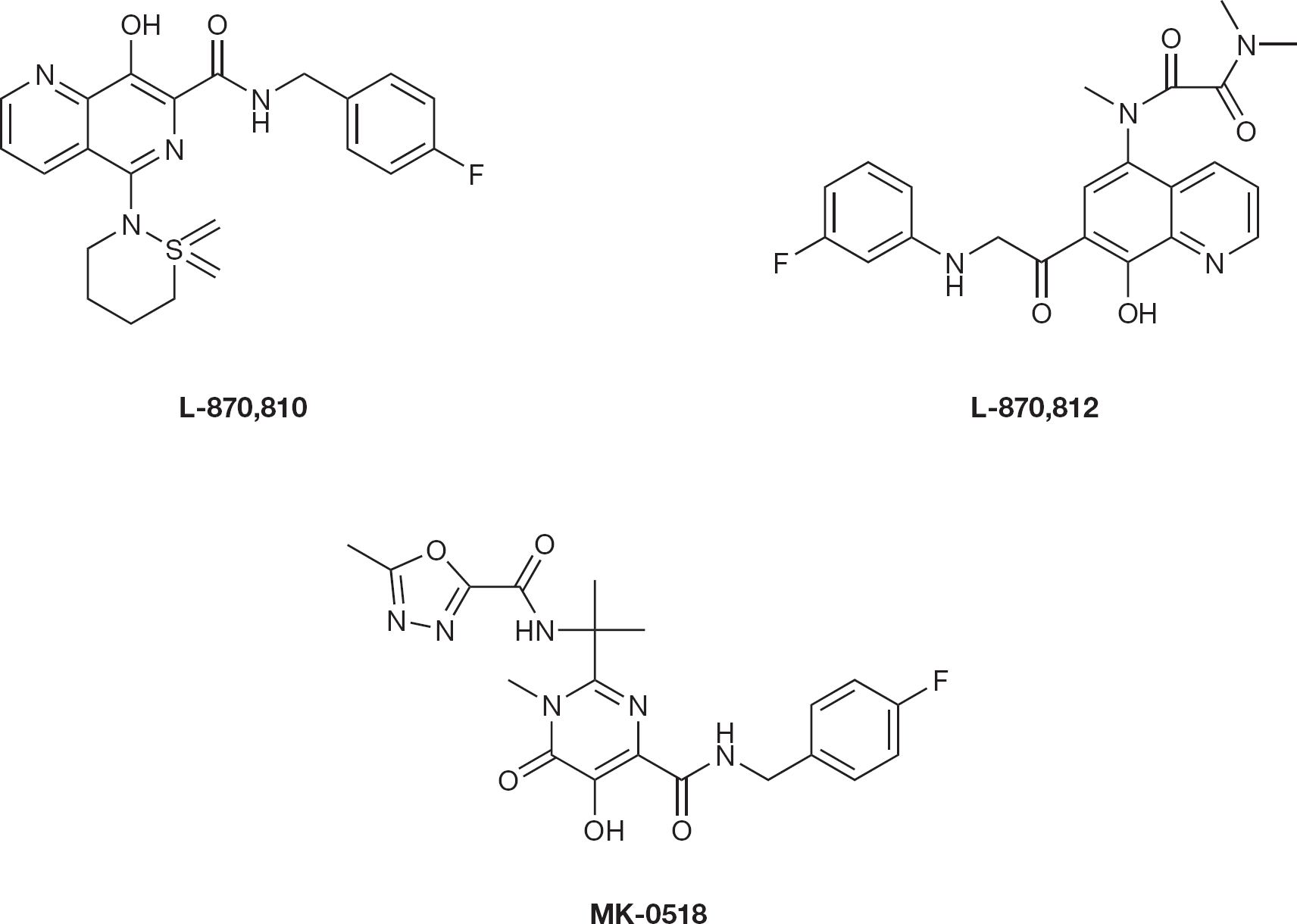
HIV type-1 integrase inhibitors
Guare et al. [52] synthesized a series of 5-amino derivatives of 8-hydroxyl-[1,6]-naphthyridine-7-car-boxamide exhibiting submicromolar potency against replication of HIV-1 in cell culture. One of these analogues, L-870,812 (Figure 9), displayed excellent phar-macokinetic properties. This work by Merck has led to the discovery of 8-hydroxy-[1,6]-naphthyridines as a suitable replacement for the 1,3-diketo moiety. Clinical trials of L-870,812 were terminated recently. Despite this, another potent compound by Merck, MK-0518 (raltegravir; Figure 9) with 27 nM through strand transfer inhibition has been approved by the US Food and Drug Administration in 2007.
Pannecouque et al. [53] have identified a novel series of selective inhibitors of HIV-1 replication, the 5-(4-substituted-phenyl)-5H-pyrano [2,3-d:-6,5-d'] dipyrimidines (PDP), with a novel mode of action. The most potent congener, and prototype of the IN B inhibitors, 5-(4-nitrophenyl)-2,8-dithiol-4,6-dihy-droxy-5H-pyrano [2,3-d:-6,5-d'] dipyrimidine (V-165; Figure 10), inhibited the replication of different HIV-1 strains, HIV type-2 and simian immunodeficiency virus (SIV), at EC50 values ranging from 3.7 to 30 μM. However, in enzymatic assays, PDPs inhibited both reverse transcription and integration reactions, indicating that PDPs seems to be non-specific inhibitors.
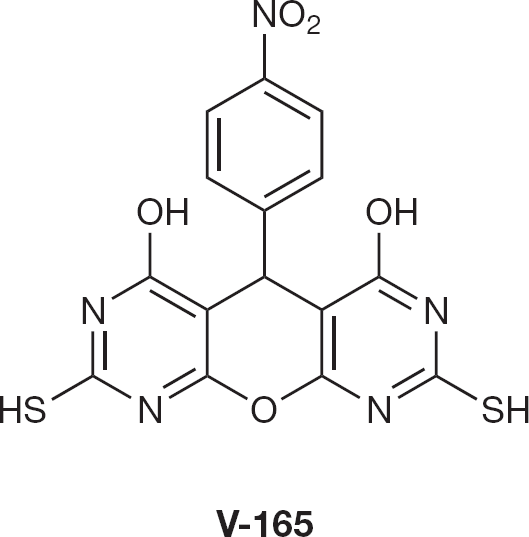
Integrase inhibitor V-165
Resistance to integrase inhibitors
Fikkert et al. [54] first studied the development of antiviral resistance to L-708,906 by growing HIV-1 strains in the presence of increasing concentrations of the compound. The mutations T66I, L74M and S230R emerged successively in the IN gene. The viruses with these three mutations were found to show 10-fold less susceptibility to L-708,906. Raltegravir (RAL), has been associated with early drug resistance development and experimental data suggest an even greater resistance to elvitegravir (EVG). Resistance to RAL occurs through mutations that decrease the sensitivity of the IN to its inhibitors, single point mutations in the IN are enough for reduction of activity [55–57].
Virological failure was generally associated with the mutations at one of three residues (Tyr143, Gln148 or Asp155), usually in combination with at least one other mutation. Clinical studies revealed two major mutational pathways for the acquisition of RAL resistance: N155H (sometimes associated with L74M, E92Q, T97A, G136R and/or V151I) and Q148K/R/H, usually associated with E138K or with G140A/S [58,59].
E92Q enhances the RAL resistance by N155H. Similar to RAL, EVG has been shown to select for T66I and E92Q, as well as substitutions in the flexible loop (Q146P and S147G) [60]. Molecular modelling studies based on docking IN inhibitors in the crystal structure of the catalytic core of the enzyme predict that Thr66, Glu92, Tyr143, Gln148 and Asn155 contribute to RAL binding, in agreement with phenotypic drug susceptibility data of HIV-1 variants carrying amino acid substitutions at those positions [61]. In addition, EVG is expected to interact with THR66, Glu92, Tyr143, gln148 and Asp116, Glu152 of the DDE motif.
To date, approximately 42 mutations within the HIV-1 IN gene have been associated with resistance to IN inhibitors. Naturally occurring IN gene polymorphisms may have some implications for emergence of resistance. In total, 21 of the 42 substitutions closely associated with IN inhibitor resistance, are observed as natural polymorphisms: V72I, L74I, T97A, T112I, A128T, E138K, Q148H, V151I, S153Y/A, M154I, N155H, K156N, E157Q, G163R, V165I, V201I, I203M, T206S, S230N and R263K [62]. Primary resistant mutations to IN inhibitors are located within the catalytic domain and extended active sites associated with the high level resistance to the strand transfer inhibitors (STIs). Primary mutations for RAL or EVG are rare in IN inhibitor-naive patients, while secondary or resistance-associated mutations are observed more frequently. Interestingly, some secondary mutations appear to influence drug resistance development [62–64].
The Shionogi-GSK joint venture tested the first IN inhibitor S-1360 in Phase IIa studies and further produced S/GSK-364735 for clinical trial. While the use of S/GSK-364735 in HIV-infected adults showed promising efficacy in early clinical trials, the development of this compound has been discontinued because of adverse effects observed in monkeys, in addition to long-term toxicity. Q148R and F121Y were the predominant single mutations isolated in the presence of S/GSK-364735. S/GSK-364735-resistant viruses showed strong cross resistance to other IN inhibitors to a different extent.
Conclusions
There has been remarkable progress since the identification of HIV IN as a rational therapeutic target for HIV infection. Inhibitors for IN that are believed to have no counterpart in humans are expected to be selective and safe. In the development of IN inhibitors, in vitro enzymatic assays that are convenient for high throughput have produced reports of false-positive leads, indicating the importance of virological confirmation, for example, cell-based assays. The lack of diversity in the chemical structures of currently identified HIV-1 IN inhibitors can lead to cross-resistance, suggesting that further development of novel structures with different pharmacophores are warranted. The successful development of IN inhibitors with novel structures or mechanisms of inhibition might provide new treatment options for safe and effective therapy.
Footnotes
The authors declare no competing interests.