
Abstract
Background:
Pyrimidine nucleoside analogues represent an established class of clinically useful antiviral agents. Once inside the cell, they are activated by a series of intracellular phosphorylation steps to produce 5′-triphosphate derivatives. In many cases, nucleoside analogues are poor substrates for the cellular kinases needed for their activation. It is clear that intracellular introduction of nucleoside analogues as phosphorylated metabolites (so called pronucleotides) could circumvent difficulties associated with the use of non-phosphorylated nucleoside analogues.
Methods:
Among the current diverse pronucleotide approaches, nucleoside phosphoramidate derivatives appear to be an interesting class of potential antiviral agents because of the known relatively low stability of the P–N bond in cellular media. On the basis of oxathiaphospholane chemistry, a series of novel conjugates of 5′-O-phosphorylated zidovudine (AZT) and stavudine (d4T) with amino acids carboxamidates were obtained. The synthesis was performed using N-(2-thiono-1,3,2-oxathiaphospholane) derivatives of amino acids carboxamides as precursors.
Results:
All synthesized compounds were studied against DNA and RNA viruses. Specific antiviral activities were only detected against HIV type-1 and HIV type-2 in MT-4 cell cultures at compound concentrations that were equally active or slightly inferior to the activity of their parent drugs (2- to 20-fold for the AZT prodrugs and 6- to 40-fold for the d4T prodrugs). The compounds were also evaluated for their anti-HIV activity in CEM and in CEM thymidine-kinase-deficient (CEM/TK−) cell cultures.
Conclusions:
Loss of compound antiviral potency in the CEM/TK− cells suggested an eventual conversion of the test compounds to the free nucleosides prior to further phosphorylation to the active 5′-triphosphate metabolite.
Introduction
Since the recognition of the first HIV/AIDS case >25 years ago, a number of emerging and re-emerging infectious diseases of humans and animals have been reported, including severe acute respiratory syndrome, avian flu, West Nile fever and Ebola [1]. Interestingly, all these infectious diseases are caused by viral infections and, in particular, RNA viruses.
With the elucidation of virus-specific events that can be used as chemotherapeutic targets, selective treatment can be achieved and virus replication can be suppressed without deleterious effects on the host. Any of the steps involved in the viral replication cycle could potentially be targets for chemotherapeutic intervention [2].
Notably, viral polymerases appear to be one of the most promising targets for antivirals against viruses that utilize their own polymerases for replication. For example, the HIV type-1 (HIV-1)-encoded reverse transcriptase is a promising target because human cells lack RNA-dependent DNA polymerase activity. Other viruses might also code for their own polymerases, such as the RNA-dependent RNA polymerase of flaviviruses [3]. Therefore, it is not surprising that out of the >30 compounds that are currently marketed in the US for treatment of viral infections, 15 are polymerase inhibitors, most of which are nucleoside analogues [4]. Among them, ribavirin is a particularly important drug that possesses broad-spectrum antiviral activity against several RNA and DNA viruses [5]. It should be noted that ribavirin might have several mechanisms of action. Ribavirin is phosphorylated by cellular adenosine kinase to ribavirin-5′-monophosphate, which is a potent inhibitor of inosine monophosphate (IMP) dehydrogenase, resulting in a decrease of cellular GTP pools and inhibition of viral RNA synthesis [6]. However, it is also an effective inhibitor of viral RNA guanylyl transferase and (guanine-7N)-methyl transferase, which results in a defective 5′-cap structure of viral transcripts and insufficient translation. The 5′-triphosphate form of ribavirin has been implicated as a direct inhibitor of the viral RNA polymerase. Because it is a nucleotide analogue, ribavirin-5′-triphosphate, the major metabolite accumulating in vivo [7], can also be incorporated into the viral genome, inducing an error catastrophe that will result in the accumulation of mutations in the viral genome (for example, polio virus) [8]. Ribavirin, in combination with pegylated interferon, has become the standard therapy for the treatment of HCV infections [9].
In many cases, nucleoside analogues are poor substrates for the cellular kinases needed for their activation [10]. Also, the presence and activity of the intracellular enzymes necessary for activation of nucleoside analogues are highly dependent on the host species, the cell type and the stage of the cell cycle. It is worth noting that among the three successive activating phosphorylation steps, the first step that is responsible for formation of the nucleoside analogue monophosphate is often regarded as the bottleneck, although, in other cases, the second phosphorylation step can also be rate-limiting (for example, conversion of AZT [zidovudine] monophosphate to AZT diphosphate) [11,12]. It is thus clear that intracellular introduction of nucleoside analogues as phosphorylated metabolites could circumvent difficulties associated with the use of non-phosphorylated nucleoside analogues and could even activate inactive compounds or could increase the activity of the nucleoside analogues. However, polarity (hydrophilicity) and a ready degradation by phosphatases make the use of free nucleotide analogues impractical. Therefore, much of the recent efforts have been focused on finding suitable prodrugs of nucleoside analogue monophosphates (pronucleotides) and a number of pronucleotide approaches have been developed [13–16]. In general, these methods rely on remote chemical or enzymatic activation of the masking moiety, followed by degradation of an unstable intermediate and release of the nucleotide [17].
Among the current diverse prodrug approaches, nucleoside phosphoramidate derivatives appear to be an interesting class of antiviral agents [13,16]. In several cases, these compounds exhibited enhanced antiviral activity and reduced cytotoxicity when compared with the parent nucleoside [18]. Recently, Iyer et al. [19] demonstrated that nucleoside amino acid phosphoramidate monoesters, despite carrying one formal charge on the phosphate moiety, fulfill requirements of lipophilicity for passive diffusion through the membrane barrier and non-toxicity after hydrolytic or enzymatic release in the target cell. For example, corresponding AZT derivatives were less lipophilic than AZT (logP=0.24), with logP values ranging from −0.8 to −1.8.
We recently described a new approach to the preparation of O-alkyl-N-acylphosphoramidothioates and corresponding phosphoramidates [20]. This method relies upon oxathiaphospholane chemistry, which we have found to be applicable to the synthesis of a wide variety of organophosphorus compounds [21]. On the basis of these results, the goal of this study was to synthesize a series of conjugates of 5′-O-phosphorylated antiviral nucleosides with amino acid carboxamidates. To our knowledge, O-alkyl-N-acylphosphoramidates have not been tested as potential prodrugs, and their synthesis are of interest because of the known relatively low stability of the P–N bond in cellular media. Although the enzyme responsible for hydrolysis of the P–N bond in phosphoramidates has not been established [22], involvement of putative phosphoramidase (Hint-1) in the intracellular release of nucleoside 5′-O-phosphate has been demonstrated [23].
Methods
General procedure for the synthesis of N-(2-thiono-1,3,2-oxathiaphospholanyl)amino acid carboxamides To a solution of amino acid carboxamide
Physicochemical characteristics of compounds
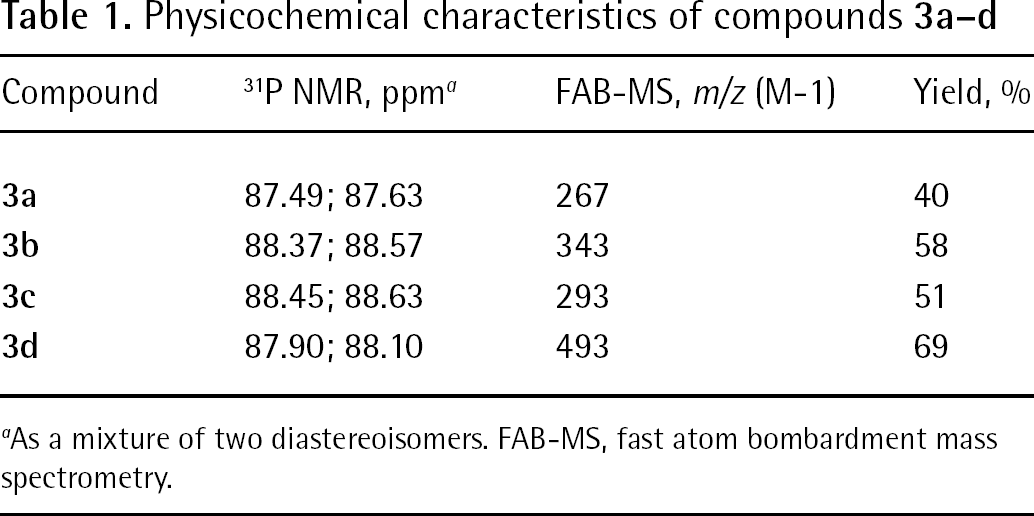
As a mixture of two diastereoisomers. FAB-MS, fast atom bombardment mass spectrometry.
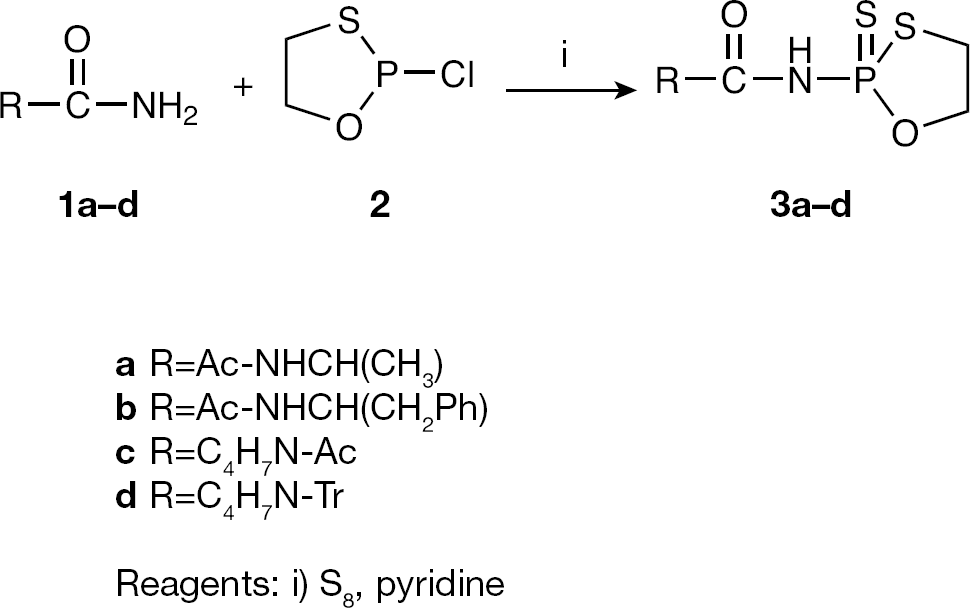
Synthesis of compounds
General procedure for the synthesis of nucleoside N-acylphosphoramidates
Corresponding compound
Compounds
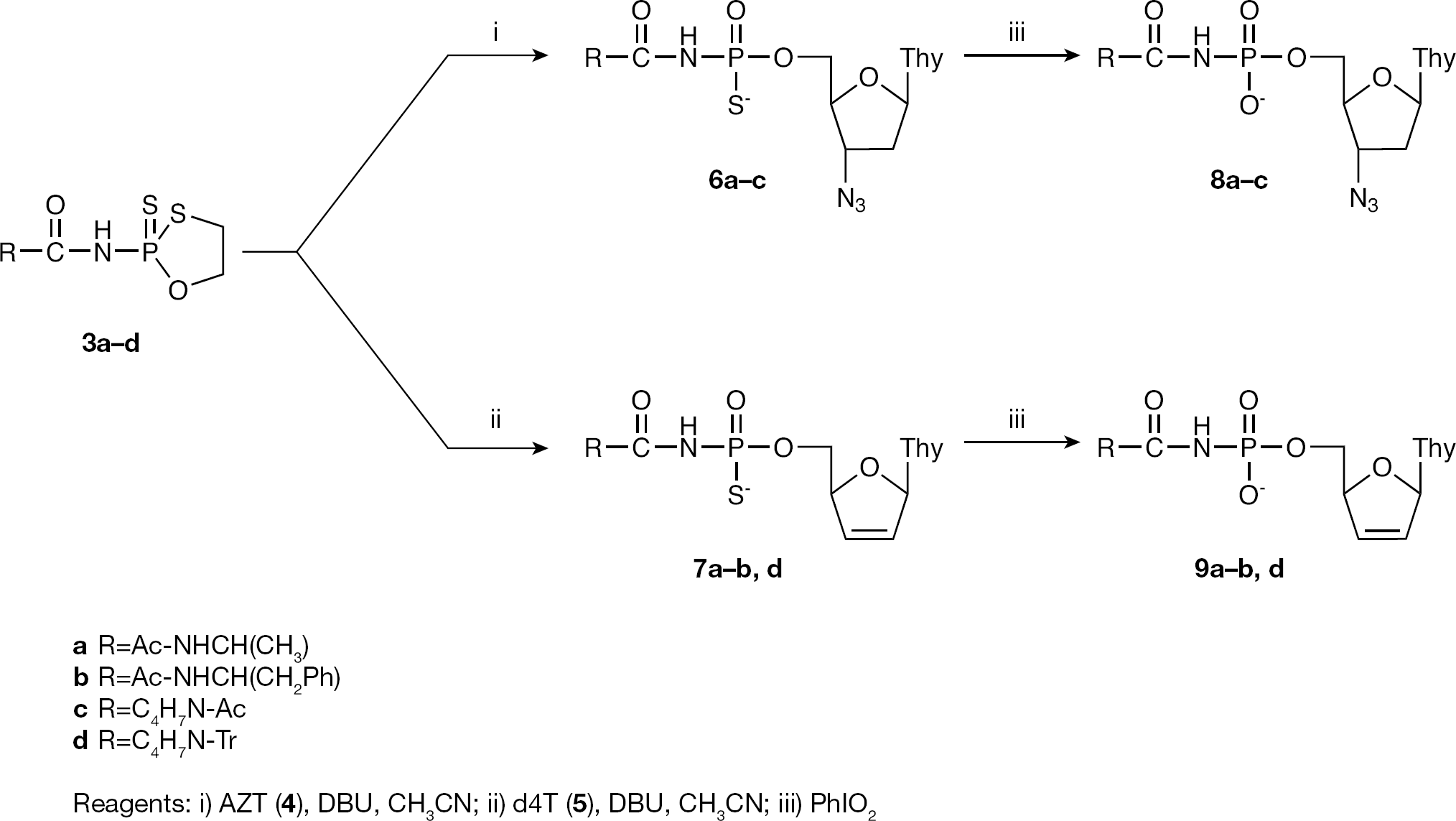
Synthesis of nucleoside 5′-O-(N-acyl)phosphoramidates
3′-Azido-3′-deoxythymidine-5′-O-[N-(α-N-acetylalanyl)] phosphoramidothioate (6a)
Yield 47%. 31P MMR (D2O) δ: 46.83; 47.19. 1H MNR (D2O) δ: 7.73 (s, 1H), 6.17 (t, 1H), 4.30 (m, 1H), 4.22 (m, 1H), 4.06 (m, 3H), 2.44 (t, 2H), 1.92 (s, 3H), 1.86 (s, 3H), 1.32 (d, 3H). FAB-MS m/z: (M-1) 474.
3′-Azido-3′-deoxythymidine-5′-O-[N-(α-N-acetylphenyloalanyl)] phosphoramidothioate (6b)
Yield 47%. 31P MMR (D2O) δ: 46.83; 47.03. 1H MNR (D2O) δ: 7.62 (s, 1H), 7.20 (m, 5H), 6.21 (t, 1H), 4.34 (m, 2H), 4.04 (m, 3H), 3.14 (m, 1H), 2.94 (m, 1H), 2.41 (m, 2H), 1.92 (s, 3H), 1.82 (s, 3H). FAB-MS m/z: (M-1) 550.
3′-Azido-3′-deoxythymidine-5′-O-[N-(α-N-acetylprolyl)] phosphoramidothioate (6c)
Yield 64%. 31P MMR (D2O) δ: 46.57; 47.04. 1H MNR (D2O) δ: 7.74 (s, 1H), 6.17 (t, 1H), 4.40 (m, 2H), 4.11 (m, 2H), 3.53 (m, 2H), 2.40 (m, 2H), 2.19 (m, 1H), 2.01 (s, 3H), 1.86 (m, 4H), 1.85 (s, 3H). FAB-MS m/z: (M-1) 500.
3′-Deoxy-2′,3′-didehydrothymidine-5′-O-[N-(α-N-acetylalanyl)] phosphoramidothioate (7a)
Yield 52%. 31P MMR (D2O) δ: 46.73; 47.21. 1H MNR (D2O) δ: 7.54 (s, 1H), 6.88 (m, 1H), 6.29 (d, 1H), 5.80 (d, 1H), 4.92 (m, 1H), 4.16 (m, 1H), 3.92 (m, 2H), 1.96 (s, 3H), 1.77 (s, 3H), 1.24 (d, 3H). FAB-MS m/z: (M-1) 431.
3′-Deoxy-2′,3′-didehydrothymidine-5′-O-[N-(α-N-acetylphenyloalanyl)] phosphoramidothioate (7b)
Yield 46%. 31P MMR (D2O) δ: 46.64; 47.01. 1H MNR (D2O) δ: 7.50 (s, 1H), 7.27 (m, 5H), 6.84 (m, 1H), 5.85 (d, 1H), 4.93 (m, 1H), 4.64 (m, 1H), 3.95 (m, 2H), 3.08 (m, 1H), 2.91 (m, 1H), 1.81 (s, 3H), 1.79 (s, 3H). FAB-MS m/z: (M-1) 507.
3′-Deoxy-2′,3′-didehydrothymidine-5′-O-(N-prolyl) phosphoramidothioate (7d)
Yield 23%. 31P MMR (D2O) δ: 46.60; 48.80. 1H MNR (D2O) δ: 7.48 (m, 1H), 6.83 (m, 1H), 6.35 (d, 1H), 5.87 (d, 1H), 4.99 (br, 1H), 4.04 (m, 3H), 3.16 (m, 2H), 2.26 (m, 1H), 1.81 (m, 3H), 1.80 (s, 3H). FAB-MS m/z: (M-1) 415.
3′-Azido-3′-deoxythymidine-5′-O-[N-(α-N-acetylalanyl)] phosphoramidate (8a)
Yield 18%. 31P MMR (D2O) δ: −5.18. 1H MNR (D2O) δ: 7.70 (s, 1H), 6.17 (t, 1H), 4.35 (m, 1H), 4.19 (m, 1H), 4.04 (m, 3H), 2.40 (t, 2H), 1.94 (s, 3H), 1.84 (s, 3H), 1.32 (d, 3H). FAB-MS m/z: (M-1) 458.
3′-Azido-3′-deoxythymidine-5′-O-[N-(α-N-acetylphenyloalanyl)] phosphoramidate (8b)
Yield 13%. 31P MMR (D2O) δ: −5.45. 1H MNR (D2O) δ: 7.67 (s, 1H), 7.27 (m, 5H), 6.20 (t, 1H), 4.34 (m, 2H), 4.01 (m, 3H), 3.11 (m, 1H), 2.96 (m, 1H), 2.41 (m, 2H), 1.90 (s, 3H), 1.85 (s, 3H). FAB-MS m/z: (M-1) 534.
3′-Azido-3′-deoxythymidine-5′-O-[N-(α-N-acetylprolyl)] phosphoramidate (8c)
Yield 35%. 31P MMR (D2O) δ: −5.26. 1H MNR (D2O) δ: 7.71 (s, 1H), 6.17 (t, 1H), 4.36 (m, 2H), 4.08 (m, 2H), 3.53 (m, 2H), 2.37 (m, 2H), 2.34 (m, 1H), 2.01 (s, 3H), 1.87 (m, 4H), 1.82 (s, 3H). FAB-MS m/z: (M-1) 484.
3′-Deoxy-2′,3′-didehydrothymidine-5′-O-[N-(α-N-acetylalanyl)] phosphoramidate (9a)
Yield 34%. 31P MMR (D2O) δ: −4.07. 1H MNR (D2O) δ: 7.58 (s, 1H), 6.85 (m, 1H), 6.29 (d, 1H), 5.82 (d, 1H), 4.96 (br, 1H), 4.16 (m, 1H), 3.97 (m, 2H), 1.92 (s, 3H), 1.79 (s, 3H), 1.24 (d, 3H). FAB-MS m/z: (M-1) 415.
3′-Deoxy-2′,3′-didehydrothymidine-5′-O-[N-(α-N-acetylphenyloalanyl)] phosphoramidate (9b)
Yield 15%. 31P MMR (D2O) δ: −5.53. 1H MNR (D2O) δ: 7.53 (s, 1H), 7.22 (m, 5H), 6.87 (br, 1H), 5.85 (d, 1H), 4.97 (br, 1H), 4.61 (m, 1H), 3.95 (m, 2H), 3.04 (m, 1H), 2.89 (m, 1H), 1.85 (s, 3H), 1.79 (s, 3H). FAB-MS m/z: (M-1) 491.
3′-Deoxy-2′,3′-didehydrothymidine-5′-O-(N-prolyl) phosphoramidate (9d)
Yield 19%. 31P MMR (D2O) δ: −5.88. 1H MNR (D2O) δ: 7.52 (s, 1H), 6.86 (d, 1H), 6.36 (d, 1H), 5.89 (d, 1H), 5.00 (m, 1H), 4.03 (m, 1H), 4.00 (m, 2H), 3.22 (m, 2H), 2.28 (m, 1H), 1.89 (m, 3H), 1.81 (s, 3H). FAB-MS m/z: (M-1) 399.
Antiviral and cytotoxic activity assays
The antiviral assays (with the exception of anti-HIV-1 assays) were based on the inhibition of virus-induced cytopathic effect in HEL (herpes simplex virus type-1 [HSV-1], herpes simplex virus type-2 strain G [HSV-2(G)], vaccinia virus and vesicular stomatitis virus), Vero (parainfluenza-3, reovirus-1, Sindbis, Coxsackie virus B4 and Punta Toro virus), Crandell–Rees feline kidney cells (CrFK; feline herpesvirus and feline infectious peritonitis virus [feline coronavirus]) or HeLa (vesicular stomatitis virus, Coxsackie virus B4 and respiratory syncytial virus) cell cultures. Confluent cell cultures in microtitre 96-well plates were inoculated with 100 cell culture inhibitory dose 50% (CCID50) of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures). After a 1 h virus adsorption period, residual virus was removed and the cell cultures were incubated in the presence of varying concentrations (200, 40 and 8 μM) of the test compounds. Viral cytopathic effect was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. The anti-HIV activity and cytotoxicity of the compounds were evaluated against wild-type HIV-1 strain IIIB and HIV type-2 (HIV-2) strain ROD in MT-4 cell cultures using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method [24]. Briefly, virus stocks were titrated in MT-4 cells and expressed as the CCID50. MT-4 cells were suspended in culture medium at 1×105 cells/ml and infected with HIV at a multiplicity of infection of 0.02. Immediately after viral infection, 100 μl of the cell suspension was placed in each well of a flat-bottomed microtitre tray containing various concentrations of the test compounds. The test compounds were dissolved in dimethyl sulfoxide (Sigma–Aldrich) at 50 mM (stock solution). After 4 days of incubation at 37°C, the number of viable cells was determined using the MTT method. Compounds were tested in parallel for cytotoxic effects in uninfected MT-4 cells. For the antiviral activity assays in peripheral blood mononuclear cells, 106 cells/ml were plated in the presence of serial dilutions of the test compounds and were infected with HIV stocks at 1,000 CCID50 per ml (titrated on MT-4 cells). At day 4 post-infection, 125 μl of the supernatant of the infected cultures was removed and replaced by 150 μl of fresh medium containing the test compound at the appropriate concentration. At 7 days after plating the cells, p24 antigen levels were determined in the culture supernatant by the HIV-1 p24 ELISA kit (NEN, Brussels, Belgium).
Results
Chemistry
A series of conjugates of 5′-O-phosphorylated AZT and d4T with amino acids carboxamidates were obtained. The synthesis was performed based on oxathiaphospholane chemistry using N-(2-thiono-1,3,2-oxathiaphospholane) derivatives of amino acids carboxamides (
These compounds, which are key intermediates in the synthesis of nucleoside 5′-O-(N-acyl)phosphoramidates, were obtained in low yield (23–56%) because the DBU-assisted condensation reaction that provided these compounds was accompanied by the production of the by-product diazophospholane-type (compound
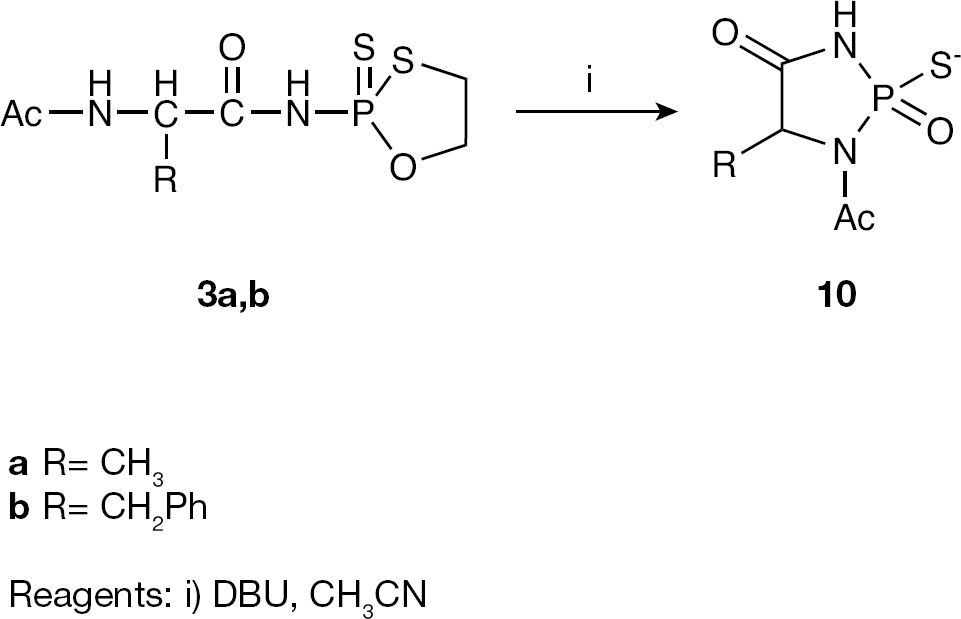
Diazophopholane-type by-product
After isolation by ion-exchange chromatography, compounds
The final products
Virology
Results on the virological assays can be found in Tables 2, 3, 4, 5 and 6.
Cytotoxicity and anti-HIV activity of compounds
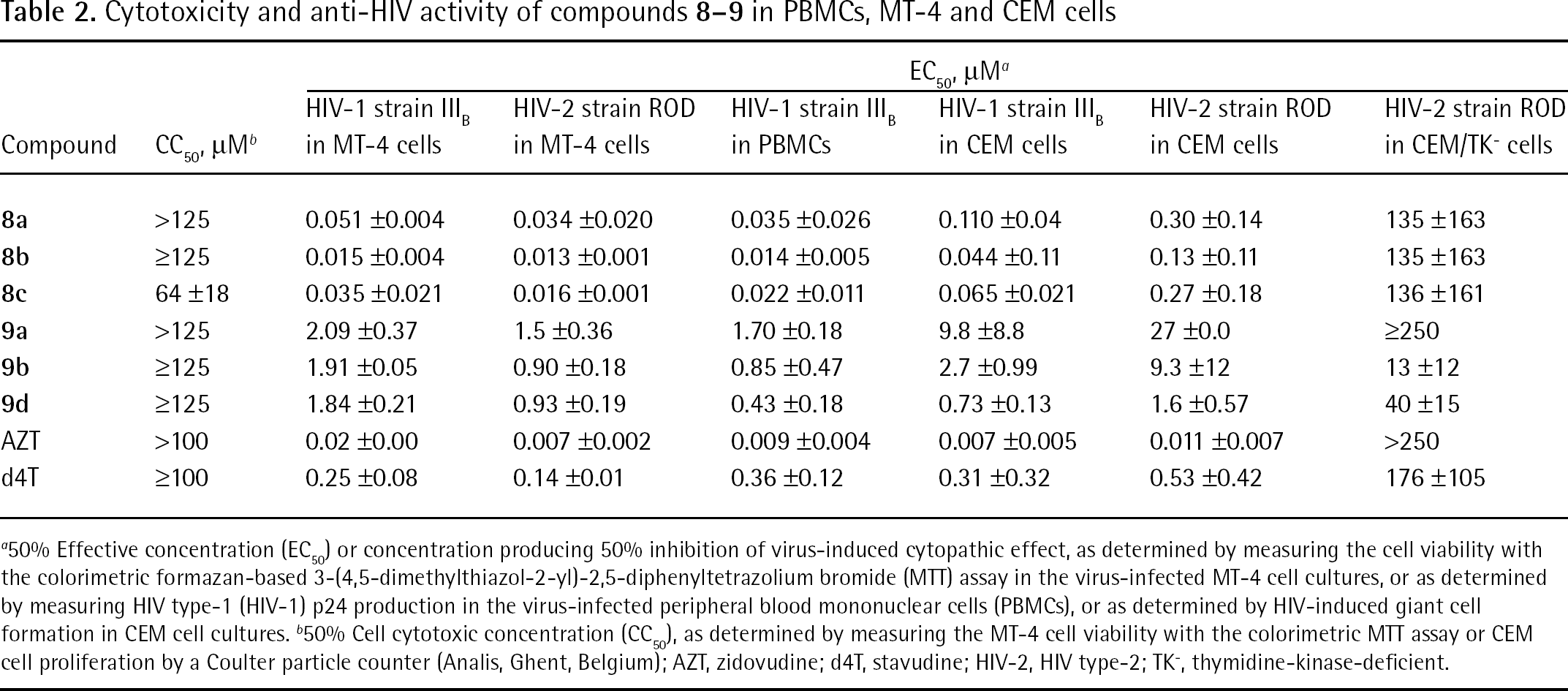
50% Effective concentration (EC50) or concentration producing 50% inhibition of virus-induced cytopathic effect, as determined by measuring the cell viability with the colorimetric formazan-based 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in the virus-infected MT-4 cell cultures, or as determined by measuring HIV type-1 (HIV-1) p24 production in the virus-infected peripheral blood mononuclear cells (PBMCs), or as determined by HIV-induced giant cell formation in CEM cell cultures.
50% Cell cytotoxic concentration (CC50), as determined by measuring the MT-4 cell viability with the colorimetric MTT assay or CEM cell proliferation by a Coulter particle counter (Analis, Ghent, Belgium); AZT, zidovudine; d4T, stavudine; HIV-2, HIV type-2; TK−, thymidine-kinase-deficient.
Cytotoxicity and antiviral activity of compounds
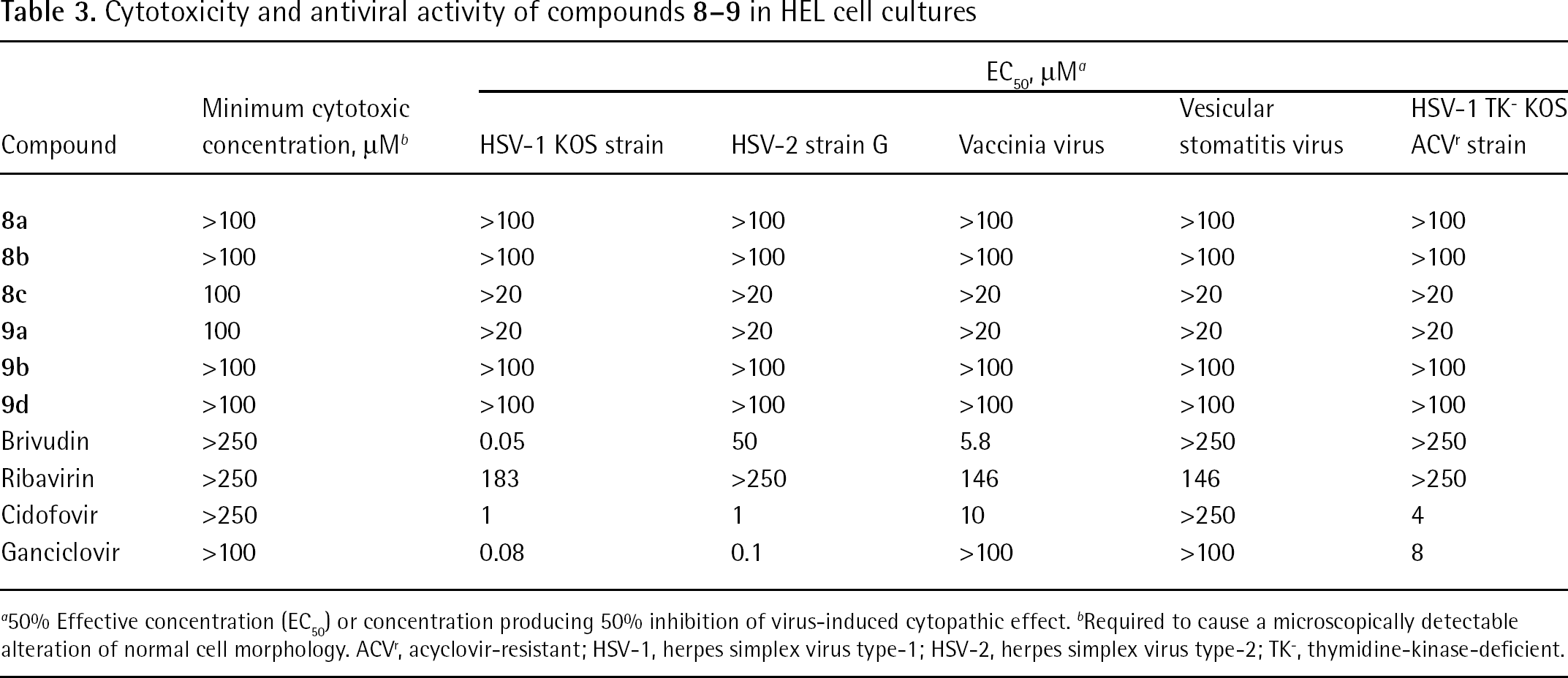
50% Effective concentration (EC50) or concentration producing 50% inhibition of virus-induced cytopathic effect.
Required to cause a microscopically detectable alteration of normal cell morphology. ACVr, acyclovir-resistant; HSV-1, herpes simplex virus type-1; HSV-2, herpes simplex virus type-2; TK−, thymidine-kinase-deficient.
Cytotoxicity and antiviral activity of compounds

50% Effective concentration (EC50) or concentration producing 50% inhibition of virus-induced cytopathic effect.
Required to cause a microscopically detectable alteration of normal cell morphology.
μg/ml. (S)-DHPA, (S)-g-(2,3-dihydroxypropyl)adenine.
Cytotoxicity and antiviral activity of compounds
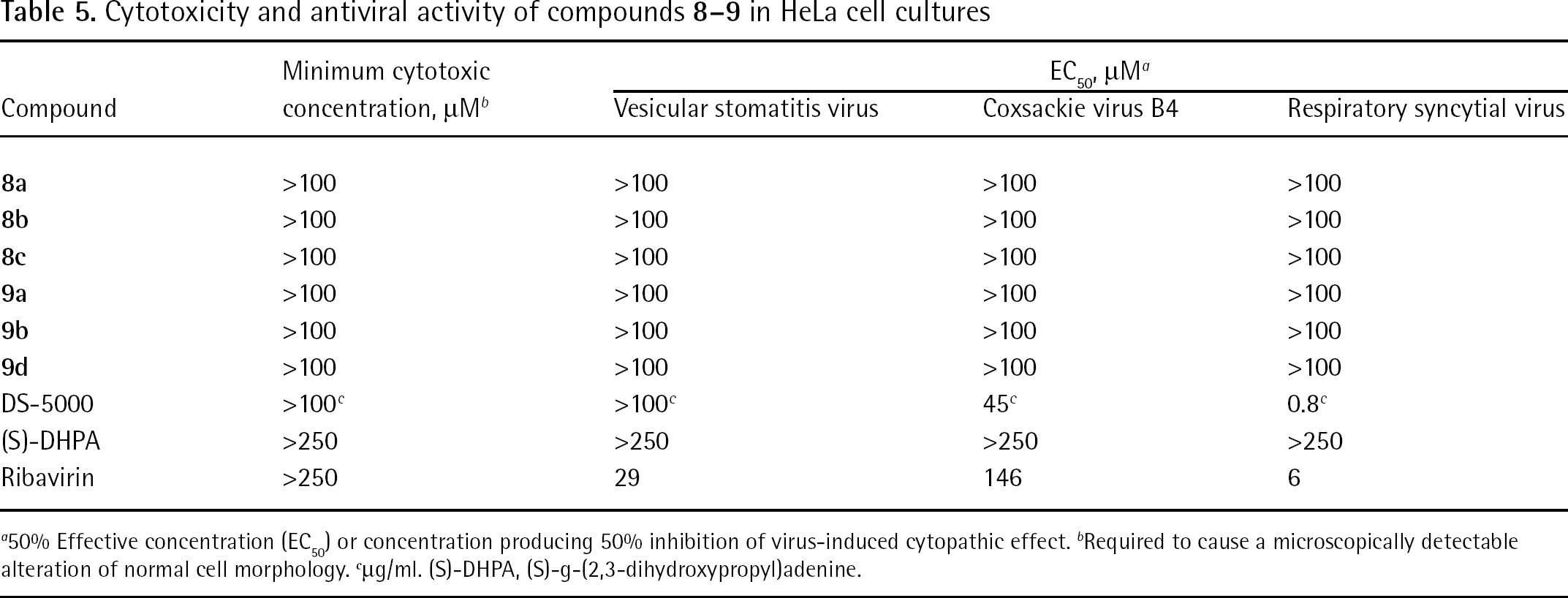
50% Effective concentration (EC50) or concentration producing 50% inhibition of virus-induced cytopathic effect.
Required to cause a microscopically detectable alteration of normal cell morphology.
μg/ml. (S)-DHPA, (S)-g-(2,3-dihydroxypropyl)adenine.
Cytotoxicity and antiviral activity of compounds
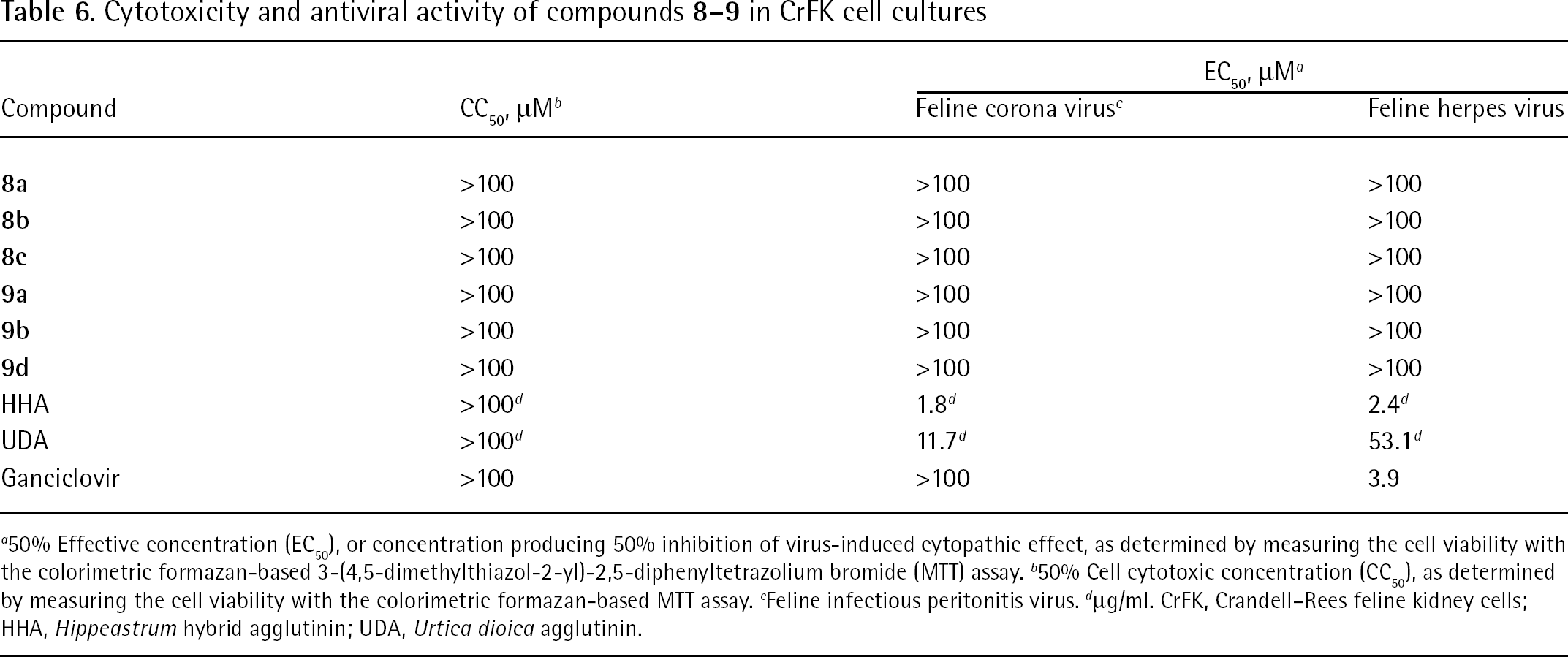
50% Effective concentration (EC50), or concentration producing 50% inhibition of virus-induced cytopathic effect, as determined by measuring the cell viability with the colorimetric formazan-based 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
50% Cell cytotoxic concentration (CC50), as determined by measuring the cell viability with the colorimetric formazan-based MTT assay.
Feline infectious peritonitis virus. d μg/ml. CrFK, Crandell–Rees feline kidney cells; HHA, Hippeastrum hybrid agglutinin; UDA, Urtica dioica agglutinin.
Discussion
Both the d4T and the AZT prodrugs markedly inhibited HIV-1 and HIV-2 replication in MT-4 cell cultures at compound concentrations that were equally active or inferior to the activity of their parental free nucleosides (2- to 20-fold for the AZT prodrugs and 6- to 40-fold for the d4T prodrugs). The pronounced antiviral activity noticed in the MT-4 cell cultures was also confirmed for HIV-1 in PBMCs, in which the EC50 values were even more pronounced than those found in the MT-4 cell cultures (Table 2). The compounds were also evaluated for their anti-HIV activity in CEM and in CEM thymidine-kinase-deficient (CEM/TK−) cell cultures. They substantially lost antiviral potency in the CEM/TK− cells, suggesting an eventual conversion of the test compounds to the free nucleoside prior to further phosphorylation to the active 5′-triphosphate metabolite.
Unfortunately none of the tested compounds showed antiviral activity against a broad range of DNA and RNA viruses at subtoxic concentrations, except for HIV-1 and HIV-2 (Tables 2, 3, 4, 5 and 6).
It would now be interesting to investigate whether the d4T and AZT prodrugs have more favourable pharmacokinetics compared with the parent drugs in vivo. Also, it would be worth synthesizing similar types of nucleotide prodrugs containing other bases, such as cytosine or (substituted) purines.
Footnotes
Acknowledgements
These studies were supported financially by the Polish Ministry of Science and Higher Education (projects N 204 075 32/2063 and PBZ-MNiSW-07/I/2007) and Katholieke Universiteit Leuven (GOA 10/14). We thank Leentje Persoons, Frieda De Meyer, Leen Ingels, Kristien Erven and Kris Uyttersprot for excellent technical assistance with the antiviral assays.
The authors declare no competing interests.