
Abstract
Background:
HIV-1 integrase is a clinically validated therapeutic target for the treatment of HIV-1 infection, with one approved therapeutic currently on the market. This enzyme represents an attractive target for the development of new inhibitors to HIV-1 that are effective against the current resistance mutations.
Methods:
A fragment-based screening method employing surface plasmon resonance and NMR was initially used to detect interactions between integrase and fragments. The binding sites of the fragments were elucidated by crystallography and the structural information used to design and synthesize improved ligands.
Results:
The location of binding of fragments to the catalytic core of integrase was found to be in a previously undescribed binding site, adjacent to the mobile loop. Enzyme assays confirmed that formation of enzyme–fragment complexes inhibits the catalytic activity of integrase and the structural data was utilized to further develop these fragments into more potent novel enzyme inhibitors.
Conclusions:
We have defined a new site in integrase as a valid region for the structure-based design of allosteric integrase inhibitors. Using a structure-based design process we have improved the activity of the initial fragments 45-fold.
Introduction
Integration of viral DNA into the host cell genome is important for viral replication and is achieved by the two steps of 3′-processing (3′P) that involve excision of 3′-GT bases and strand transfer (ST), both carried out by HIV-1 integrase. These reactions occur in a complex that has been postulated to resemble a Holliday junction complex [1] containing integrase and components of the pre-integration complex, including the lens epithelium-derived growth factor (LEDGF) protein (which acts to anchor the complex to chromatin), the viral DNA termini and the host DNA. Enzymatic activity of integrase requires two magnesium ions to be coordinated by a DDE motif (D64, D116, E152) in the active site of integrase [2], with the position of E152 modulated by the ‘mobile loop’ (residues G140 to G149), suggesting that the loop plays a role in positioning the metals. Furthermore, mutagenesis studies have demonstrated that the mobility of this loop is important for catalytic activity [3] and different conformations of the loop and active site have been observed in model systems with and without integrase inhibitors [4].
Raltegravir, launched in 2007, was the first inhibitor to validate integrase as a therapeutic target [5]. The metal binding pyrimidinone scaffold of raltegravir was initially shown to bind to the magnesium ions present in hepatitis C NS5b polymerase, and subsequently optimized for binding to the magnesium ions bound to the catalytic triad DDE of integrase [6]. Both in vitro studies and clinical use of raltegravir have given rise to mutations in the integrase protein [7], in particular the residues Q148R/H, G140S/N/H/E/Q and Y143R, all of which are within the mobile loop. Recent structures of the integrase protein from the foamy virus (PFV) [4], including one with the protein complexed with two magnesium ions, viral DNA and raltegravir (PDB code 3L2T) show an interaction between Y143 (Y212 in PFV) and the inhibitor, which is significant because Y143R is a primary resistance mutation seen in response to raltegravir [8,9]. The Pommier group carried out a convincing study [10] of variants of the 3′-CA overhang which implicated a direct interaction of the Q148 residue with the cytosine base during the catalytic process. Detailed kinetic studies by Langley et al. [11] led to a model in which the adenine opens or closes the active site for DNA processing. Mutation of Q148 to R or H would interfere with these actions and has recently been shown to interfere with both 3′P and ST reactions [12]. NMR studies of the loop have indicated stable clusters of conformations even in the absence of the DNA ligand, suggesting functionally favoured orientations [13]. Modelling studies [14] of the mobile loop suggest that the G140S/Q148H double mutant blocks raltegravir binding to the active site directly by reducing the number of possible docking orientations. Taken together, these observations indicate a role for structural re-arrangement of the integrase complex and more specifically the requirement for mobility of mobile loop residues, including Q148. The requirement for mobility of this loop and the numerous reported crystal structures represents a potential target for the design of allosteric inhibitors. Structure-based design of integrase inhibitors has been complicated by the lack of crystal structures of small ligands bound to the active site of the isolated protein or the complex. The only known active site complex of HIV-1 integrase with a ligand is of 1-(5-chloroindol-3-yl)-3-hydroxy-3-(2H tetrazol-5-yl)-propenone [15] and this is of limited descriptive power because the ligand's orientation is influenced by a symmetry-related protomer in the crystal. For the avian sarcoma virus (ASV), allosteric inhibitors of its integrase protein, compounds
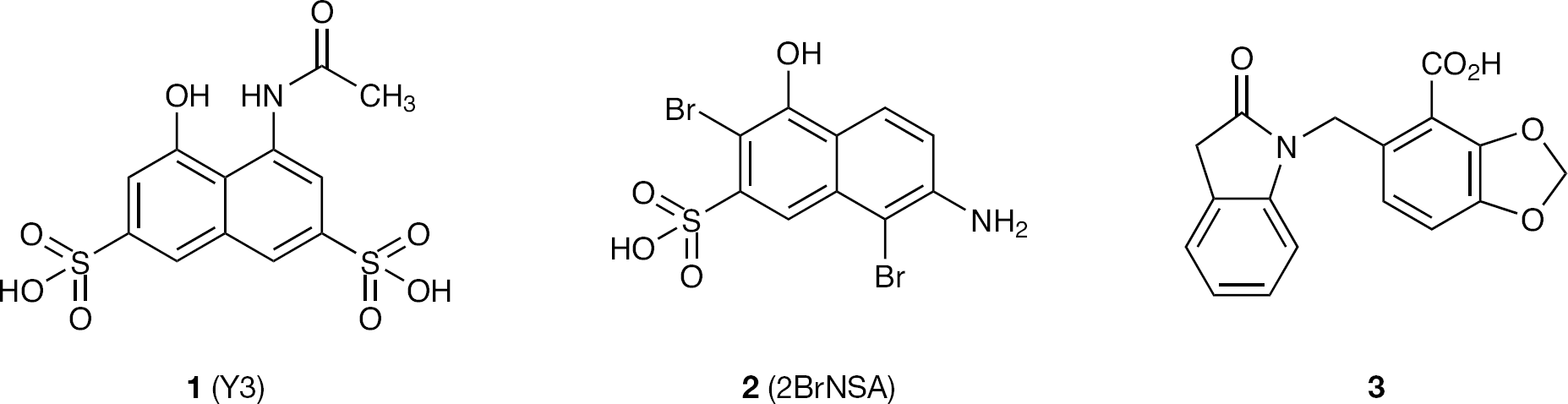
Structures of compounds
A fragment-based screen of HIV-1 integrase by surface plasmon resonance (SPR) and saturation transfer difference NMR identified a number of small molecule ligands that bind to one or more sites on the integrase catalytic core domain (CCD; DIR, unpublished observations). By crystallography we determined some of these to be at a site adjacent to the mobile loop. We used these as a basis for structure-based design of novel allosteric inhibitors of HIV-1 integrase with the potential to overcome resistance to raltegravir binding.
Methods
Protein preparation
N-terminally hexa-His tagged CCD integrase (residues 50 to 210) containing the mutations C56S, F139D and F185H (core3H) was cloned into the E. coli expression vector pET28b(+) (Novagen, Madison, WI, USA) and expressed and purified essentially as described for core4H in Wielens et al. [17], however the current studies retained the His tag.
3′-processing strand transfer combined assay An assay procedure similar to that already published [18,19] was used to determine the inhibitory effect of the compounds. The assay was adapted to a 96-well plate format. Briefly, 400 ng integrase was incubated with 30 nM substrate DNA, consisting of annealed U5 LTR sequence oligonucleotides containing a digoxigenin (DIG) tag at the 3′-end (5′-ACTGCTAGAG ATTTTCCACACTGACTAAAAGGGTC-DIG-3′) or biotin (Bio; 5′-Bio-GACCCTTTTAGTCAGTGTGGA AAATCTCTAGCAGT-3′) so that each substrate has either a DIG or Bio tag on opposite strands. Reactions were carried out for 2 h at 37°C and products generated as a result of 3′P and ST activity were bound to streptavidin plates and, following denaturation, were detected using an anti-DIG-alkaline phosphatase conjugate (Roche Diagnostics, Mannheim, Germany) and p-nitro phenyl phosphate substrate (Sigma–Aldrich, St Louis, MO, USA). In this assay system raltegravir (IsentressTM) returns a 50% inhibitory concentration (IC50) of 0.13 μM (n=8) which compares to the Merck ST only assay system giving a published value of IC50 15 nM [5]. The second control compound, the diketo acid (L731,988) [20] gives an IC50 of 0.18 μM.
Chemistry
In an SPR, NMR and crystallography screening programme the N-benzyl indolinone
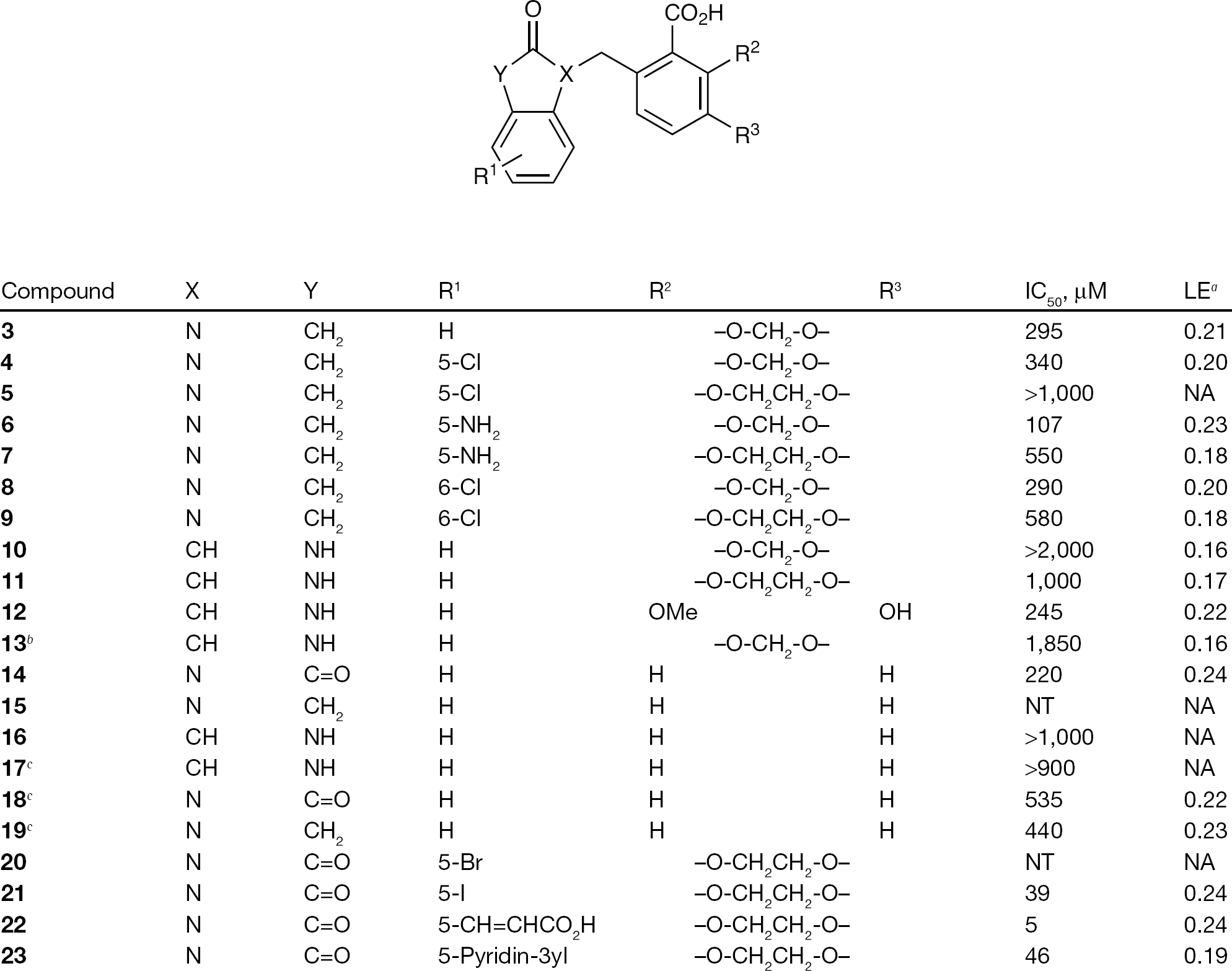
Compounds
General procedure for N-benzyl isatin
Indoline-2,3-dione (isatin), and its ring-substituted analogues, could be conveniently alkylated with the appropriate benzyl bromide, using piperidine as a base, in 60–70% yield. The resulting ester
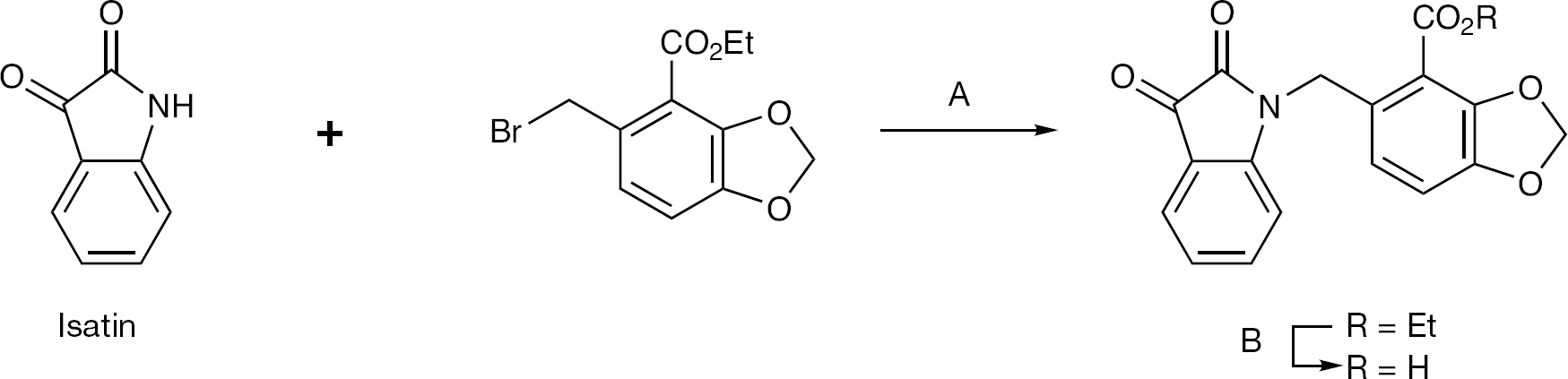
Alkylation of isatin core
General procedure for N-benzyl indolin-2-ones
An attempt of direct N-alkylation of indolin-2-one via deprotonation with lithium diisopropyl amide or sodium hydride failed, giving instead dialkylation of the 3-methylene group of the indoline. We therefore used an alternative route via the isatin derivatives previously prepared. Wolff–Kishner reduction with hydrazine produced the required N-benzyl indolin-2-ones, usually in good yield. In some cases, most notably compound
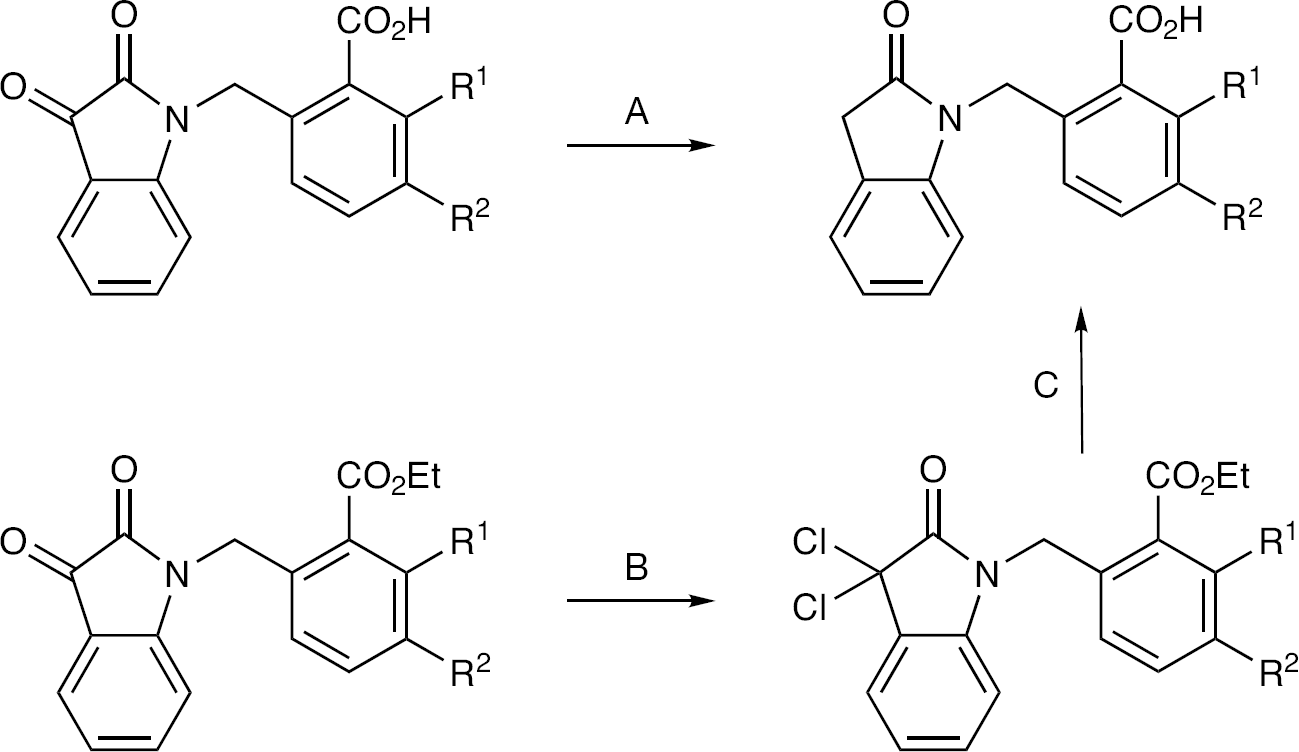
General routes to N-benzyl indolin-2-ones
General procedure for 3-benzyl indolin-2-ones
Compounds
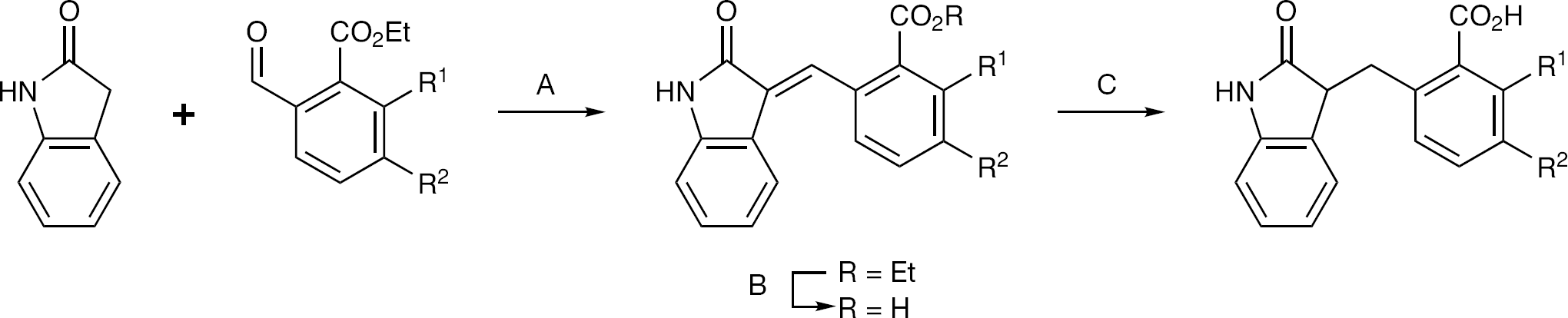
Synthesis of C-alkylated 3-benzyl indolin-2-ones
Ring-substituted isatins
To introduce acidic and basic functionality into the isatin ring we investigated metal-mediated coupling with 5-halosubstituted analogues. A palladium catalyzed Heck reaction of
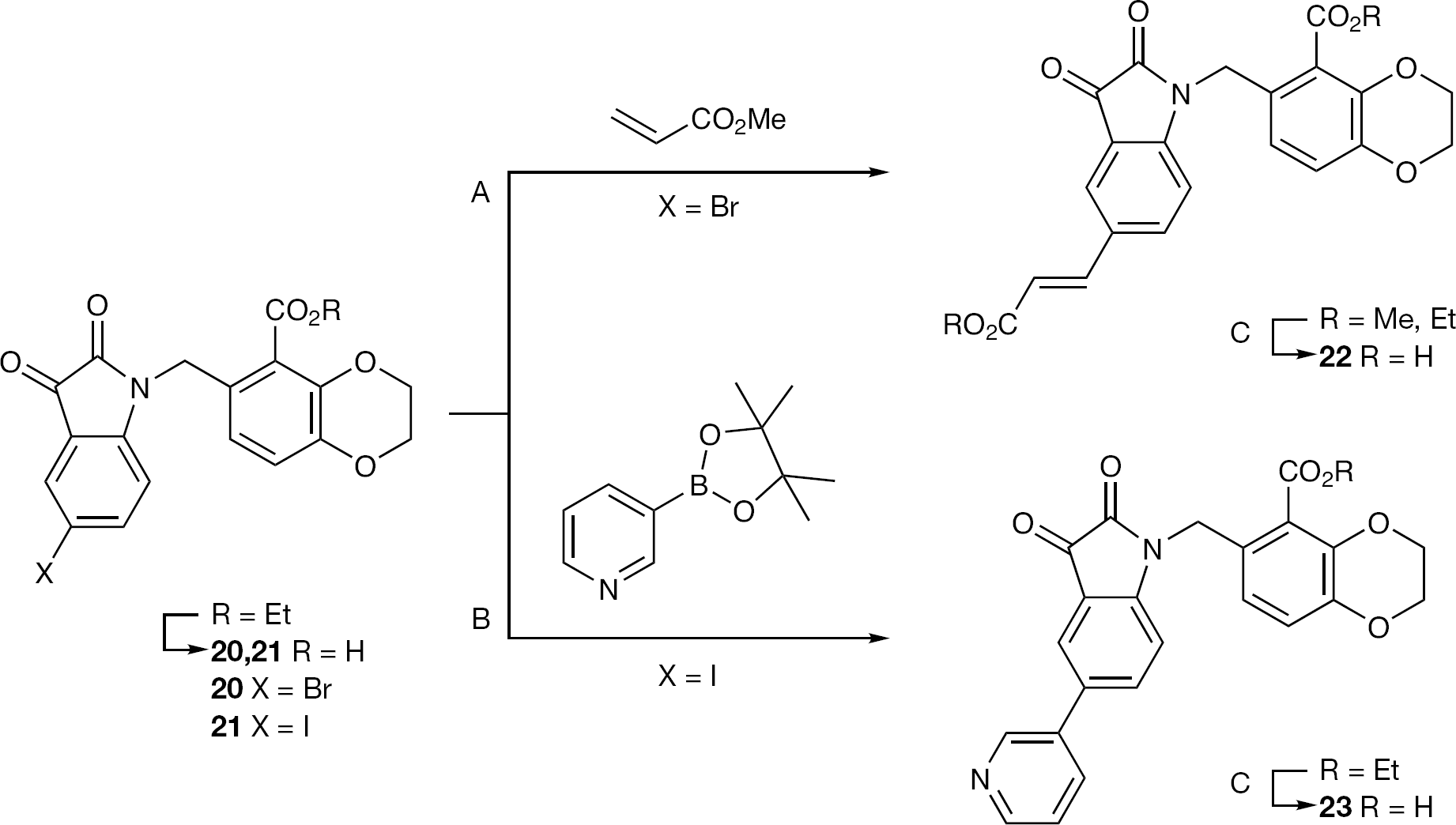
Synthesis of compounds
Analytical HPLC was performed on a Waters Alliance 2690 system (Waters, Milford, MA, USA) with 996 PDA and Phenomenex PFP Luna PFP 2×150 mm 5 μ, 100 Å column (Phenomenex, Torrance, CA, USA), LCMS (compounds
5-((2-Oxindolin-1-yl)methyl)benzo[d] [1,3] dioxole-4-carboxylic acid 3
To a stirred mixture of isatin (52 mg, 0.35 mmol), K2CO3 (73 mg, 0.53 mmol) and KI (10 mg) in DMF (4 ml) was added ethyl 5-(bromomethyl)benzo[d] [1,3] dioxole-4-carboxylate [22] (110 mg, 0.34 mmol). The mixture was heated to 80°C for 1 h, after which H2 O (40 ml) was added and the mixture extracted with EtOAc (2×50 ml). The combined extract was washed with saturated brine and dried (Na2SO4), filtered then concentrated under reduced pressure to give the crude product, ethyl 5-((2,3-dioxoindolin-1-yl)methyl) benzo[d] [1,3] dioxole-4-carboxylate, which was purified by flash chromatography (eluant: EtOAc/Pet. ether:1/2, v/v, silica gel, Rf 0.40) to give the intermediate in 82% yield. 1H-NMR: 1.45 (t, 3H, J=7.2 Hz), 4.60 (q, 2H, J=7.2 Hz), 5.22 (s, 2H), 6.10 (s, 2H), 6.72 (d, 1H, J=8.0 Hz), 6.80–6.84 (m, 2H), 7.13 (t, 1H, J=8.0 H), 7.50 (td, 1H, J=8.0, 1.2 Hz), 7.65 (dd, 1H, J=8.0, 1.2 Hz).
To a stirred mixture of ethyl 5-((2,3-dioxindolin-1-yl)methyl)benzo[d] [1,3] dioxole-4-carboxylate (50 mg, 0.14 mmol) in benzene (1 ml) was added PCl5 (67 mg, 0.32 mmol). The mixture was stirred for 24 h at room temperature, then water (10 ml) was added and the mixture was extracted with EtOAc (2×10 ml). The combined organic extract was washed with saturated brine, dried (Na2SO4), filtered and concentrated under reduced pressure to give a solid, which was purified by preparative TLC to give ethyl 5-((3,3-dichloro-2-oxindolin-1-yl)methyl)benzo[d] [1,3] dioxole-4-carboxylate (26 mg, yield 45%) as a light yellow solid (petroleum ether/EtOAc:4/1, v/v, Rf=0.40). To a stirred mixture of ethyl 5-((3,3-dichloro-2-oxindolin-1-yl)methyl)benzo[d] [1,3] dioxole-4-carboxylate (20 mg, 0.05 mmol) in AcOH (1 ml) was added zinc powder (32 mg, 0.5 mmol) The mixture was stirred for a further 5 min at room temperature after which TLC analysis indicated the starting material had been consumed. The zinc powder was filtered off and the filter cake was washed with EtOAc. Water (10 ml) was added into the filtrate and the mixture was extracted with EtOAc (2×10 ml). The organic layers were combined, washed with saturated NaHCO3, dried (Na2SO4) and concentrated under reduced pressure to give ethyl 5-((2-oxindolin-1-yl)methyl)benzo[d] [1,3] dioxole-4-carboxylate (17 mg, yield 100%) as a light yellow solid (petroleum ether/EtOAc: 6/1, v/v, Rf=0.20). The ester (4.0 mg, 0.01 mmol) was taken up in AcOH (0.5 ml) and HCl (6 M, 0.5 ml) and the mixture was heated to reflux. After 5 h the reaction mixture was neutralized with 1 M NaOH and extracted with EtOAc (2×10 ml). The combined organic extract was washed with saturated brine, dried (Na2SO4) and concentrated under reduced pressure to give 5-((2-oxoindolin-1-yl) methyl)benzo[d] [1,3] dioxole-4-carboxylic acid
3-Hydroxy-2-methoxy-6-((2-oxindolin-3-yl)methyl) benzoic acid 12
To a mixture of ethyl 6-formyl-2-methoxy-3-(methoxymethoxy)benzoate (450 mg, 1.68 mmol) and indolin-2-one (436 mg, 3.28 mmol) in EtOH (10 ml) was added piperidine (2 drops) under an atmosphere of N2. The mixture was heated to reflux overnight and then allowed to cool forming a precipitate. The condensation product, ethyl 2-methoxy-3-(methoxymethoxy)-6-((2-oxindolin-3-ylidene) methyl)benzoate, was obtained (517 mg, 45.3%) as a yellow solid by recrystallization from EtOH. A sample of this material (130 mg, 0.34 mmol) in MeOH (3 ml) was treated with 2 M NaOH (0.42 ml, 0.84 mmol) at 40°C for 2 days and then the mixture was concentrated. The residue was dissolved in EtOAc (5 ml) and washed by water (5 ml). The aqueous phase was separated and adjusted to pH 4–5, and then extracted with EtOAc (2×5 ml). The combined organic extracts were washed with brine, dried (Na2SO4), filtered and concentrated to give 3-hydroxy-2-methoxy-6-((2-oxindolin-3-ylidene)methyl)benzoic acid (76 mg, 63.3%, crude) as a yellow solid (TLC: DCM/MeOH:10:1, v/v, Rf=0.2). To the acid (70 mg, 0.22 mmol) was added Pd/C (10 mg) in MeOH and the reaction mixture was stirred at room temperature under H2 overnight. Pd/C was removed by filtration (celite) and the filtrate was concentrated to give the crude product. It was purified by preparative TLC to obtain title compound 3-hydroxy-2-methoxy-6-((2-oxindolin3-yl)methyl)benzoic acid
(E)-6-((5-(2-Carboxyvinyl)-2,3-dioxoindolin-1-yl)methyl)-2,3-dihydrobenzo[b] [1,4]dioxin-5-carboxylic acid 22
Ethyl 6-((5-bromo-2,3-dioxoindolin-1-yl)methyl)-2,3-dihydrobenzo[b] [1,4] dioxin-5-carboxylate was prepared using the method described in step one for compound
6-((2,3-Dioxo-5-(pyridin-3-yl)indolin-1-yl)methyl)-2,3-dihydrobenzo[b] [1,4] dioxine-5-carboxylic acid 23
Ethyl 6-((5-iodo-2,3-dioxoindolin-1-yl)methyl)-2,3-dihydrobenzo[b] [1,4] dioxine-5-carboxylate was prepared using the method described in step one for compound
Crystallography
For crystallization, the protein sample was concentrated to 5.5 mg/ml and exchanged into 40 mM Tris-Cl pH 8.0, 250 mM sodium chloride, 30 mM magnesium chloride and 5 mM DTT. Crystals were generated at the Bio21 Collaborative Crystallization Centre (C3; Parkville, Australia), with initial screening focused around previously published conditions. All drops were set up in SD-2 (IDEX Corp, Lake Forest, CA, USA) sitting drop plates using a Phoenix robot (Art Robbins Industries, Sunnyvale, CA, USA) with 50 μl of crystallant in the reservoir and droplets consisting of 200 nl of the reservoir and 200 nl of the protein sample. For large-scale production, the final conditions were: 100 mM sodium acetate at pH 5.0–5.5 and 1.6–2.0 M ammonium sulfate. A cryosolution was made up of 100 mM sodium acetate pH 5.5, 1.75 M ammonium sulfate, 25% ethylene glycol and 5% compound in neat DMSO. Twenty-four h prior to data collection 1.5 μl of the cryosolution was added to the crystal containing droplet and the crystallization plate was resealed. When compounds as dry powders were used for soaking, the cryosolution was the same as above but without compound, and the compound was ‘sprinkled’ on top of the drop after the addition of the cryosolution. For data collection, the crystal was gently looped out using MicroLoops (MiTeGen, Ithica, NY, USA) and cryo-cooled in the nitrogen stream. All data collection was done at 100 K. Typically 181 frames of data were collected with an oscillation angle of 1 degree. All data were indexed with Mosflm [23] scaled with SCALA (CCP4) [24] and molecular replacement was done using Phaser (CCP4) [24]. Manual rebuilding was done with the molecular graphics programme Coot [25] and the compounds were initially fitted into the density using the programme Afitt [26]. Subsequent refinement was done using Refmac [27]. All figures were prepared using Afitt [26], except Figure 7, which was made using the programme PyMol (Schrödinger, New York, NY, USA). Refined and validated structure data are reported in Table 1.
Structure refinement and model validation
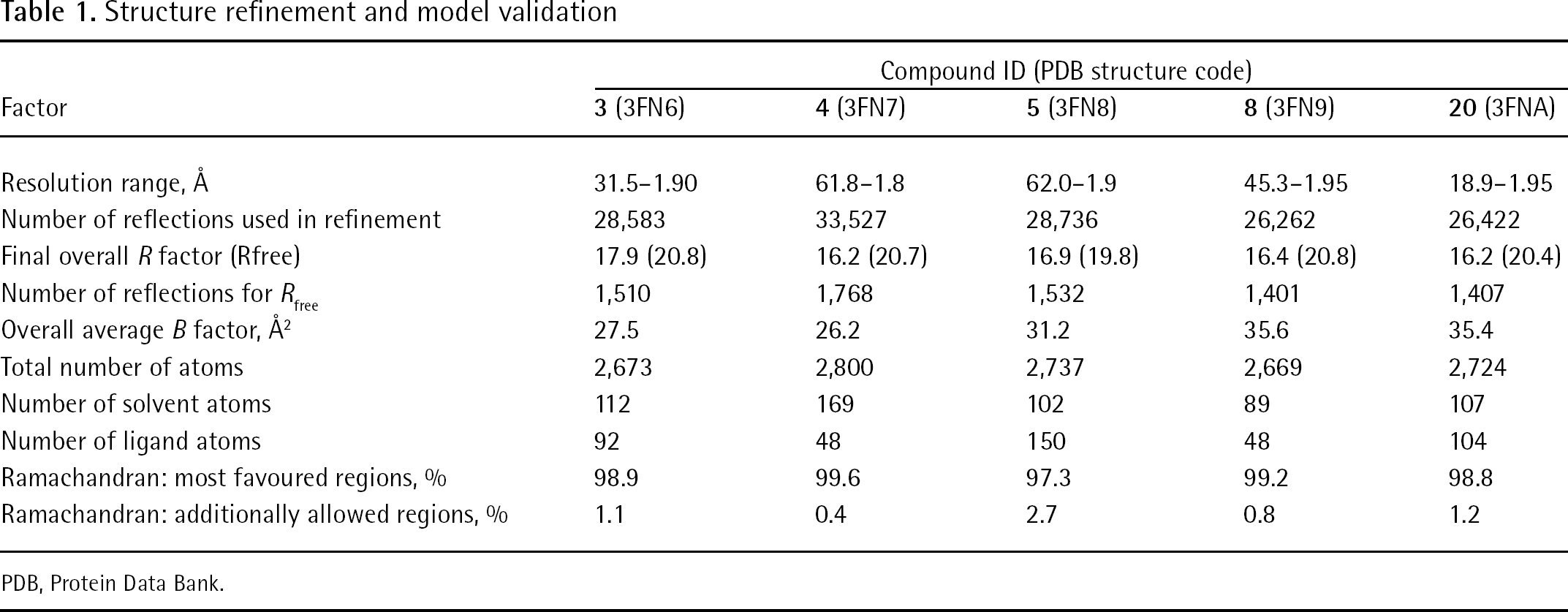
PDB, Protein Data Bank.
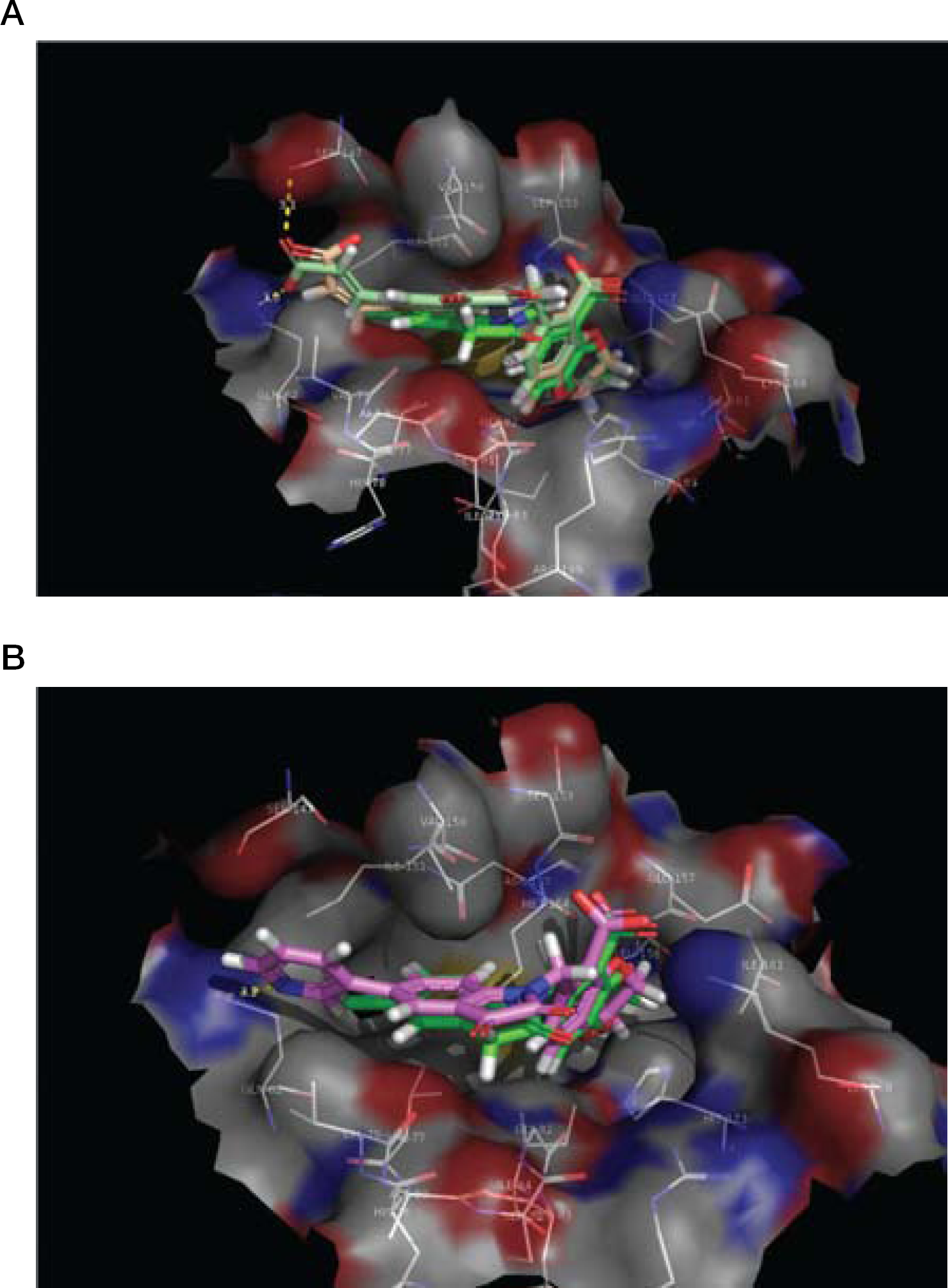
Modelling of compunds
Structures of the compounds in complex with integrase have been deposited at the Protein Data Bank (PDB) with codes 3NF6, 3NF7, 3NF8, 3NF9 and 3NFA for compounds
Modelling studies
Compounds
Following the initial docking, 60 ns of molecular dynamics was performed for both stereo isomers of compound
Results
Several constructs of the integrase CCD were made and tested for their ability to yield diffraction quality crystals. These constructs were made with two goals in mind, to solubilize the protein and to keep its activity intact. Two constructs, core3H and core4H (core3H containing C56S, F139D and F185H mutations [15] and core4H containing a W131D mutation in addition to these three), both containing a hexa-his tag at the N-terminus, were found to crystallize. During optimization of the crystallization process for integrase CCD, we observed that the core4H crystallized in a space group (P212121) where the active site is occluded by symmetry-related molecules. However, the core3H construct crystallized in a different space group (P31), with an accessible active site and relatively ordered mobile loop.
A complex of compound
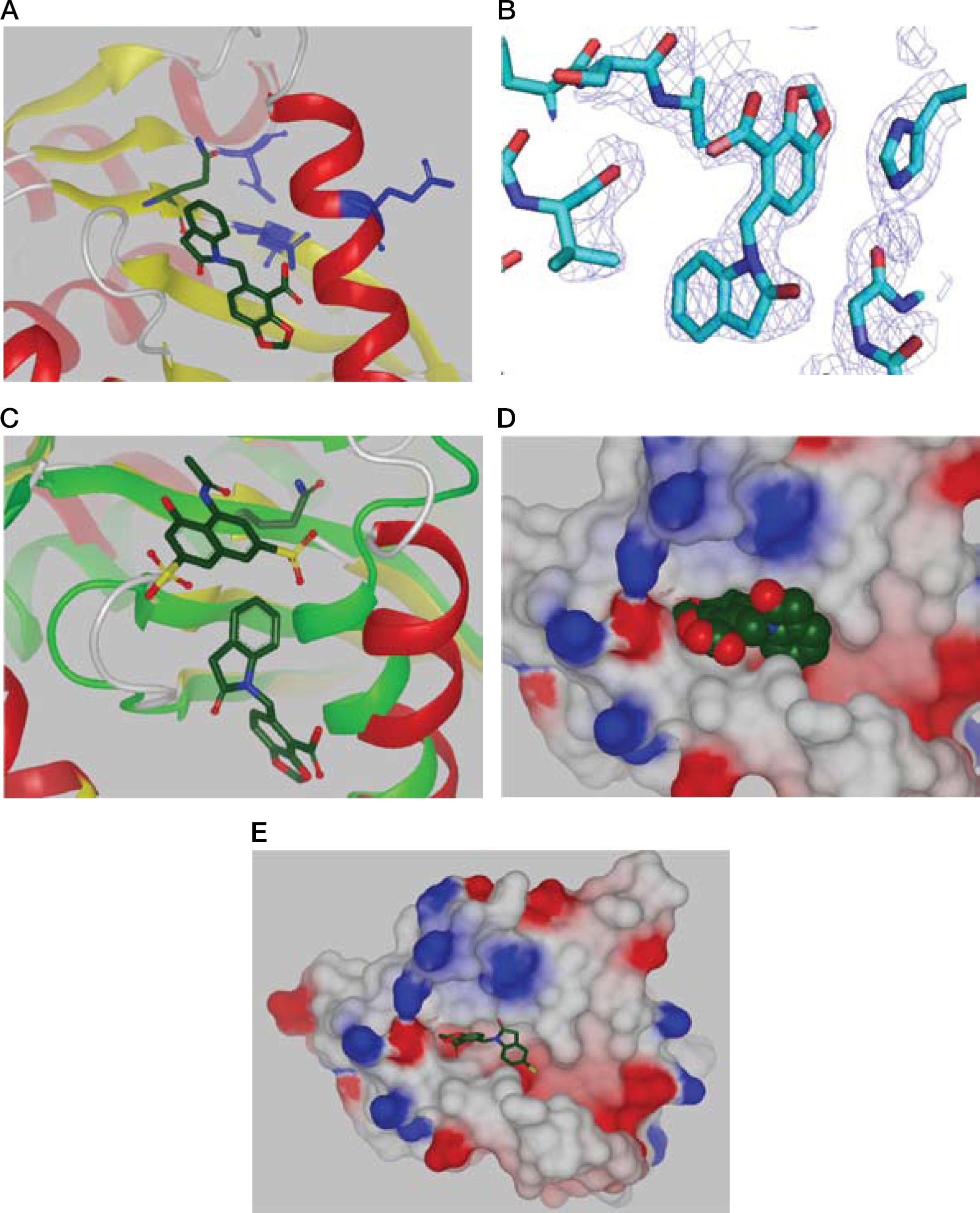
HIV-1 integrase in complex with compound
Structural information for full length HIV-1 integrase is not available and we have not found the full length protein to be amenable to the SPR and NMR studies conducted here with the CCD constructs. Therefore, in order to determine whether this newly identified pocket is likely to be present in full length integrases we overlayed the structures of the protein and ligands generated in this study with the only full length structure available, that of the PFV integrases [4]. As anticipated, a high degree of structural homology was observed together with the existence of the pocket in the full length protein with space to accommodate the binding of ligands (TSP, data not shown). The observation of high structural homology and the data from the 3′ST assays showing ligands that bind to this pocket in the CCD inhibit the activity of full length enzyme is consistent with the pocket being present in the full length HIV-1 integrase.
A molecule, compound
As expected, crystallographic studies showed that analogues
Examination of the complexes of compounds
In an attempt to target the Q62 residue for H-bonding interactions, and an anticipated improvement in compound IC50, a further series of analogues were prepared with either acidic (
Discussion
We initiated this study by firstly successfully identifying a construct, hexa-his tagged core3H, and crystallization conditions that enabled the generation of crystals with a defined loop and accessible active site suitable for use in identifying fragments binding to pockets in the HIV-1 integrase CCD. These crystals were then utilized with fragment hits from an SPR and NMR screening campaign to identify a new site in the CCD of HIV-1 integrase, where ligands act as allosteric inhibitors of the catalytic activity of the enzyme.
Crystallographic analysis and modelling of a series of compounds identified in this study has in more detail elucidated a pocket identified previously in ASV integrase and further extends this with a basis for inhibitor design, capitalizing on interactions with H183, Q62 and S147. We initially identified an isatin analogue bound in this pocket which is a member of a class of molecules with previously described biological activities such as antitumor and antiviral activities [49]. By using structure-based design we developed a compound,
Footnotes
Acknowledgements
The work was supported by a commercial ready grant COMO4229 from the Commonwealth of Australia, Department of Innovation, Industry, Science and Research to Avexa Ltd. We thank the Australian Synchrotron and the beamline scientists at MX1 and MX2 for their help in data collection. We also thank the Bio21 Collaborative Crystallisation Centre (Parkville, Australia) for producing the protein crystals used in this study. In addition, we thank Roger Mulder and Jo Cosgriff for being more than accommodating in the timely collection and workup of the 13C NMR spectra and HRMS data. Compounds
The authors declare no competing interests.