
Abstract
HCV infection is a significant human disease, leading to liver cirrhosis and cancer, and killing >10,000 people in the US annually. Translation of the viral RNA genome is initiated by ribosomal binding to a highly structured RNA element, the internal ribosomal entry site (IRES), which presents a novel target for therapeutic intervention. We will first discuss studies of oligonucleotide therapeutics targeting various regions of the 340-nucleotide IRES, many of which have effectively blocked IRES function in vitro and are active against virus replication in cell culture. Although low nanomolar potencies have been obtained for DNA- and RNA-based molecules, stability and drug delivery challenges remain to be addressed for these particular HCV compounds. Several classes of small molecule inhibitors have been identified from screening protocols or designed from established RNA therapeutic scaffolds. In particular, small molecule IRES inhibitors based on a benzimidazole scaffold bind specifically to the IRES, and inhibit viral replication in cell culture at micromolar concentrations with low toxicity. The structure of the RNA target in complex with a representative member of these small molecule inhibitors demonstrates that a large RNA conformational change occurs upon inhibitor binding. The RNA complex shows how the inhibitor alters the global RNA structure and provides a framework for structure-based drug design of novel HCV therapeutics.
Introduction
HCV infection is a major public health problem, with as many as 170 million people infected worldwide and 2–3 million new cases per year. In western countries, HCV infection causes the majority of cases of liver cancer and is the major cause of liver transplantations. In the US, approximately 1.6%, or 4 million people, are HCV-infected, resulting in the death of approximately 10,000–12,000 individuals each year [1–3]. In the developing world, the disease is readily transmitted where controls over the blood supply are still lax and as a result of infection spread among intraveneous drug users. Currently, the most effective treatment combining ribavirin with pegylated interferon-α is only effective in approximately 50% of patients; furthermore, therapy is expensive and has frequent and troublesome side effects [4,5]. In the developing world, hepatitis C is essentially untreated and, because of its transmission through contaminated blood, patients are frequently infected with both HCV and HIV [6]. There is a clear need for new, effective therapeutics to treat HCV infection.
HCV is a plus-strand single-stranded RNA Hepacivirus in the Flaviviridae family of which yellow fever virus and dengue virus are related viral pathogens [7]. The HCV viral life cycle presents a number of potential targets for therapeutic development. The RNA-dependent RNA polymerase (NS5B) is the target of several nucleoside inhibitors, and also a group of non-nucleoside allosteric inhibitors [1,8]. Compounds in these classes of polymerase inhibitors are analogous to effective treatments for HIV. Likewise, HCV encodes a protease (NS3-4A), which processes the initial polyprotein into functional enzymes and structural components. Inhibition of the protease blocks replication [9], and this activity has been shown to be effective in human patients [10]. In 2010, both Merck & Co., Inc. and Vertex Pharmaceuticals announced positive Phase III results for their boceprevir and telaprevir protease inhibitors. In addition to targeting the enzymes of HCV, other strategies included targeting the genome with nucleic acid drugs using either antisense oligonucleotides, or small interfering RNAs [8]. The limited coding capacity of viral genomes requires that cellular factors are often recruited to perform key roles in viral replication. An intriguing development in HCV biology is the demonstration that high levels of the miR-122 microRNA in liver are necessary for efficient viral replication; there are two binding sites for miR-122 in the HCV 5′-untranslated region (UTR) [11,12]. A report of therapeutic efficacy in chronically infected chimpanzees for a specific miR-122-targeted antagomir oligonucleotide represents a novel therapeutic approach that would be complementary to existing treatment [13]. The common theme of these diverse approaches is to target a characteristic of HCV biology that is unique to the virus, and thus can be selectively inhibited without toxicity.
HCV translation initiation – a novel process
HCV replicates by first using its single-stranded RNA genome as the messenger RNA for production of the viral encoded proteins, including the NS5B polymerase and the NS3-4A protease. The 5′-UTR of the HCV RNA contains a large structured domain that serves as an internal ribosomal entry site (IRES). The IRES of HCV is one of the best characterized among IRES elements from many different viruses and of IRES elements found in cellular genes [14]. Upon HCV infection, the normal cap-dependent translation process is strongly inhibited, resulting in the infected cell devoting most of the protein synthesis capacity toward making viral proteins. The HCV IRES recruits the 40S ribosomal subunit directly without the requirement for several translation initiation factors (Figure 1). Zhao and Wimmer [15] performed an early analysis of 124 HCV sequences and found that 83 folded into the consensus secondary structure shown schematically in Figure 1. Most of the mutations they found were conservative Watson–Crick swaps that would maintain the secondary structure in stem regions. The updated HCV sequence databases confirm that the 5′- and 3′-UTR are the most conserved sequence regions in the entire genome, with essentially no significant sequence variation observed for the IRES region [16]. Because IRES-mediated translation is required for HCV replication [17,18], and because this is distinct from how most cellular proteins are made, the conserved HCV IRES has been recognized as an attractive therapeutic target [19].
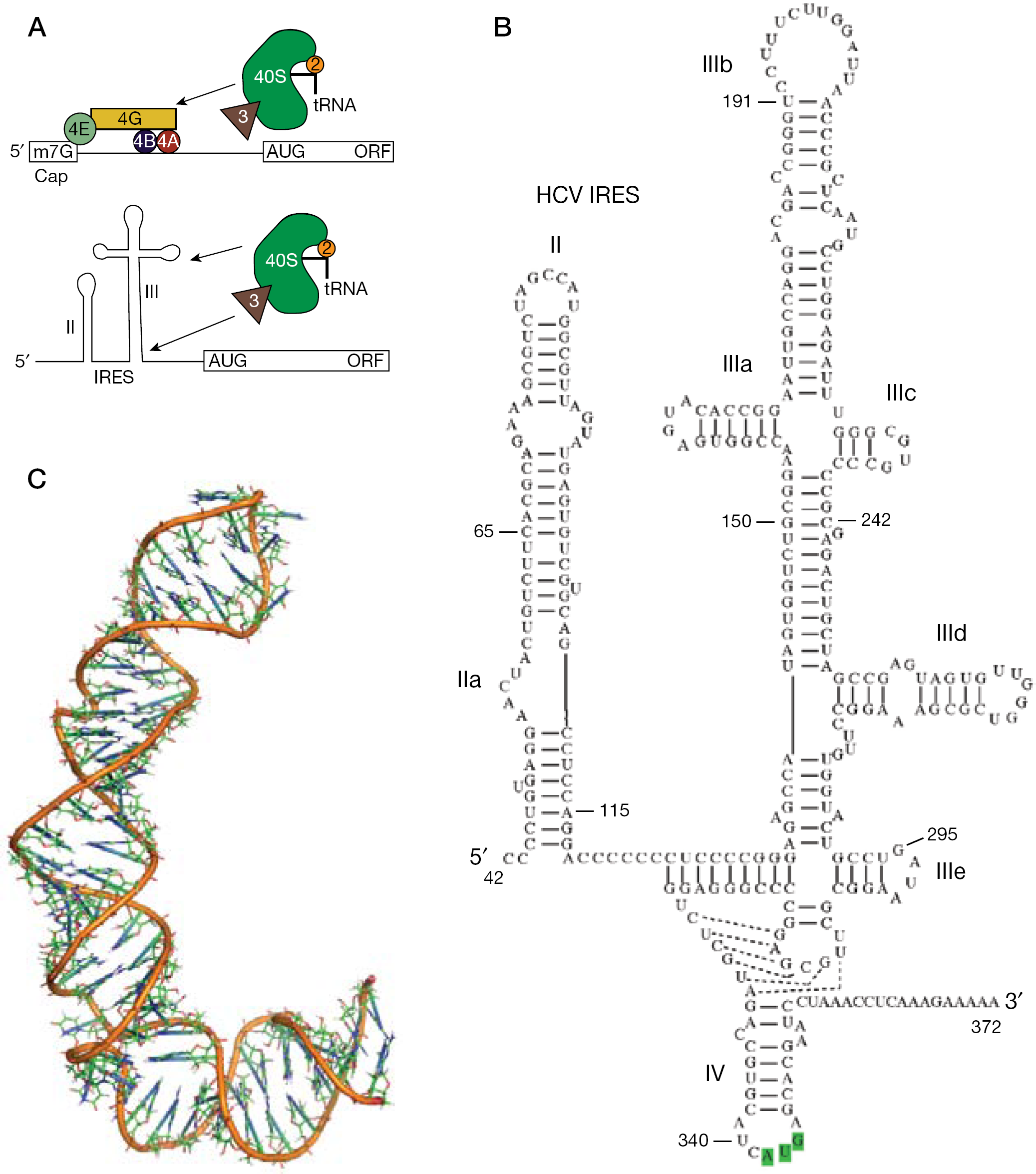
IRES-mediated translation initiation
Inhibition of RNA functional domains with oligonucleotides
The IRES element in the 5′-UTR of HCV is highly structured and is composed of four major independently folded regions with domains II, III and IV each containing functionally relevant subdomains or key nucleotide sequences [15]. Domains II and III are highly structured and each is necessary for IRES function, with specific regions of domain III making the largest contributions to ribosome binding [20–22]. Domain II appears to serve a ‘catalytic’ role wherein the proper positioning of the apical loop of domain II serves to catalyse GTP hydrolysis in the eIF2–GTP complex, a key step in the assembly of the complete, translationally competent 80S ribosome [23,24]. A small 5′ RNA hairpin defines domain I, which is disposable for IRES function. Domain IV has a structurally simple hairpin subregion containing the functionally crucial AUG initiation codon, whereas the upstream region of domain IV forms a complicated pseudoknot structure [25].
The presence of conserved RNA sequence elements within functionally important regions has proven attractive for targeting with various complementary oligonucleotides (Table 1). Modified antisense oligonucleotides have been targeted to regions throughout the IRES [26–28], and first generation full phosphorothioate 20-mer antisense oligonucleotides targeting domain IV containing the start codon progressed to the Phase I trial stage. The Isis Pharmaceuticals antisense inhibitor was shown to be effective in reducing HCV IRES driven luciferase gene expression in vaccinia-virus-infected mice [26] and a Phase I trial indicated transient HCV reduction in a small number of patients, although efficacy concerns for this first-generation antisense compound likely prevented this oligonucleotide from further consideration [29]. Antisense oligonucleotides often have quite impressive 50% inhibitory concentration (IC50) values in cell culture assays with lipid-assisted transfection, achieving the low nanomolar potency levels desired for selective therapeutics. The incorporation of modified nucleotides such as 2′-OMe and locked nucleic acid improve binding affinities for RNA targets [28,30], and peptide nucleic acids have also been used effectively to inhibit IRES activity [30,31]. The RNA inhibitor identified by Tallet-Lopez et al. [28] against the IIId region using an RNA selection procedure was found to act by an antisense mechanism where the inhibitor competed with the 40S ribosome for the HCV IRES; the investigators were able to improve the potency to the low nanomolar range by incorporating 2′-OMe modifications [28]. The activity of this oligonucleotide was shown to be somewhat poorer (30 nM) in a salt-optimized rabbit reticulocyte system that was also used to show that 2′-OMe antisense oligonucleotides could be effective against the highly structured domain III–IV pseudoknot target [25].
Activity summary of representative RNA- and oligonucleotide-based HCV IRES inhibitors
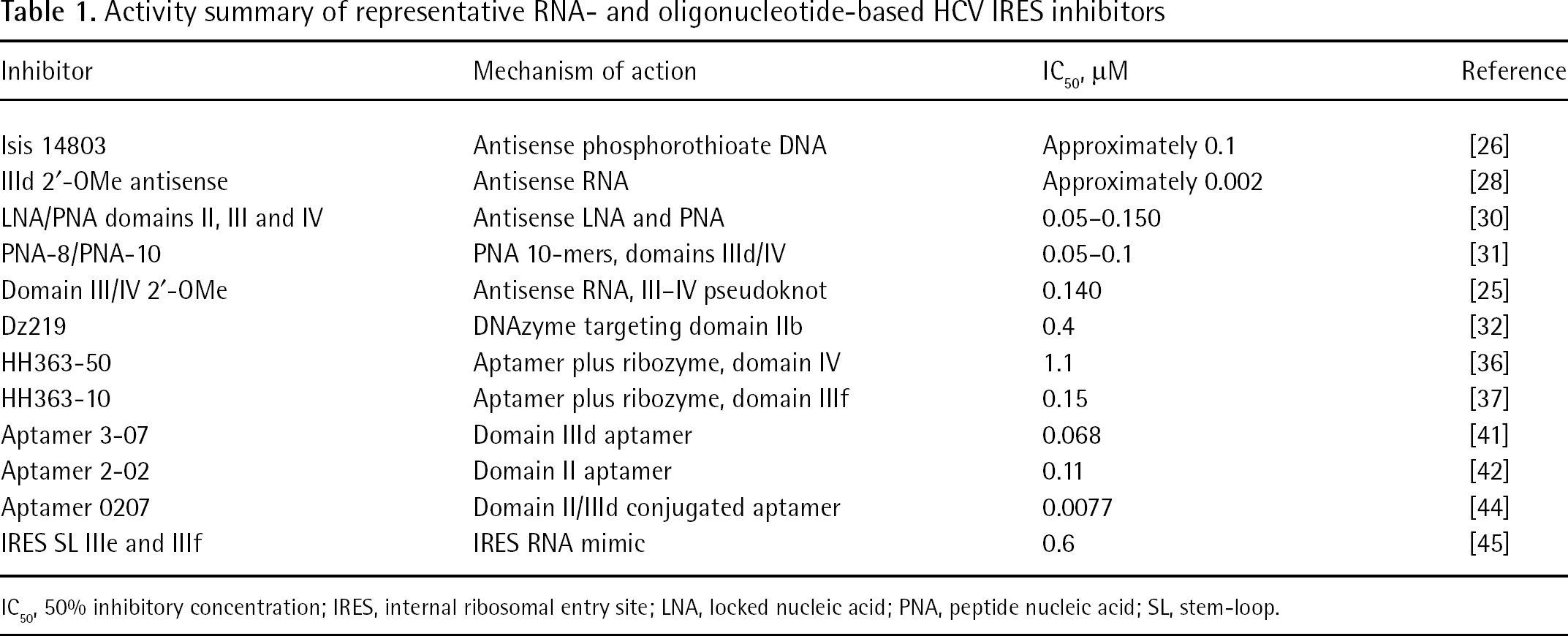
IC50, 50% inhibitory concentration; IRES, internal ribosomal entry site; LNA, locked nucleic acid; PNA, peptide nucleic acid; SL, stem-loop.
Roy et al. [32] used DNAzymes incorporating the 10–23 hairpin motif described by Santoro and Joyce [33,34] to target each of the major domains of the HCV IRES RNA. The most potent DNAzymes were those targeting domain III regions in the HCV IRES; however, even the best of that subset achieved only a 70% reduction of viral RNA levels assayed ex vivo using an HCV genotype 1a replicon and 0.8 μM concentration of the transfected DNAzyme. Mutant DNAzymes were ineffective, and no activity was seen for the genotype 1a-specfic DNA against genotype 1b, which has a different RNA sequence in the target region, demonstrating conclusively that activity was mediated by sequence-specific recognition of IRES RNA. DNAzymes have a number of advantages over RNA-based oligonucleotide therapeutics, including ease of synthesis, nuclease resistance and better cell penetration. DNAzymes also tolerate chemical modification in the ‘wing’ regions that improve target affinity and provide for even better nuclease resistance [35]. The relatively modest HCV antiviral response at nearly micromolar levels of transfected DNA, however, suggests that significant design improvements would be required for such oligonucleotide therapeutics to be considered as clinical candidates.
Aptamers and decoys
Romero-Lopez et al. [36–38] used ‘systematic evolution of ligands by exponential enrichment’ (SELEX) methods to identify RNA aptamers specific for domain IV containing the IRES initiation codon, and also identified aptamers specific for the IIIf pseudoknot domain. The aptamer recognition in both cases was mediated by loop–loop recognition motifs; in the HH363-50 molecule, an anchoring G:A pair was selected for that has been shown to be particularly advantageous not only for HCV-RNA-recognizing aptamers, but also in the well-studied HIV Tar–Tar* complex [36,39,40]. To facilitate the IRES inhibition activity of the HCV-targeted aptamers, the loop-recognizing motif was subsequently fused with a hammerhead ribozyme motif, potentially adding catalytic cleavage activity to the RNA therapeutic. Mutation experiments of the recognition and catalytic domains indicated that each contributed to activity, and that the fused RNAs were more potent than the two independent domains administered together. The in vitro IC50 value of the domain IV ribozyme aptamer was approximately 1 μM as assayed by luciferase gene expression driven by the HCV IRES, whereas the aptamer targeted against the IIIf domain was more potent, having an IC50 of 150 nM. Large RNA aptamers might present a number of challenges for drug delivery; therefore, despite some attractive IC50 values, it is difficult to see how these could be seriously considered as clinical candidates.
In vitro RNA selection has also been used by Kikuchi et al. [41,42] to identify HCV IRES inhibitors directed against domains II and IIId. The most potent RNAs had KD values in the 50–100 nM range. These rather large inhibitor RNAs, of 60–75 nucleotides in length, interact with targets in the HCV IRES via loop–loop interactions in a fashion similar to those identified by Da Rocha Gomes et al. [43], and by Romero-Lopez et al. [36]. The consistent selection of such molecules would support a hypothesis that hairpin loop sequences are more accessible targets for therapeutics and do not have the disadvantage that intramolecular interactions within the target must be overcome, as would be the case if the duplex regions were targeted. Kikuchi et al. [44] took this principle one step further, and were able to show that modest increases in affinity could be obtained by linking two loop-recognizing aptamers together, thereby obtaining RNA-based inhibitors having low nanomolar KD values; however, the resulting molecules were >130 residues long, likely making them impractical for legitimate therapeutic consideration.
Synthetic RNAs corresponding to functionally important regions of the IRES itself also represent a strategy for inhibiting IRES-meditated translation. Domains III and IV represent the IRES regions most responsible for ribosome binding recognition. RNA transcripts comprising those regions would compete with the IRES for cellular factors – IRES-transacting factors, ribosomal proteins and intact ribosomes – blocking IRES-mediated translation. Of the various possible IRES domains that could serve as decoys, an RNA stem-loop comprising regions IIIe and IIIf was found to selectively inhibit IRES activity with an IC50 of approximately 0.6 μM and was linked mechanistically to binding of the ribosomal protein S5 [45]. Previous work had shown that ribosomal protein S5 does not contact the messenger RNA directly in cap-dependent translation [46], but S5 is cross-linked to IRES domain IIIe [47], explaining the observation that the stem-loop IIIe and IIIf RNA inhibitor interferes with IRES-dependent, but not cap-dependent translation.
HCV IRES RNA structures: Targets for structure-based drug design
Non-canonical RNA regions are known to be potential binding sites for small molecule therapeutics [48], with the ribosome representing by far the richest target for RNA interacting drugs [49–51]. A few examples studied by NMR include paromomycin bound to the ribosomal A-site [52], phenothiazines bound to the HIV TAR loop [53] and also by aromatic amines [54,55], peptides bound to the HIV Rev-responsive element and to HIV TAR [56,57], and doxorubicin bound to the HIV frameshift site [58]. The HCV IRES structure has been studied in detail by X-ray crystallography, cryo-electron microscopy (EM) and by NMR. Regions of domain III and domain II have high-resolution NMR structures [20–22], and the overall bent structure of domain II seen by NMR (Figure 1C) has been confirmed in the ribosome-bound form using cryo-EM [59]. X-ray crystal structures of domain IIa have confirmed the bent structure of that domain, and confirmed key metal ion binding sites suggested by earlier NMR work [60,61]. The bent structure of domain II is conserved among the IRES elements for related viruses; HCV and classical swine fever virus each employ an internal bulge in domain II that is necessary for IRES function [23]. These viral IRES allow 43S ribosomal complexes to attach directly to the initiation codon without ribosomal scanning [24]. The Dicistroviridae intergenic region IRES are even more elaborate, using an all-RNA mechanism to position the 80S ribosome in order to initiate translation [62], whereas many cellular IRES are minimal RNA structures requiring an extensive set of cellular factors that more closely resembles the ‘land and scan’ mechanism of canonical translation initiation [63].
Identification and optimization of small molecule inhibitors
The function of the HCV IRES depends not only on the direct interaction of conserved structured domains with the ribosomal machinery, but also with defined internal structures of the IRES, and possibly RNA conformational dynamics [24,64]. Small molecules might have difficulty competing with direct interactions between the ribosome and the IRES RNA because of the large surface areas involved, but could be effective in modulating the IRES structure and dynamics. Furthermore, although the oligonucleotide inhibitors described in this review are clearly potent at nanomolar levels in vitro and in cell culture with cationic lipid transfection, only one has progressed into clinical trials. The design of oligonucleotide-based therapeutics and methods for their delivery have progressed tremendously [65], but the development of small-molecule HCV inhibitors remains an attractive therapeutic avenue.
Aminoglycoside antibiotics are the prototypical RNA-targeted therapeutic class. Zhou et al. [66] and Carnevali et al. [67] proposed that the 2-deoxystreptamine (DOS) structural core of natural aminoglycosides would provide a rigid framework upon which to develop a modular recognition platform for RNA recognition, and 3,5-diaminopiperidine (DAP) heterocycles were then developed as a structural mimic of the 2-DOS pharmacophore (Figure 2). This group had previously determined the X-ray structure of IRES domain IIa and made fluorescently tagged versions of the RNA for drug screening. They chose IIa as a target to screen a series of modular DAP ligands. Several of the analogues caused a fluorescence decrease when titrated into 2-aminopurine-labelled RNA, suggesting a binding mode wherein the conserved IIa loop becomes stabilized [67]. Evaluation of the same compounds in a fluorescence resonance energy transfer (FRET) assay showed no change in the FRET signal, suggesting that the global RNA structure was unaffected by DAP compound binding. By contrast, a large conformational change occurs when benzimidazole inhibitors bind the domain IIa loop [68,69]. DAP ligand binding was also rather sensitive to magnesium concentration. At 100 μM Mg+2, where the RNA is folded, the compounds had low micromolar affinity; however, increasing the Mg+2 further to 1 mM caused a >10-fold affinity decrease. An additional affinity decrease was seen with 100 mM Na+, suggesting that DAP binding has a strong electrostatic component.
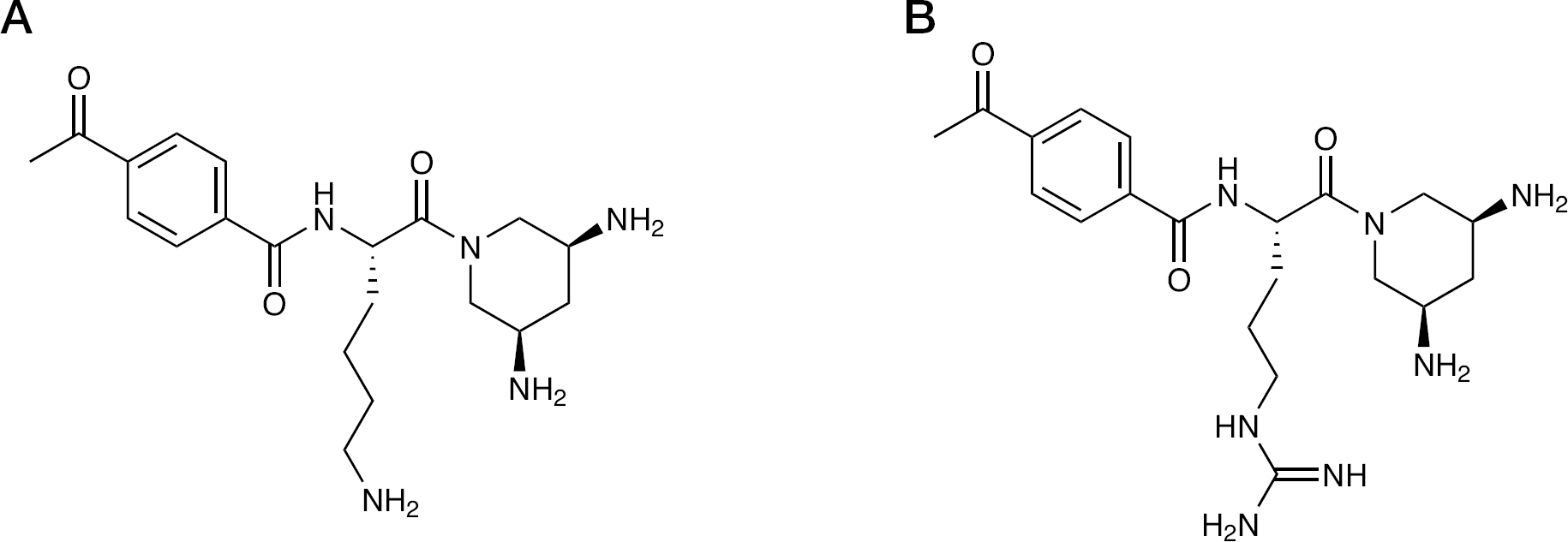
DAP compounds targeting IRES domain IIa containing lysine- and arginine-derived side chains
High-throughput screens have identified several small-molecule hits that inhibit HCV IRES activity either in vitro or in cell culture [70–73]. Gooding et al. [70] were able to identify approximately six compounds with RNA KD values of 1–50 μM out of a library of 300,000 screened against the simple stem-loop RNA from domain IIIe. Five additional compounds with weaker RNA binding, but still meeting the cutoff for mass spectrometry detection, were confirmed to have biological activity. The chemical classes were not described in detail, but included peptides and aminoglycosides, as well as amines. Wang et al. [71] screened 160,000 compounds in cell culture to identify a phenazine molecule with an approximately 100 nM IC50; unfortunately, none of the subsequent derivatives they synthesized had comparable activity, and there was no indication that the inhibitor directly affected the IRES RNA.
Using the dicistronic IRES reporter system of Honda et al. [73,74], another class of aromatic inhibitors, biaryl guanidines, was discovered in a screen of approximately 180,000 RNA-biased compounds [73]. Structure–activity relationship (SAR) studies indicated that the basic guanidino functionality was crucial for IRES inhibition (Figure 3). Replacing the guanidine with a urea/thiourea moiety or introducing methyl groups completely abolished activity. Similarly, replacing the pyrimidine ring with a benzene or a pyridine ring abolished activity. The above SAR suggested that the guanidino and the pyrimidine nitrogen atoms might be involved in H-bond type interactions with the RNA. Alternatively, the basic guanidine and the pyrimidine nitrogens might participate in an intramolecular H-bonding network that would rigidify a particular conformation for H-bonding or stacking interactions with the RNA. Optimal HCV IRES inhibition was seen when the alkyl tether between the guanidine and the phenyl ring was 2-carbon atoms. In general, introducing substituent groups on the phenyl ring or replacing the phenyl ring with other aromatic rings reduced IRES inhibition. Substitution along the tether between the guanidine and the phenyl ring was also poorly tolerated, except for a hydroxyl group in the (S) configuration at the benzylic position. Introducing a methyl or a methoxy group at C-5 of the pyrimidine ring maintained IRES inhibition. Introduction of a dimethylaminopropyloxy group at the C-5 position resulted in a three- to fourfold improvement in IRES inhibition. This suggested that the basic amino functionality might be involved in interactions with the RNA.
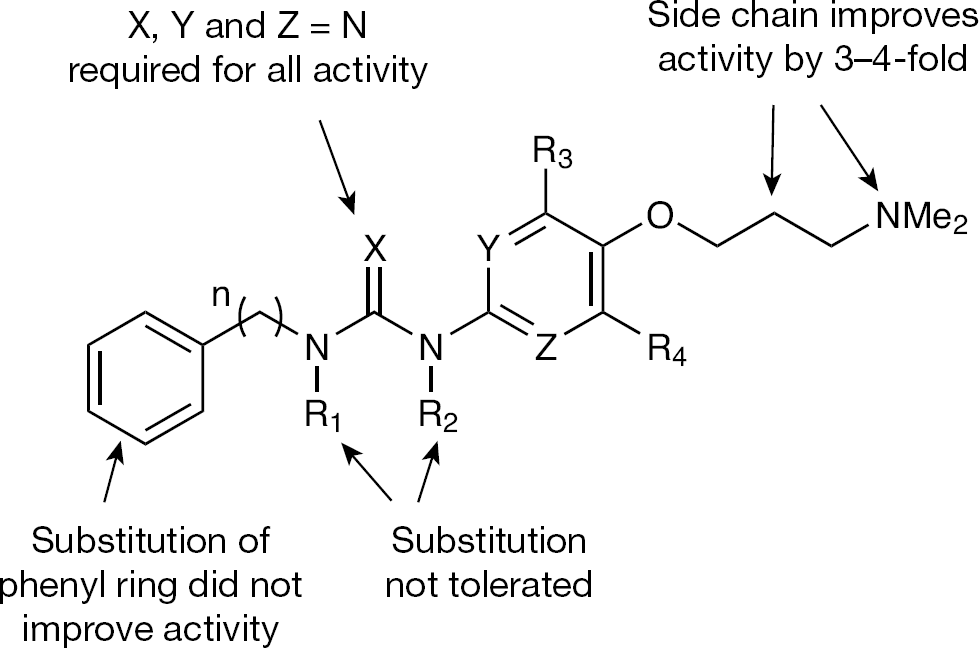
Structure–activity relationship of biaryl guanidine HCV internal ribosomal entry site inhibitors
Mass-spectrometry-based screening against the IRES domain IIa RNA starting from a library of approximately 180,000 compounds provided a benzimidazole hit having an RNA KD of approximately 100 μM, and a threefold selectivity preference over a control ribosomal RNA [72]. Further SAR studies showed that the dimethylaminopropyl chain at N-1 and the amino group at N-2 were absolutely essential for binding to the RNA (Figure 4). Introducing electron-withdrawing or electron-donating substituent groups at the C-4 or C-5 position of the benzimidazole ring reduced binding significantly. The only benzimidazole ring substituent that improved binding was a methoxy group at the C-6 or C-7-position. Replacing the C-6 methoxy group with a dimethylaminopropyloxy chain improved binding affinity two- to fourfold. To probe the specificity of the binding interaction, the dimethylaminopropyloxy group was moved to the C-5 position and/or the 2-amino group was removed. Both analogues showed substantially reduced binding affinity for the RNA, indicating that the improved affinity was not a result of non-specific interactions of the cationic side chains with the negatively charged RNA backbone. Replacing the dimethylamino head groups with polyamino groups improved binding affinity; however, these groups were deemed too polar for potential therapeutic applications.
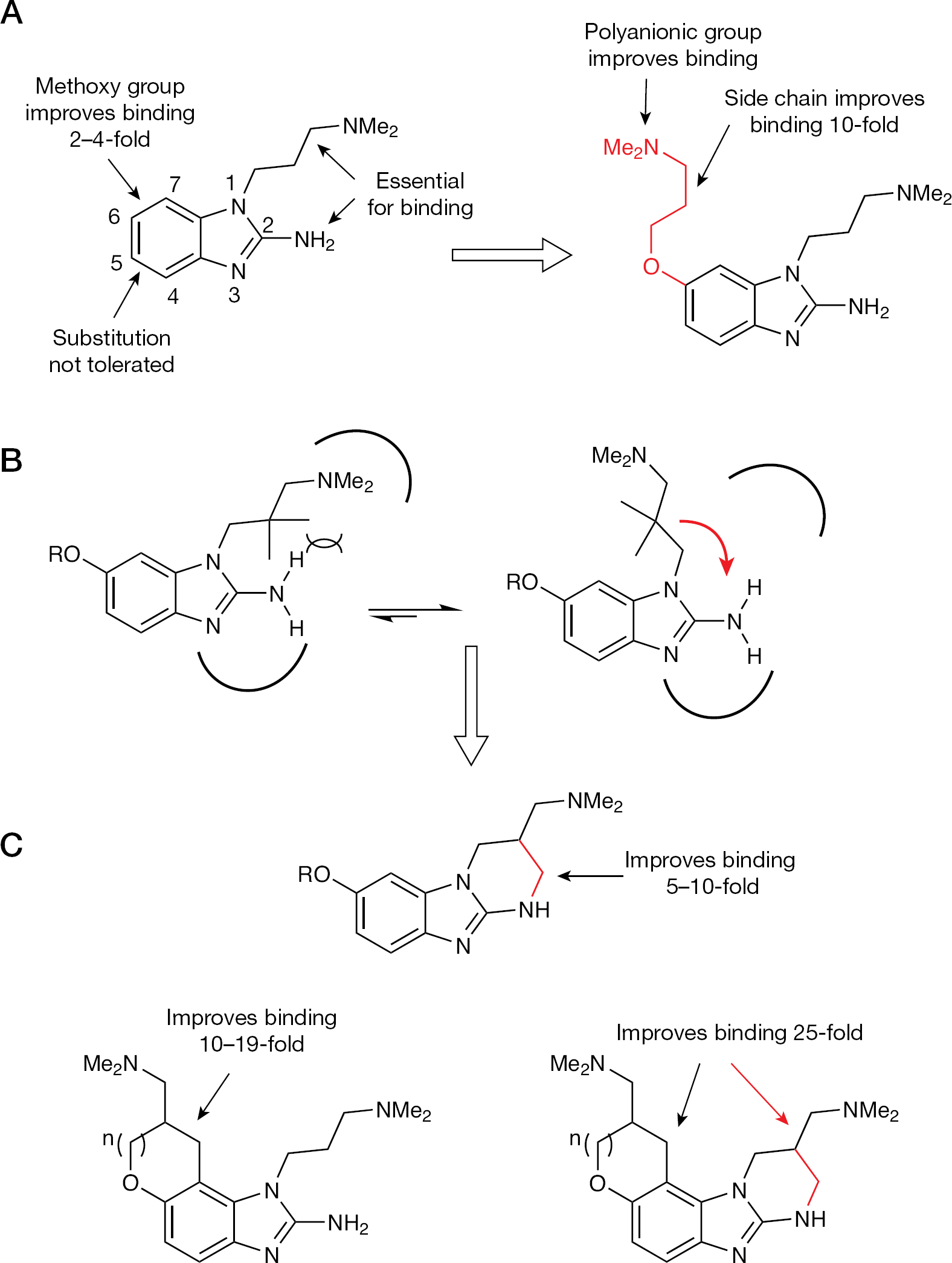
Structure–activity relationship of the benzimidazole class of HCV internal ribosomal entry site inhibitors
Interestingly, the introduction of methyl groups along the N-1 chain abolished binding completely. One way to rationalize this observation was that the 2-amino group and the N-1 dimethylamino group were making specific contacts with the RNA (Figure 4). Introducing steric bulk along the N-1 side chain could result in steric clash with the 2-amino group and change the conformational preference of the side chain such that it can no longer productively interact with the RNA. To test this hypothesis, the N-1 side chain was constrained into the 2-amino group so as to restrict it in the putative RNA binding conformation. This strategy of conformational restriction was successful and resulted in a 5–10-fold improvement in binding affinity. To test if the strategy of conformational restriction could be applied to the C-6 dimethyaminopropyloxy side chain, additional analogues were synthesized and evaluated for RNA binding (Figure 4C). Once again, the strategy was successful and resulted in a 10–19-fold improvement in binding affinity. Further conformational restriction of both the N-1 and the C-6 side chains produced a 25-fold improvement in binding affinity relative to the unconstrained analogue and a 140-fold improvement relative to the initial benzimidazole hit. We recently showed that there is a stereochemical activity preference; separation of the Isis-11 enantiomers by chiral HPLC and subsequent RNA binding studies using 2-aminopurine-labelled RNA showed a threefold binding difference for the isomers [68].
To investigate the pharmacokinetic properties of the benzimidazole class of compounds, two representative benzimidazole analogues (Figure 5) were evaluated in rat. Both compounds displayed excellent pharmacokinetic properties, and compound
Pharmacokinetics of heterocycle modified benzimidazoles
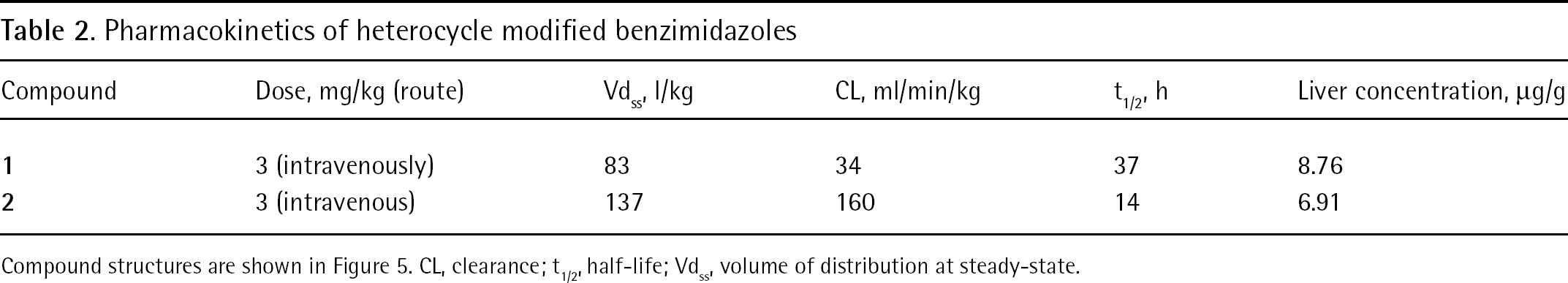
Compound structures are shown in Figure 5. CL, clearance; t1/2, half-life; Vdss, volume of distribution at steady-state.
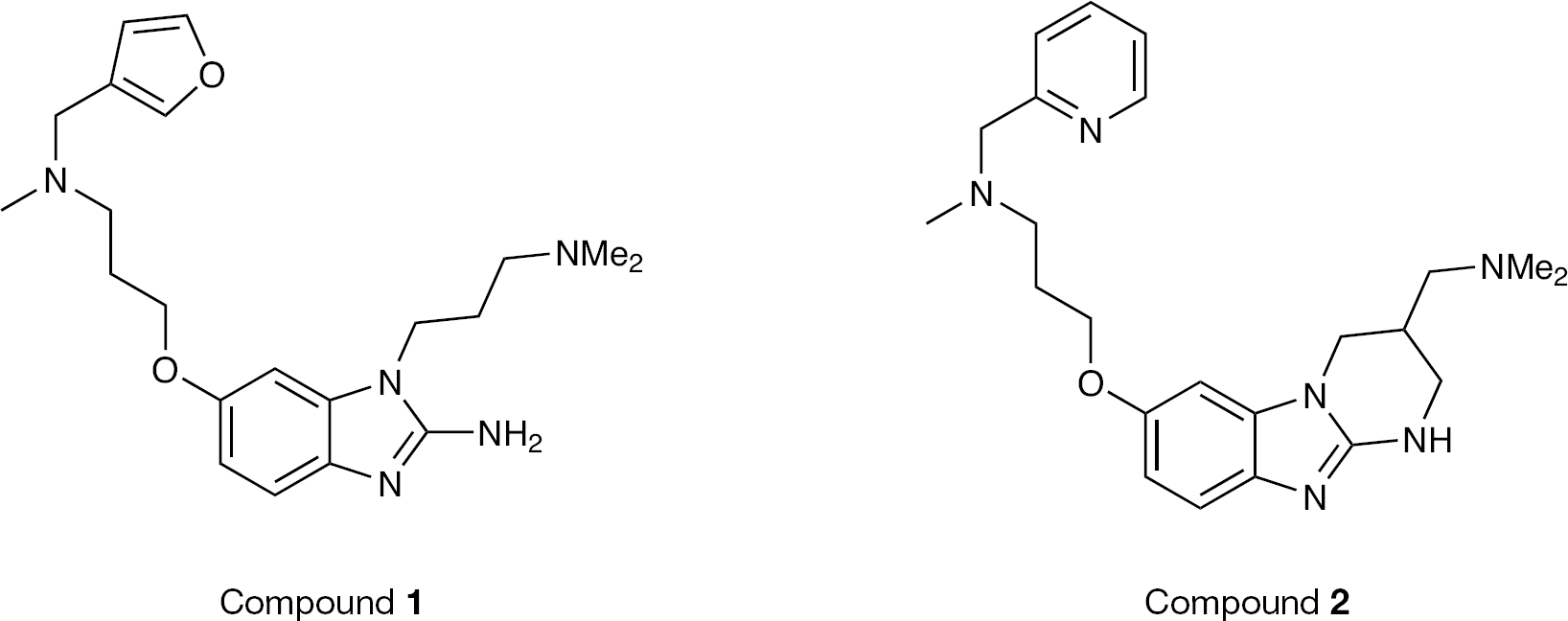
Modification of benzimidazole inhibitors with tethered heterocycles resulted in compounds with good pharmacokinetic properties
Structural studies of RNA-bound inhibitor complexes
Domain II of the HCV IRES mediates the release of a key initiation factor, eIF2, from the 40S subunit, to promote 80S ribosomal assembly [24]. The bent structure of domain II is thought to be conserved, and is crucial to the biological role of this RNA element [23]. Seth et al. [72] had shown that the benzimidazole inhibitors interacted specifically with domain IIa, and Parsons et al. [69] used FRET measurements to show that the Isis-13 doubly constrained benzimidazole compound induced a large conformational change. We initiated a structural study of the inhibitor complex in anticipation that NMR methods might provide insight into the binding mechanism, and that a detailed structure of the complex might inform on the synthesis of more potent derivatives. An RNA hairpin containing the domain IIa bend-inducing bulge, capped with the ultra-stable UUCG tetraloop motif, was used for NMR screening of the racemic Isis-11 inhibitor and subsequent high-resolution structure determination (Figures 6 and 7) [68]. Inhibitor-induced chemical shift changes of pyrimidine residues were conveniently monitored using a sensitive two-dimensional total correlation spectroscopy (TOCSY) NMR experiment, demonstrating that residues within the bulge experienced a significant change in structural environment upon inhibitor binding. Two-dimensional 1H-13C heteronuclear single quantum coherence (HSQC) NMR spectra of the purine H8–C8 cross-peak region showed that G:A residues within and near the bulge likewise experienced a conformational change, an effect confirmed by fluorescence spectroscopy of 2-aminopurine-substituted RNAs. The overall effect of Isis-11 binding to the domain IIa bulge is to displace two conserved adenosines, resulting in an alteration of the RNA helical axis from a 90° bent conformation, shown to be necessary for IRES function, toward a nearly straight conformation shown by mutagenesis to be inactive (Figure 6). The bound and free RNA structures have similar free energies according to molecular simulations using AMBER [75], suggesting a hypothesis that the inhibitor might selectively bind to a rarely sampled conformational form similar to the bound state, as opposed to a mechanism where the bound form is ‘induced’ by inhibitor binding [76].
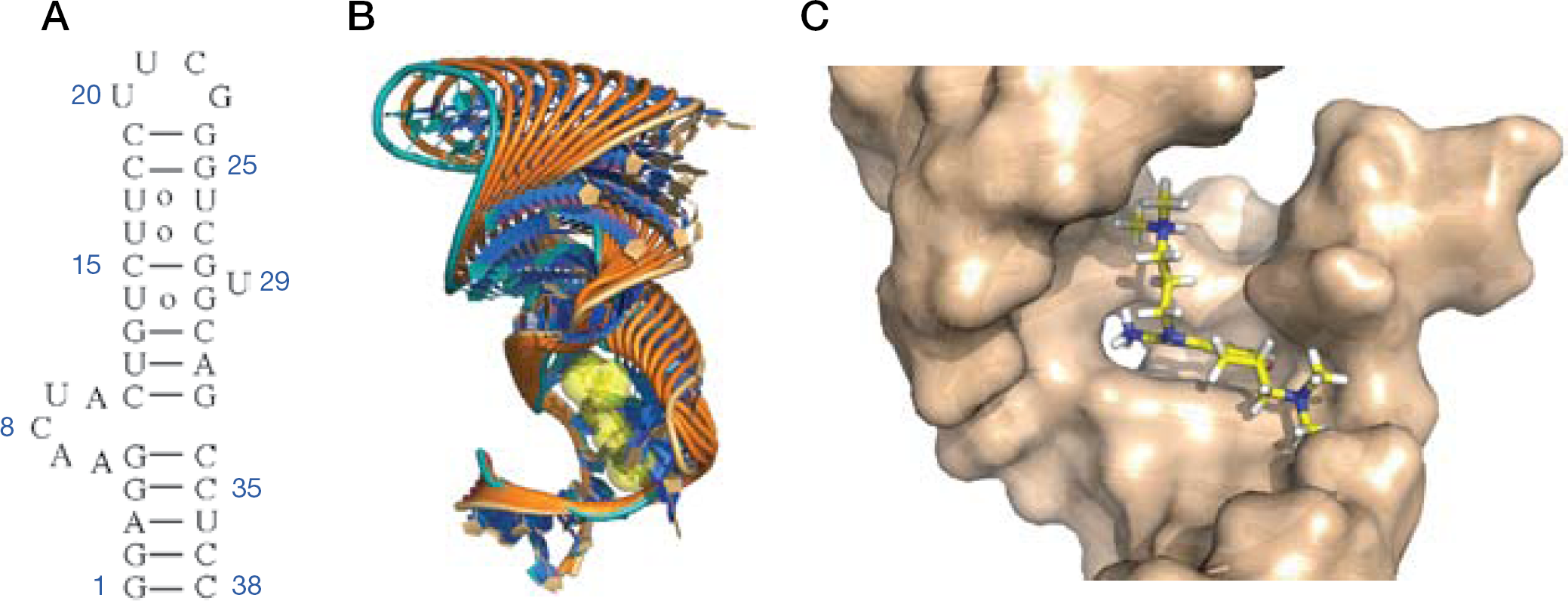
Isis-11 binding to HCV internal ribosomal entry site domain IIa RNA
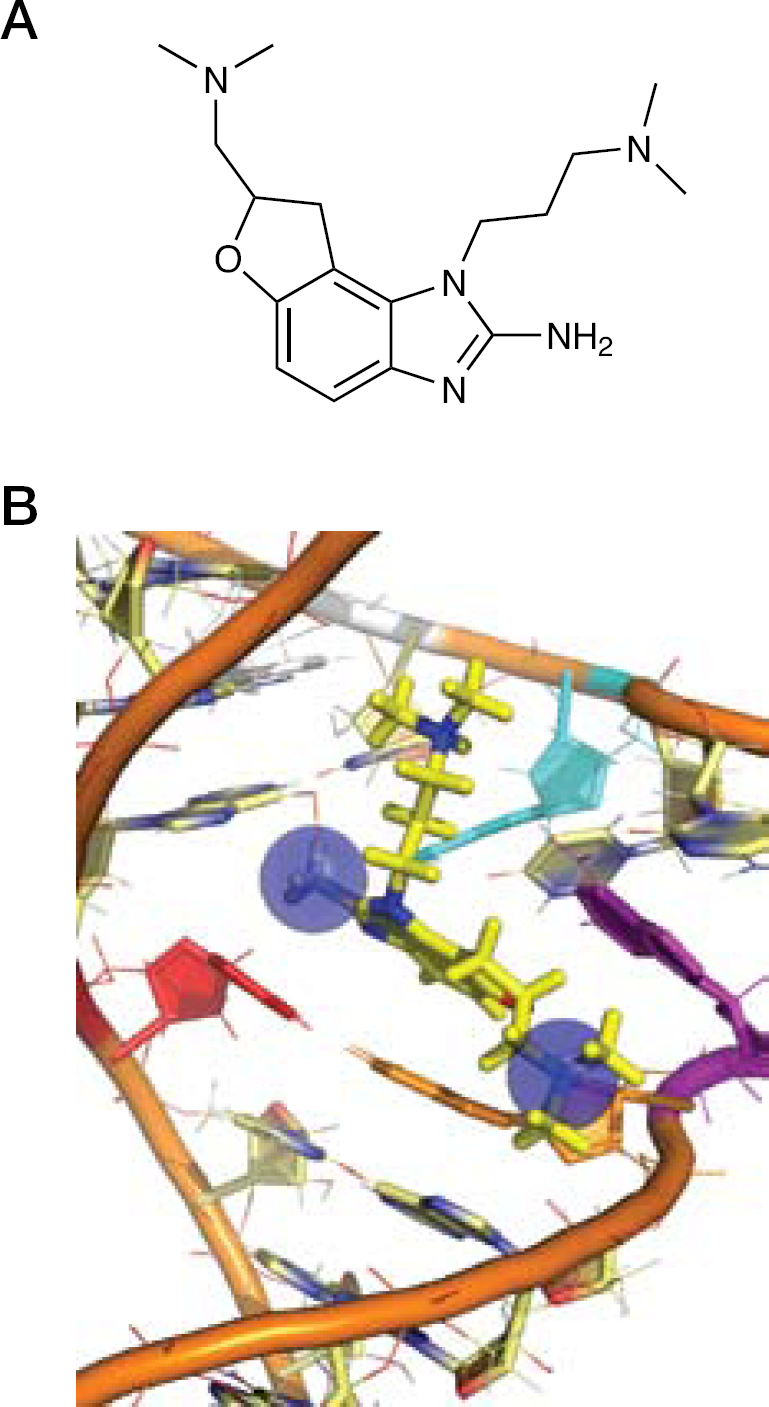
Details of isis-11 binding
The domain IIa complex provided some insight into principles that might be useful for further development of this inhibitor class, and for RNA-targeted therapeutics in general. An electrostatic surface map of the bound RNA showed that the two dimethylamino groups interact with local regions of partial negative change within the RNA interior. This somewhat counterintuitive principle of RNA–ligand interaction has been reported previously [55], and suggests that specific RNA inhibitors be designed to orient cationic groups to buried complementary sites, rather than targeting the phosphate backbone that is effectively neutralized at physiological salt concentrations. Aliphatic substituted amine groups might be particularly effective in this regard as they can interact with electrostatic ‘hot spots’ as well as participate in cation–π interactions with RNA bases (Figure 7) [68]. The general development of RNA-targeted therapeutics has been particularly challenging, in part because the principles for drug design are still unclear. The one prototypical chemical class, aminoglycoside antibiotics, have not been generalized to RNA targets outside of the ribosome despite extensive efforts [51]. These flexible, polar and often highly charged molecules have not proven amenable to rational design, and the well-established therapeutic classes used to target proteins have not been readily adapted to target RNA; however, the benzimidazole HCV inhibitors demonstrate that small molecules with few functional groups are capable of modulating RNA structure and can be active at the micromolar level. With an inhibited structure in hand, there is now the opportunity to rationally elaborate these compounds, engineering in additional RNA contacts to provide low nanomolar inhibitors. In principle, this could be accomplished without the large increases in molecular weight and lipophilicity that is the unfortunate outcome of many hit-to-lead therapeutic development projects using protein targets. The benzimidazole binding pocket in the IRES domain IIa RNA might not look like a traditional hydrophobic protein target (Figure 6C), but the target site is replete with polar functional groups that could be specifically recruited for increased inhibitor affinity.
Conclusions and future directions
Two recent screens of the LOPAC library [77] containing 1,280 pharmacologically active compounds and of the NIH Clinical Collection [78] containing 446 clinically relevant compounds identified >50 HCV inhibitors. These compounds were shown to interfere with viral replication by a variety of mechanisms, although none appear to inhibit IRES function, and none have IC50 values significantly below the 1 μM level. Therefore, although the screening platforms described in these reports are impressive, no highly potent compounds were discovered among these clinical compounds. The further development of HCV-IRES-targeted compounds seems attractive because the benzimidazoles already have low micromolar activities and structural information is available to facilitate further development. Protease inhibitors are emerging as additions to the standard of care for HCV treatment, but the experience with HIV treatment would suggest that a cocktail of drugs against diverse targets would also have advantages for HCV therapy. The IRES is the most sequence-conserved region of the HCV genome, suggesting that clinical resistance against IRES inhibitors might be slow to develop. Small molecule therapeutics targeting the IRES RNA represent a novel therapeutic strategy, and structure-based chemical optimization could provide a paradigm for small molecule drug development against a wide range of intriguing RNA targets.
Footnotes
The authors declare no competing interests.