
Abstract
There are currently no licensed antivirals available for the treatment of dengue virus (DENV), which causes significant morbidity and mortality throughout tropical areas of the world and is now encroaching on the southern United States. Recent improvements in existing animal models and cell culture systems have been very important in elucidating the mechanisms of DENV pathogenesis in humans, including the identification of potential viral and host proteins that might be targeted for the treatment of DENV infection. The AG129 mouse model is a major advance in the development of antiviral and vaccine candidates for clinical use. It allows for testing of potential therapeutics in a relevant system that exhibits some aspects of disease that are similar to those observed in humans. This review focuses on recent developments in the AG129 mouse model and discusses compounds that have been found to be active in available cell and animal model systems within the past year.
Introduction
Dengue disease is caused by four related and antigenically distinct viruses, which are designated dengue virus (DENV) 1–4. DENV is transmitted from person to person via mosquito vectors (particularly Aedes sp). Approximately 2.5 billion people are at risk of contracting dengue in endemic areas, and as many as 50 million people are infected with DENV each year [1]. DENV infection generally results in dengue fever (DF), a relatively mild and self-limiting disease associated with fever, malaise and other non-specific symptoms. However, in hyperendemic areas with multiple circulating serotypes, the more severe, potentially lethal disease manifestations of dengue haemorrhagic fever (DHF)/dengue shock syndrome (DSS) can occur. The hallmark of DHF/DSS is plasma leakage, which is commonly associated with a cytokine storm, thrombocytopaenia and intestinal haemorrhage [2]. Several risk factors, including race, age, sex and host genetic factors (for example, human leukocyte antigen [HLA], dendritic-cell-specific intracellular adhesion molecule-3-grabbing non-integrin [DC-SIGN], tumour necrosis factor [TNF]-α, transforming growth factor [TGF]-β1 and interleukin [IL]-10) appear to be associated with severe disease [3]; however, the single greatest risk factor is the presence of subneutralizing levels of antibodies with specificity to another strain of DENV acquired either in utero from mother to child or from repeat infection by another serotype [4].
Three major hypotheses have been formulated to explain DENV pathogenesis: antibody-dependent enhancement of disease (ADE), aberrant T-cell response and viral virulence factors. According to the ADE hypothesis, cross-reactive antibodies generated against a previous infecting serotype of DENV facilitate the Fc-mediated uptake of a subsequent infecting heterologous strain by Fc-receptor-positive cells. This ultimately results in a more severe pathological response to the secondary virus infection [5]. The involvement of ADE as a cause of severe DENV-associated disease is further supported by the observation of non-neutralizing maternal antibodies to DENV, which could persist for up to 12 months in infants born to immune mothers [6] and might be associated with increased frequency of severe disease in such cases [7]. According to the aberrant T-cell response hypothesis, serotype cross-reactive memory T-cells contribute to the pathogenesis of DHF/DSS. In support of this hypothesis, inappropriate T-cell responses have also been observed with higher frequency in DENV-infected patients with more severe symptoms [8]. Finally, the viral virulence hypothesis postulates that viral genotype might play an important role in DENV pathogenesis, as specific viral genotypes have been shown to be associated with more severe disease outbreaks [9], supporting the contribution of viral determinants in DENV pathogenesis. Recent research has further characterized the host response on a genetic level to give insight into the differences in the development of DSS as compared with less severe disease phenotypes [10].
Currently, fluid replacement, with or without blood factors, is the only course of action for the management of plasma leakage associated with DHF/DSS [11–14]. Children have the highest risk of developing severe disease manifestations, further underscoring the need for effective care. Most primary infections resolve with a relatively low mortality rate and do not require treatment. In hyperendemic areas, many infected patients must be hospitalized, which puts a tremendous financial burden on these countries. A recent estimate placed the burden of DENV in eight countries between 587 million and 1.8 billion USD annually, which does not include the cost of surveillance and vector control [15]. An effective antiviral could reduce morbidity and thereby ease the financial burden.
Challenges faced in the development of DENV-specific antiviral therapies are numerous and include development of a low-cost rapid diagnostic technique that is predictive of disease severity, and identifying a compound that is safe, targets multiple serotypes and is effective even after the onset of severe clinical disease. These are not easy goals, but recent advances in the understanding of DENV biology and improvements in animal and cell culture models should contribute towards the discovery and advancement of potential anti-DENV agents for clinical use. Here, we will briefly review recent advances in animal models of DENV and discuss, in more detail, the anti-DENV agents that have shown potential for use in the treatment of this disease.
Recent advances in animal models of DENV
The use of a small animal model of disease is crucial to the advancement of antiviral agents toward clinical use, as compounds that possess antiviral properties in vitro might not have the same effect in an in vivo model. For example, the antiviral prodrug ribavirin inhibits DENV replication in vitro [16–18], but had no effect on viraemia when evaluated in two separate in vivo model systems [19,20]. Optimally, animal models should reflect the clinical pathology of infection observed in humans. Development of an animal model for DENV has been difficult because the virus does not replicate well in species other than its natural hosts, that is, humans and mosquitoes. Some strains have been adapted to cause neurological disease in mice, which might have some utility in the evaluation of antiviral therapies [21,22], but these neuropathological mouse models lack many important and relevant disease features that are seen in human infection cases.
Non-human primate models of DENV infection have been examined (reviewed in [23]). In these models, only transient viraemia is detected and the infected animals do not generally exhibit outward pathology, with the exception of a recent study showing skin rash/haemorrhage in DENV-infected rhesus macaques [24]. Thus, the development of a non-human primate model with relevant disease features is a difficult prospect. At present, the lack of key DHF/DSS manifestations (for example, plasma leakages and cytokine storm), combined with the high cost and ethical considerations associated with conducting studies in higher mammals, has limited the use of primates as model systems for antiviral testing.
A key advance in the field of anti-DENV research is the recent development of a useful mouse model of disease. This allows for the testing of potential antiviral compounds in a small animal model with many disease features that are similar to those observed in humans. Some recent reviews provide a history of the development of mouse models of DENV infection [23,25,26], which will only be addressed briefly here.
129/Sv mice lacking both the type-I and type-II interferon (IFN) receptors (AG129) are highly susceptible to infection with various mouse-adapted and clinical DENV strains, including all four serotypes [20,27,28]. Infection of AG129 mice with sufficient titres of these DENV strains generally results in viraemia and presence of infectious virus in other tissues, splenomegaly, detectable levels of serum NS1 protein and paralysis between 8–12 days post-virus challenge [20,29]. Such models allow the evaluation of potential antiviral agents in the treatment of mild disease and reduction of viraemia, but are complicated by neurological involvement during the final stages of disease, which rarely occurs in humans [30,31].
The development of an AG129 infection model that uses D2S10, a DENV-2 strain alternately passaged through mouse and insect cells has been reported [28]. In this model, animals infected via the intravenous route display an early death phenotype (4–6 days post-infection [dpi]) that does not involve paralysis and includes additional parameters relevant to human infection, such as increased vascular permeability and cytokine responses. A similar disease phenotype is observed with S221, a triple-plaque-purified clone isolated from D2S10. A non-mouse-adapted strain of DENV-2 (D2Y98P) was recently reported to cause similar severe disease manifestations after intraperitoneal administration of 106–107 plaque-forming units of virus, which included cytokine storm, vascular leak, haemorrhage and early death (5–6 dpi) in AG129 mice [32]. Inoculation with lower doses of D2Y98P DENV resulted in viral replication in relevant tissues, tissue damage, increased vascular permeability without haemorrhage and death a few days after virus clearance. This represents a model of less severe disease following infection with a relatively low viral challenge dose. The AG129 mouse model of primary DENV infection has shown utility in antiviral studies evaluating the efficacy of various compounds, including α-glucosidase inhibitors, nucleoside inhibitors and sulfated polysaccharides. Parameters used in these studies included survival, viraemia, levels of inflammatory cytokines and splenomegaly [20,33–35].
Greater utility of the AG129 mouse model was found after the first demonstration of ADE in vivo using the D2S10 and S221 viruses. Severe DHF/DSS is 40x more frequent during secondary infection as compared with primary infection and is likely to be caused by the presence of enhancing antibodies generated against the heterologous serotype [36]. Therefore, the role of ADE in the context of antiviral therapy of DENV infection is a very important issue to consider. Evaluation of human antibodies generated during natural DENV infection revealed that the majority had specificity to the precursor membrane (prM) protein, with strong enhancing and weak neutralization characteristics [37]. Both Zellweger et al. [29] and Balsitis et al. [38] independently demonstrated that administration of heterologous or subprotective levels of homologous anti-DENV antibodies prior to DENV challenge resulted in an enhancement of disease severity. The S221–AG129 model of ADE-induced disease featured all the hallmarks of severe dengue disease in humans, including increased hematocrit levels, cytokine storm, low platelet count, increased vascular permeability, haemorrhagic manifestations and shock-induced death. Additionally, in this model, only a specific cell subset, liver sinusoidal endothelial cells (LSECs), was found to permit ADE of DENV infection in vivo. On the basis of in vitro studies, macrophages and dendritic cells have been presumed to support ADE, although there is no consensus in the field about cellular tropism of DENV in humans, and no study has defined the cell types that support the ADE model of DENV infection in humans. Hepatitis is more frequently observed in DENV-infected patients that succumb to disease [39], which is consistent with the observation of increased infection of LSECs. Thus, despite the caveat associated with the immunocompromised status of AG129 mice, this finding provides the impetus towards determining the precise cell types that are infected in humans. Importantly, both the S221–AG129 and D2S10–AG129 models of ADE-induced disease are well-suited for evaluating antiviral compounds. Indeed, the D2S10–AG129 model has already been used to demonstrate that a recombinant anti-DENV antibody that does not bind the Fc-γ receptor has prophylactic and therapeutic efficacy against lethal DENV infection [38]. The publication of additional studies evaluating antiviral therapies in these models of ADE-induced disease is anticipated in the near future.
Therapeutic targets
Approximately 2.5 billion people are at risk of contracting DENV; therefore, it is vitally important that antivirals that specifically or broadly inhibit DENV are developed. The development of antiviral treatments for DENV is complicated, as an effective therapy for the treatment of dengue must be safe, inexpensive and active when administered at or soon after the onset of disease. Other factors that reduce the incentive to develop anti-DENV treatments include the short duration of disease, an often mild clinical course and a high survival rate. The National Institute of Allergy and Infectious Disease has supported programmes to encourage the discovery of antiviral therapies for DENV and other important human viral diseases [40].
Intuitively, direct and specific inhibition of the virus would be the most attractive route of therapeutic intervention. Antiviral agents could target structural and non-structural proteins produced during viral infection. Once viral replication is inhibited by host responses, immunopathological events might be more important in later stage disease [41]; hence, reversal or inhibition of immunopathological disease might be a valid therapeutic strategy to pursue. Recent advances in the inhibition of viral and cellular targets, including specific anti-DENV agents, will be discussed below.
Viral protein targets
Similar to other flaviviruses, DENV is composed of a single positive-sense genomic RNA (gRNA) that codes for a polyprotein (Figure 1), which is processed by host and viral proteases into various intermediate and mature structural and non-structural proteins. Recent findings on the cellular and molecular mechanisms of DENV provide insight on how best to inhibit the replicative cycle and pathogenicity through the use of antivirals [42,43]. An in-depth review of specific viral targets and their potential inhibitors was recently published [44]. In the current review, we present a discussion of more recent discoveries in the area of viral targets and newly identified inhibitors (Table 1).
Recently discovered agents with anti-DENV activity
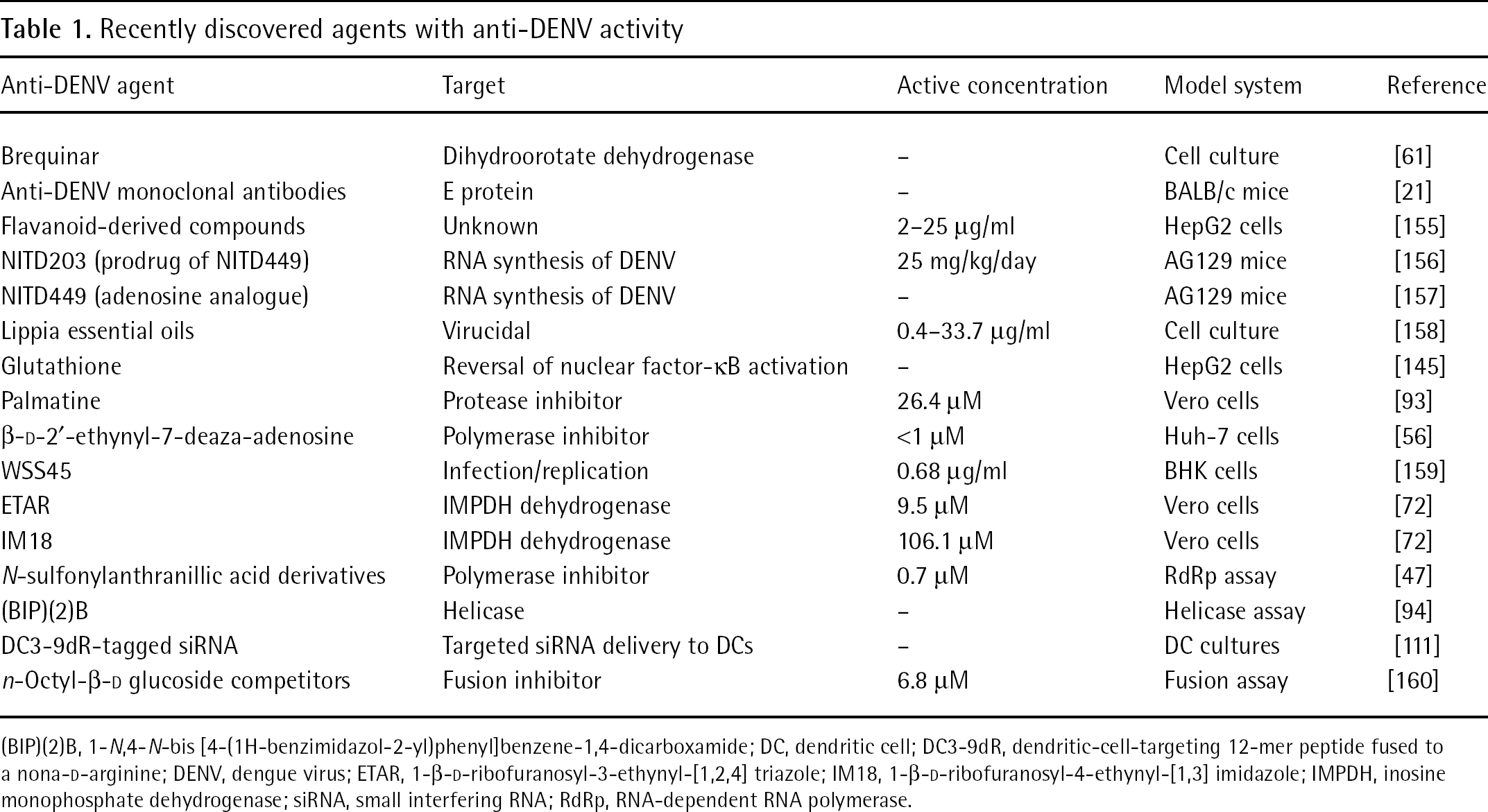
(BIP)(2)B, 1-N,4-N-bis [4-(1H-benzimidazol-2-yl)phenyl]benzene-1,4-dicarboxamide; DC, dendritic cell; DC3-9dR, dendritic-cell-targeting 12-mer peptide fused to a nona-D-arginine; DENV, dengue virus; ETAR, 1-β-D-ribofuranosyl-3-ethynyl-[1,2,4] triazole; IM18, 1-β-D-ribofuranosyl-4-ethynyl-[1,3] imidazole; IMPDH, inosine monophosphate dehydrogenase; siRNA, small interfering RNA; RdRp, RNA-dependent RNA polymerase.

A graphical representation of the flavivirus polyprotein
NS5
During DENV infection, all virus-encoded enzymatic processes are accomplished by the NS3 and NS5 proteins, which make these non-structural components attractive targets for antiviral therapy [45]. The flavivirus NS5 protein performs key roles in virus replication and in the modulation of host immune response [46], presenting multiple potential targets because it is a viral polymerase (Figure 2) [46–48], has methyltransferase function [49,50], binds to various host proteins (forming interactions that are necessary for replication) [51,52] and is involved in inhibition of the antiviral response of the cell [53,54]. Therefore, targeting NS5 will inhibit various processes that the virus uses to establish replication and cause disease.
DENV envelope, polymerase and protease
Great effort has been undertaken to identify inhibitors of the polymerase function of NS5, including large screening programmes that utilize RNA-dependent RNA polymerase (RdRp) [55], virus-like particle (VLP) [56] and replicon [57] assays. Utilization of these assays in antiviral compound research has resulted in the discovery of several inhibitors of NS5 polymerase function, and will continue to be important in discovering inhibitors in the future.
NITD008, which has broad-spectrum activity against several flaviviruses via the inhibition of NS5, was effective in cell culture as well as in a mouse model of disease in the treatment of DENV-2 [35]. Pyrazine carboxamides, including T-705 and T-1106, might also be useful for the treatment of DENV, having shown efficacy in animal models of yellow fever virus (YFV) [58,59], West Nile virus (WNV) [60] and several other RNA viruses [61–63]. These compounds target the viral polymerase [61], but might also have other modes of activity. The compound β-D-2′-ethynyl-7-deaza-adenosine triphosphate targets DENV polymerase and causes chain termination during replication in cell culture [64].
NS5-associated methyltransferase activity [50] is another potential target for antiviral development. Methylation of the RNA cap of DENV by this enzyme occurs in two sequential steps and the inhibition of either or both methylation events will attenuate or abolish DENV replication, respectively [49]. Resistance to methyltransferase inhibitors is, however, a concern [44,65], and resistance mutations have been reported [66].
A virtual screening programme identified 263 compounds with favourable NS5 binding capability [67]. Out of these candidates, six were confirmed with 50% inhibitory concentration (IC50) values <100 μM and four with IC50<10 μM in in vitro assays. It will be interesting to see if methyltransferase inhibitors with in vitro activity are effective in animal models of DENV disease.
Depletion of the components necessary for nucleic acid synthesis might also inhibit DENV replication via indirect inhibition of viral polymerase. Treatment with brequinar, an inhibitor of dihydroorotate dehydrogenase, inhibits DENV RNA synthesis in vitro by reducing pyrimidine pools, and might also inhibit assembly or release of the virus [68]. The compound 2′-C-methylcytidine is also active against DENV in cell culture [69] and has been shown to work through a similar mechanism against the related flavivirus, YFV, in a hamster model [70]. Other compounds that inhibit DENV through the reduction of nucleotide pools include mycophenolic acid and ribavirin [16,17,71]. Ribavirin is a broad-spectrum antiviral compound that is active against many different flaviviruses in cell culture [72–74]. This compound has many modes of action, including inhibition of viral polymerase; however, against flaviviruses, the mode of action is largely caused by the inhibition of inosine monophosphate dehydrogenase with a resulting depletion of GTP pools [71]. Ribavirin has been shown to be active against YFV in a hamster model [75,76]. Also, this compound has been shown to have activity in various primary cells and cell lines infected with DENV [16–18]. Despite activity in cell culture, however, ribavirin did not show any activity in a mouse model of DENV disease [20] or in a primate model of DENV viraemia [19]. A similar phenomenon, where no activity was observed in animal models after observable activity in cell culture, has also been demonstrated in a hamster model of WNV disease [77]. Ribavirin 2′,3′,5′-triacetate was effective in a mouse model of DENV [78] and further investigation might be warranted. The ribavirin analogues, 1-β-D-ribofuranosyl-3-ethynyl-[1,2,4] triazole (ETAR) and 1-β-D-ribofuranosyl-4-ethynyl-[1,3] imidazole (IM18), have also shown activity in cell culture against several flaviviruses, including DENV [79]. Ribavirin monotherapy has no efficacy against HCV [80], but is quite effective as a part of combination therapy with IFN [81]. Such an approach might be useful in the treatment of DENV.
The NS5 protein is involved in stimulation of immune mediators, including IL-8 [52]. Some aspects of immune stimulation have been elucidated. For example, NS5 normally shuttles between the nucleus and the cytoplasm, which involves exportin proteins, and is essential for replication [51]. Inhibition of the exportin CRM1 through alteration of the nuclear localization sequences results in nuclear accumulation of NS5 and a decrease in IL-8 induction [82]. Improper stimulation of certain inflammatory cytokines has been implicated in severe disease caused by DENV [83–86]. Specifically, IL-8 has been shown to be significantly increased in cases of severe dengue in humans and might contribute to disease severity [87]. An immature form of NS5 binds to the signal transducer and activator of transcription-2 (STAT-2), causing degradation of this IFN pathway protein [47,88]. Inhibiting the protein–protein interactions of NS5 and key host proteins is therefore also a potential target strategy.
NS3
The NS3 protein, in combination with NS2, functions as a serine protease (Figure 2). This viral-encoded enzyme also functions as a helicase, presenting a second modality that could be targeted. Protease inhibitors have already been developed against other flaviviruses, such as HCV [89], reviewed in [90,91]. Work with in silico docking programmes has led to the identification of potential inhibitors of DENV NS3 protein, two of which were shown to have activity in cell culture models [92,93]. The recent structure determination of the active site of DENV protease will further allow for targeted design of inhibitors of this enzyme [94]. A cyclopeptide derived from the kalata B1 plant protein shows potent inhibition in in vitro assays against DENV protease activity [95].
A fluorescence-based quenching assay, which is sensitive and adaptable to other viral proteins, was recently developed to identify protease inhibitors [96]. Palmatine was shown to inhibit the protease of WNV and had broad-spectrum antiviral activity against several flaviviruses, including DENV, in cell culture [97]. Anti-helicase activity has been observed with the compound 1-N,4-N-bis [4-(1H-benzimidazol-2-yl)phenyl]benzene-1,4-dicarboxamide ((BIP)(4)B), with broad-spectrum activity against DENV, HCV and Japanese encephalitis virus [98].
Structural proteins
DENV structural proteins are important targets (Figure 2). Part of these proteins are found on the surface of virions and might be inhibited by therapeutic antibodies with broad-spectrum efficacy that target common epitopes shared by all four DENV serotypes [99]. A similar approach has been used for the treatment of WNV and a humanized monoclonal antibody has been shown to have potent activity in animal models [100–102]. Selective and potent antibodies, such as those discovered for WNV [103], might be generated for the treatment of severe DENV infections if the risk of ADE can be avoided [37]. However, neutralizing antibodies are often highly specific and might not be effective even for the treatment of closely-related homologous DENV serotypes, which was demonstrated recently in a mouse model [104].
The majority of antibodies generated during natural DENV infection have specificity for the prM protein and have been shown to have higher disease enhancement and lower neutralization properties [37]. This study also demonstrated that antibodies specific for the E protein generally have a better neutralization capacity, suggesting potential application of anti-E antibodies in the treatment of DENV. Similar conclusions were reached in a study evaluating the functional activity of a large panel of antibodies in cell culture as well as a BALB/c mouse model of intracranial infection. Several antibodies with specificity to various portions of the E protein were effective in the treatment of DENV, even when administered several days post-viral challenge [21,105]. Modification of the Fc portion of an antibody might eliminate the disease enhancement characteristics. Removal or inactivation of the Fc region reduced viraemia and tissue viral burden in an AG129 mouse model, demonstrating the potential for modified antibodies as potential therapeutic agents [38].
Agents with affinity to viral structural proteins can block fusion of the virion with the host cell membrane and several fusion inhibitors have been recently described. A recently described small molecule DENV inhibitor, compound
Viral RNA targets
Important structural–functional elements of the viral RNA also provide potential avenues for therapy that, if conserved across different DENV serotypes, could result in broad-spectrum inhibition. Various portions of the 5′- and 3′-ends of the DENV genome are required for replication and might represent one such target [110]. Small interfering RNAs (siRNAs) are effective against DENV and related flaviviruses in various cell culture and animal models [111–114]. The tagging of siRNA constructs with various peptides allows targeting to specific cell types or tissues, for example, dendritic cells [115]. Hammerhead ribozymes delivered by lentivirus vectors effectively reduce virus production from transduced mosquito cells [116], suggesting potential utility in vector control or vector-delivered therapy. Peptide-conjugated phosphorodiamidate morpholino oligomers have shown promise as anti-DENV agents and were found to inhibit the virus in cell culture [117].
The subgenomic flavivirus RNA (sfRNA) comprises a portion of the 3′ untranslated region and is formed after partial degradation of viral gRNA in host processing bodies [118]. The sfRNA of Kunjin, a related flavivirus, has been shown to contribute to pathogenesis in cell culture as well as in a mouse model of disease [118]. The sfRNA represents another potential target for antiviral therapy. Targeting the 3′ untranslated region could further disrupt cyclization of gRNA, which could further inhibit or impede viral replication [119,120].
Host cell targets
Host proteins play an integral role in many aspects of any viral replication cycle. Viruses, including DENV, have developed specific mechanisms to manipulate their hosts to replicate and evade immune effectors. The difficulty in targeting host proteins or pathways is the potential for toxicity or side effects. However, the potential of such compounds for screening purposes is greater because they are not DENV-specific, many are well-characterized and even compounds that have been approved by the US Food and Drug Administration might be available for testing against the virus. There are recent reviews that discuss options for targeting host cell proteins in detail [121,122]. In this review, we will only cover recent advances in host cell targeting methods for antiviral therapy (Table 1).
Cholesterol biosynthesis
Cholesterol has been shown to be integral to DENV replication. A decrease in cholesterol and very-low-density lipoprotein levels have been reported in individuals with more severe manifestations of DENV disease, which correlated with severe bleeding, hepatic dysfunction and death [123]. DENV replication was inhibited in different cell culture models after treatment with cholesterol pathway inhibitory compounds, including lovastatin, hymeglusin and zaragozic acid A [124]. This has implications in the early diagnosis of the disease and possibly in the treatment of DENV infection, although it is unclear if a similar effect will be seen in animal models.
Cytokines
Many flaviviruses, including DENV, have developed methods of blocking the IFN signalling cascade [47,125,126]. Stimulation of the IFN response or administration of exogenous IFN represents a host response manipulation that could be useful in the treatment of dengue. Recent work identified several IFN-stimulated gene products that are effective in suppressing various processes within the life cycle of DENV and WNV, including IFN-induced transmembrane protein-2 (IFITM2), IFITM3, viperin, IFN-stimulated gene 20 kDa protein (ISG20) and double-stranded RNA-activated protein kinase [127], which were similar to those involved in the response to HCV [128]. The addition of exogenous IFN reversed the effect of DENV inhibition of the cell-mediated host response [125], suggesting potential utility of IFN in the treatment of DENV. However, the AG129 mice used in DENV studies lack IFN-α/β and IFN-γ receptors, presenting an obvious impediment toward the testing of IFN and IFN stimulators against DENV in this model system. Treatment with IFN has been shown to be highly effective against HCV infection in humans [89,129] as well as in various animal models [75,77]. Treatment with pegylated IFN-α (Pegasys®) was effective in a primate model of DENV viraemia [130]. The potential use of IFN for the treatment of vascular disease is further supported by a recent study in DENV-infected human endothelial cells [131].
There is also potential for the use of IFN-γ as a therapeutic agent. IFN-γ treatment administered after virus infection was effective in treating DENV infection in dendritic cells, whereas treatment of these cells with IFN-α was ineffective because of inhibition of IFN type-1 signalling pathways [132]. It has been observed that IFN-γ treatment might increase the number of Fc receptors on promonocytic cells, which augments the uptake of DENV immune complexes in U937 cells [133], but not in peripheral blood mononuclear cells [134]. Overall, this demonstrates the varied effect of IFN-γ depending on the cell type [135].
The inflammatory cytokine, TNF-α, is implicated in triggering vascular leakage in human infection and disease models [28,136,137], suggesting inhibition of TNF stimulation could also be a viable option in the treatment of dengue. Interaction of DENV with C-type lectin domain family 5 member A (CLEC5A) on the surface of myeloid cells results in the phosphorylation of DNAX-activating protein 12 (DAP12), which then stimulates the release of proinflammatory cytokines including TNF-α. The release of TNF-α can be reduced when the CLEC5A–DENV interaction is inhibited [138]. Treatment of DENV-2-infected human umbilical vein endothelial cells with exogenous IFN reduced the production of TNF-α, and prevented virus-induced hyperpermeability [131].
Increased production of the high mobility group box 1 (HMGB1) protein (an important intracellular transcriptional regulator) has been shown to reduce DENV titres in infected dendritic cells [139], potentially suggesting another host target for the inhibition of replication. However, once released extracellularly, HMGB1 acts as a potent proinflammatory protein that might contribute to immunopathogenesis [140].
Other host cell targets
Targeting the glycoprotein-processing pathway of the host has been shown in several models to inhibit DENV. Removal of E protein glycans at residues 153/154 and 67 has been shown to enhance DENV infectivity and decrease efficient release in mammalian cells [141]. A recent review covers much of the salient information on the topic of glycosylation inhibition [122], therefore, only a brief discussion of compounds with activity against DENV is included here. Castanospermine and its derivative, 6-O-butanoyl castanospermine, are derived from the castor bean. These compounds are α-glucosidase inhibitors that disrupt N-glycosylation. They are active in rodent models of dengue disease [20,22]. N-nonyl-deoxynojirimycin, an iminosugar derivative similar to castanospermine, was also effective in reducing disease parameters in an AG129 model [20]. Geneticin is an aminoglycoside that has shown activity against DENV in cell culture, but is inactive against YFV [142]. In the same study, other aminoglycosides, including gentamicin, kanamycin and a guanidinylated geneticin, showed no activity against DENV. Mannose-binding lectin was recently shown to bind the surface of DENV, resulting in neutralization and enhanced clearance of the virus in a mouse model [143]. Host-derived surface components of DENV are useful targets for antiviral therapy.
Cyclophilins regulate cytokine production and have been shown to be involved in the efficient replication of flaviviruses [88] through an interaction with NS5 [144]. Cyclosporine and cyclosporine derivatives and analogues have been shown to be useful in the treatment of flaviviruses, including DENV, in various cell culture [88,145,146] and in the treatment of human HCV infection [147], suggesting potential for the use of such compounds in the treatment of dengue.
Y box-binding protein-1, has been shown to bind to the 3′ stem loop of DENV mediating an antiviral effect in cell culture, which is at least partially caused by the inhibition of virus translation [148].
Infection with DENV reduces intracellular glutathione concentrations, which results in activation of nuclear factor-κB by E protein domain III [149] and increased virus production [150]. The addition of exogenous glutathione reverses this effect and inhibits viral production in cell culture (Table 1).
Many intracellular proteins are used in viral replication and the correct processing of viral proteins. For example, the heat-shock proteins are involved in the life cycles of a diverse array of intracellular pathogen, and inhibition of this group of proteins might result in decreased replication of such pathogens [151]. These proteins are integral in DENV replication [152] and also play an important role in attachment and entry [153,154]. Polypyrimidine tract-binding protein is also involved in DENV replication and knockdown of this protein by siRNA targeting inhibits the production of infectious virus, although this was not observed for YFV [155]. The ubiquitin–proteasome pathway was recently shown to be important in the antiviral response [156], which could possibly be enhanced to improve viral clearance as a means of antiviral therapy.
Other anti-DENV agents that have been recently discovered in cell culture or animal models, and which are not discussed in the text, are included in Table 1. These compounds have diverse mechanisms of action, although many need to be tested in animal models to confirm cell culture activity in vivo. These compounds are included in the table for reference purposes.
Conclusions
Unfortunately, no antivirals are currently approved for the treatment of DENV in human patients. Great efforts are being made in laboratories across the world to discover an effective, safe and readily available therapy to alleviate disease caused by DENV. This effort has been accelerated through the use of various useful small animal models that have been recently developed and improved. With many potential candidates, mouse models of disease and with support of government and private funding, the prospect of an effective antiviral or therapeutic compound for the treatment of dengue is encouraging.
Footnotes
The authors declare no competing interests.