
Abstract
Background:
Macrophages serve as a depot for HIV type-1 (HIV-1) in the central nervous system. To efficiently target macrophages, we developed nanocarriers for potential brain delivery of activated nucleoside reverse transcriptase inhibitors (NRTIs) called nano-NRTIs.
Methods:
Nanogel carriers consisting of poly(ethylene glycol) (PEG)- or Pluronic-polyethylenimine (PEI) biodegradable networks, star PEG-PEI or poly(amidoamine) dendrimer-PEI-PEG dendritic networks, as well as nanogels decorated with brain-targeting peptide molecules, specifically binding to the apolipoprotein E receptor, were synthesized and evaluated. Nano-NRTIs were obtained by mixing aqueous solutions of zidovudine 5′-triphosphate or didanosine 5′-triphosphate and nanocarriers, followed by freeze-drying. Intracellular accumulation, cytotoxicity and antiviral activity of nano-NRTIs were monitored in monocyte-derived macrophages (MDMs). HIV-1 viral activity in infected MDMs was measured by a reverse transcriptase activity assay following treatment with nano-NRTIs. Mitochondrial DNA depletion in MDMs and human HepG2 cells was assessed by quantitative PCR.
Results:
Nanogels were efficiently captured by MDMs and demonstrated low cytotoxicity, and no antiviral activity without drugs. All nano-NRTIs demonstrated high efficacy of HIV-1 inhibition at drug levels as low as 1 μmol/l, representing a 4.9- to 14-fold decrease in 90% effective drug concentrations as compared with NRTIs, whereas 50% cytotoxicity effects started at 200x higher concentrations. Nano-NRTIs with a core-shell structure and decorated with brain-targeting peptides displayed the highest antiviral efficacy. Mitochondrial DNA depletion, a major cause of NRTI neurotoxicity, was reduced threefold compared with NRTIs at application of selected nano-NRTIs.
Conclusions:
Nano-NRTIs demonstrated a promising antiviral efficacy against HIV-1 in MDMs and showed strong potential as nanocarriers for delivery of antiviral drugs to macrophages harbouring in the brain.
Introduction
Latent reservoirs of HIV type-1 (HIV-1) infection represent a significant obstacle to the clearance of the virus from infected patients and might result in the emergence of drug-resistant HIV-1 strains, and a productive infection later in the disease progression. Macrophages are widely recognized as latently infected viral reservoirs and can be considered as target cells for chemotherapy [1]. Significant work has been performed to establish antiviral activity of clinically approved nucleoside reverse transcriptase inhibitors (NRTIs) in macrophages; however, the availability of many NRTIs to treat macrophages in the central nervous system (CNS) has been restricted by mechanisms of drug efflux in the blood brain barrier (BBB) [2]. Additionally, treatment with therapeutic doses of NRTIs was associated with chronic neurotoxicity and the loss of neurons, which already represents a rapidly growing problem with current highly active antiretroviral therapy (HAART) [3].
Targeted drug delivery and nanotechnology recently emerged as alternative approaches to reach HIV-1 in its natural reservoirs [4]. Although direct drug modification with brain-targeting moieties was found to be efficient in overcoming the BBB in a number of applications, it might result in a significant reduction of drug activity [5]. Fortunately, nanoparticles could safely deliver significant drug payloads and are efficiently captured by various types of macrophages following systemic administration [6]. Targeted nanoparticles also held high potential for drug delivery across the BBB to the brain [7]. Various types of nanocarriers were proposed for delivery of antiretroviral drugs, including NRTIs, although mostly in a non-phosphorylated form [8]. However, because 5′-triphosphates (5′-TPs) of NRTIs are the active drug species and are chain terminators of DNA or RNA synthesis, delivery of these compounds into HIV-1-infected cells might significantly improve the efficacy of antiviral chemotherapy compared with current NRTIs [9]. Initial attempts at liposomal delivery of 5′-TPs of NRTIs were published in the 1990s [10,11]. Red blood cells were also proposed as drug delivery vehicles for 5′-TPs of nucleoside analogues [12]. Lately, soft cationic nanogels [13,14] and polymeric nanocapsules [15,16] have been successfully evaluated as other types of nanocarriers for 5′-TPs of cytotoxic and antiviral drugs. Nanogels demonstrated efficient cellular accumulation and cytosolic drug release, and their surfaces could be decorated with various vector molecules, including brain-targeting moieties [17]. The major purpose of this study was an anti-HIV application of these innovative nanocarriers, which have previously demonstrated high efficacy in drug delivery into the brain, in order to overcome the inability of many NRTIs to cross the BBB. Here, we demonstrated, for the first time, an efficient application of hydrophilic cationic nanogels for delivery of activated antiviral drugs, 5′-TPs of NRTIs, and inhibition of HIV-1 replication in macrophages. We also found that nano-NRTIs have been generally less toxic to macrophages, major carriers of HIV-1 in the brain, than free NRTIs. In addition, as compared with free NRTIs, nano-NRTIs proved to have reduced mitochondrial toxicity, which is considered a major cause of neurotoxicity. Other aspects of chronic and neuronal toxicity of nano-NRTIs are currently under investigation. We hypothesize that nanogels can also be more efficient drug carriers for in vivo transport of NRTIs across the BBB via targeting by brain-specific peptides, in order to reach latent HIV-1 reservoirs in the CNS.
Methods
Materials
Most of the reagents and solvents, including a hydroxypropyl poly(amidoamine) (PAMAM) dendrimer G(5) and polyethylenimine (PEI) were purchased from Sigma–Aldrich (St Louis, MO, USA) and used without additional purification. [5-3H]-thymidine 5′-TP (20–25 Ci/mmol) was purchased from Moravek Radiochemicals (Brea, CA, USA). Pluronic F-68 was obtained as a gift from from BASF (Partispany, NJ, USA). The bifunctional poly(ethylene glycol) (PEG) linker with malemide and N-hydroxysuccinimide moieties (MAL-PEG5000-NHS) was purchased from GenKem Technology USA (Allen, TX, USA). 75-Armed star PEG 423 was obtained from Shearwater, Inc. (Huntsville, AL, USA). ATP BODIPY FL was purchased from Invitrogen (Carlsbad, CA, USA). Zidovudine (AZT) and didanosine (ddI) were obtained from AK Scientific (Mountain View, CA, USA). Mouse apolipoprotein E (ApoE) receptor-binding peptide (AP), CGLRKMRKRLMR with C-amide protection, was custom synthesized by Biomer Technologies (Pleasanton, CA, USA) and purified by semi-preparative reverse-phase HPLC [12].
Chemistry
Synthesis of nanogels
Cationic nanogel networks PEG-cl-(ss)PEI (NG1) and Pluronic F68-cl-(ss)PEI (NG2) were synthesized starting from biodegradable disulfide-bridged polyethylenimine, (ss)PEI, using previously described methods [13,18]. Star PEG (75 arms, molecular weight [Mw] 450,000) was activated by 1,1′-carbonyldiimidazole and then modified with an excess of branched PEI (Mw 2,000) to obtain a cationic star PEG-g-PEI network (NG3). Carboxylated PAMAM dendrimer G(5) was activated by water-soluble N-(3-dimethylaminopropyl)-N′-ethyl carbodiimide hydrochloride and then modified with an excess of PEG-g-PEI conjugate (mPEG [Mw 5,000]/branched PEI [Mw 1,200]) to obtain a cationic PAMAM-PEI-g-PEG network (NG4). Swollen nanogel networks are schematically shown in Figure 1A. All nanocarriers were fractionated by size exclusion chromatography on a column with Sephacryl S-300 (1.5×60 cm; GE Healthcare Life Sciences, Piscataway, NJ, USA), using an elution buffer containing 0.2 M sodium chloride, 20 mM sodium acetate (pH 6), 20% ethanol and a refractive index flow detector (ChromTech, Apple Valley, MN, USA). Fractions containing nanogel dispersions were collected, desalted by dialysis against water, filtered through a 0.22 μm sterile filter and freeze-dried.
AP-modified nanogels were prepared as follows: nanogel NG1 (25 mg) was dissolved in 0.5 ml water, then a solution of a MAL-PEG5000-NHS linker (10 mg/0.5 ml) was added and the mixture was incubated for 2 h at 25°C. Without isolation, a twofold excess (5 mg) of AP was added in 0.5 ml dimethylformamide and the pH was reduced to 6 by 3 M sodium acetate (pH 5.5). The reaction was continued overnight at 4°C. Peptide-modified nanogels were isolated by size-exclusion chromatography using a Sephacryl S-300 column (GE Healthcare Life Sciences) equilibrated in 0.2 M sodium chloride, 20 mM sodium acetate (pH 6), in 20% ethanol. The product, ApoE-NG1, was desalted by dialysis (Mw cutoff 8–12 kDa) and lyophilized. Peptide content was determined using an automatic aminoacid analyser (Applied Biosystems, Foster City, CA, USA).
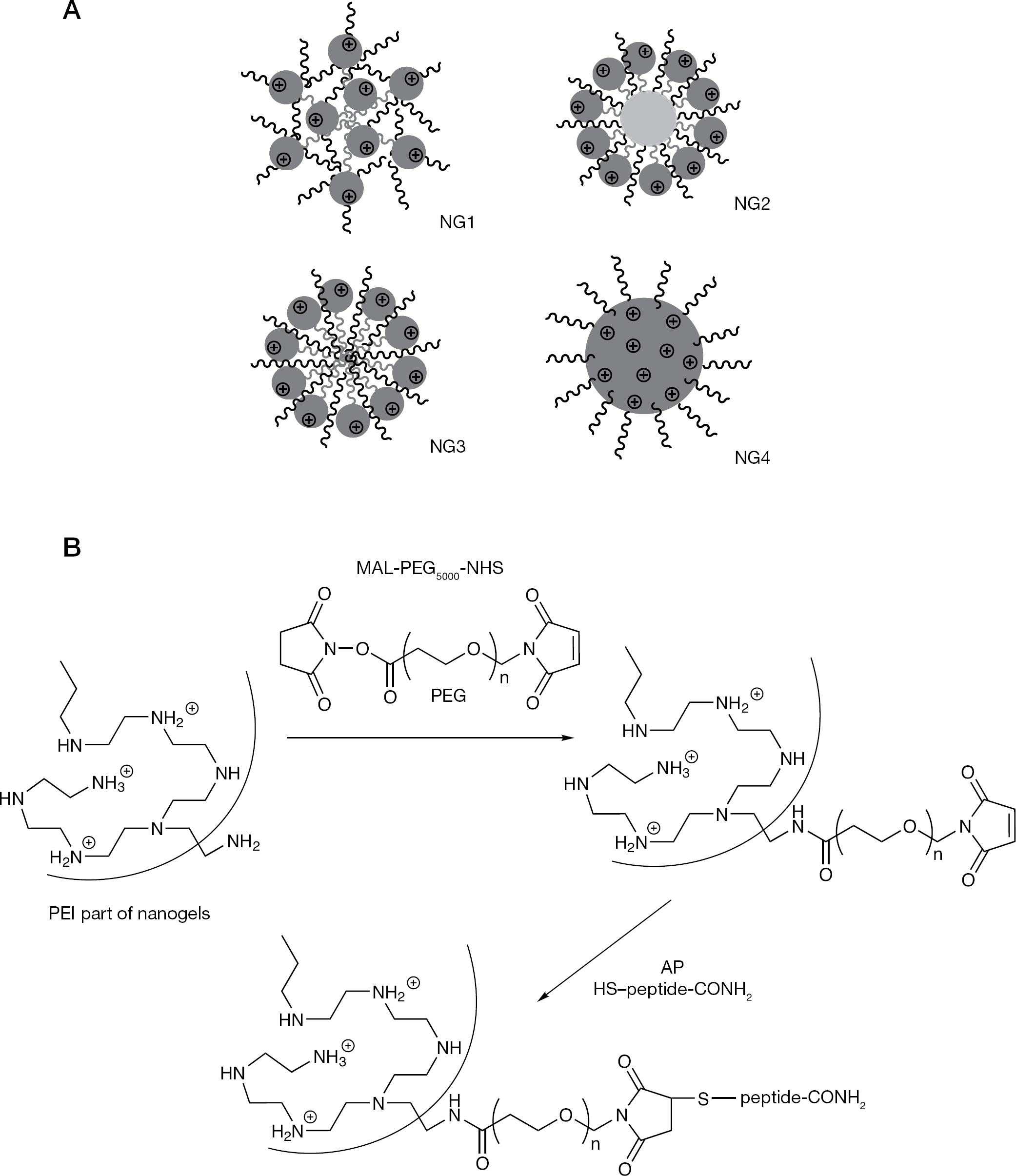
Nanocarriers used in preparation of nano-NRTIs and modification by brain-targeted peptide
Synthesis of triphosphates
AZT and ddI were phosphorylated and triphosphorylated using a previously reported one-batch method [19]. The 5′-TP products, AZT-TP and ddI-TP, were purified by anion exchange chromatography on DEAE-Sephadex A-25 (Sigma–Aldrich) with a gradient of ammonium acetate in 20% ethanol, desalted by dialysis (Mw cutoff 500 Da) using Biotech SpectraPor tubes (Spectrum Laboratories, Rancho Dominguez, CA, USA) and lyophilized. The HPLC and ultraviolet-purity of these compounds were analysed by analytical ion-pair HPLC, alkaline phosphatase hydrolysis and ultraviolet absorbance at 260 nm as previously described [19]. Spectral characteristics (proton and phosphorus NMR) and analytical HPLC profiles of these products can be found in Additional file 1.
Preparation of nano-NRTIs
Briefly, equal volumes of aqueous solutions of nanogel (80 mg/ml) and 5′-TP (20 mg/ml) were mixed together and incubated for 60 min in an ice bath. The forming nano-NRTIs were separated from non-complexed nucleosides by gel-filtration on a NAP-10 column (GE Healthcare Life Sciences), equilibrated in deionized water and freeze-dried. The calculated drug content in nano-NRTI formulation (%) was based on the measured ultraviolet absorbance at 260 nm and molar extinction coefficients (ɛ260, M−1) of the corresponding nucleosides: AZT 9,700, ddI 8,100 and cytidine 9,000 (Table 1). Nano-NRTIs were stored as dry formulations, refrigerated and used after dissolving in sterile water before treatment.
Properties of nanogels and nano-NRTIs
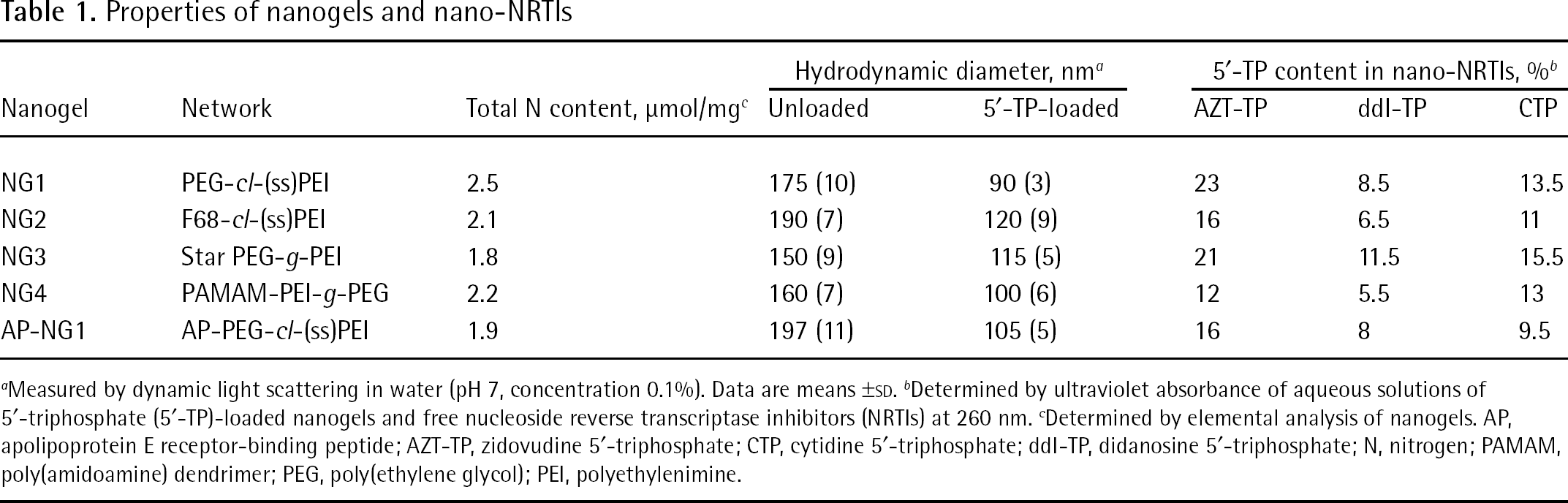
Measured by dynamic light scattering in water (pH 7, concentration 0.1%). Data are means ±SD.
Determined by ultraviolet absorbance of aqueous solutions of 5′-triphosphate (5′-TP)-loaded nanogels and free nucleoside reverse transcriptase inhibitors (NRTIs) at 260 nm.
Determined by elemental analysis of nanogels. AP, apolipoprotein E receptor-binding peptide; AZT-TP, zidovudine 5′-triphosphate; CTP, cytidine 5′-triphosphate; ddI-TP, didanosine 5′-triphosphate; N, nitrogen; PAMAM, poly(amidoamine) dendrimer; PEG, poly(ethylene glycol); PEI, polyethylenimine.
Virology
Primary human monocyte isolation and HIV type-1 infection
Monocytes were obtained from leukopheresis of HIV-1, HIV type-2 and HBV-seronegative donors and purified by countercurrent centrifugal elutriation as previously described [20]. Cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated pooled human serum and 1% glutamine (Sigma–Aldrich), 10 mg/ml ciprofloxacin (Sigma–Aldrich) and 1,000 U/ml of purified recombinant human macrophage colony-stimulating factor (R&D Systems, Minneapolis, MN, USA). A macrophage-tropic viral strain (HIV-1ADA) at a multiplicity of infection (MOI) of 0.01, was amplified as previously described [21]. This laboratory-adapted strain was selected following a prior extensive analysis of macrophage function and neurotoxicity based on strain differences. The levels of viral growth of the HIV-1ADA strain were nearly uniform and not dependent on host cell differences. This ensured that the data acquired would be reproducible from one experiment to another, regardless of the macrophage donor.
Cellular uptake
Nanogels were tagged with rhodamine (Rho) isothiocyanate and purified using a NAP-10 column (GE Healthcare Life Sciences) as described previously [17]. Fluorescent nanogels were loaded with a model cytidine 5′-TP (CTP; Table 1) to neutralize positive charges and were isolated on a NAP-10 column (GE Healthcare Life Sciences). Nanogel solutions were dispensed in full cellular medium to a 96-well plate to obtain the concentration 20 μg/ml. At appropriate time intervals, equal volumes of the monocyte-derived macrophage (MDM) suspension (1×106 cells/ml) in full medium were added. The treated cells were washed with cold phosphate-buffered saline (PBS) and centrifuged twice, resuspended with propidium iodide (10 μl) and incubated for 15 min at 4°C before the flow cytometry analysis. The percentage of live cells and mean fluorescent intensity was measured using a fluorescence-activated cell sorting (FACS) array bioanalyzer (BD Biosciences, Franklin Lakes, NJ, USA).
For confocal microscopy studies, a Rho-NG1 was loaded with a fluorescent 5′-TP, ATP BODIPY FL, as described above for CTP. MDMs were treated separately with a Rho-NG1/ATP BODIPY FL formulation, a Rho-NG1 alone (10 μg/ml) and ATP BODIPY FL (1 μg/ml) in full cellular medium for 1 h at 37°C. MDMs were washed several times with cold PBS, centrifuged and fixed with paraformaldehyde. Confocal microscopy was performed in covered chamber slides using a Zeiss 410 Confocal Laser Scanning Microscope (Carl Zeiss, Iena, Germany) equipped with an argon–krypton laser (Confocal LSM Core Facility, University of Nebraska Medical Center Omaha, NE, USA).
Antiviral effects evaluation
Human MDMs were cultured for 7 days in 96-well plates, changing culture medium every 2 days. Nano-NRTIs and NRTIs were dissolved in sterile deionized water to obtain 20x stock solutions. On day 7, 10 μl of 20x stock solutions of free NRTIs or nano-NRTIs with an equivalent drug concentration of 0.32 mg/ml were mixed in sterile 96-well plates with 190 μl of complete macrophage culture medium. Serial 1/2 dilutions (100 μl) were prepared corresponding to AZT and ddI concentrations from 16 to 0.25 μg/ml. The old medium (100 μl) from cells was then removed and supplemented with the serial dilutions of NRTIs or nano-NRTIs in full medium (100 μl). Plates were incubated for 2 or 4 h at 37°C and in 5% CO2. After incubation, supernatants were carefully aspirated, cells were washed with 200 μl of PBS, and the fresh media containing 0.01 MOI of HIV-1ADA was added for overnight (16 h) infection. MDMs were then washed, supplemented with a fresh medium and incubated for an additional 7 days. On days 5 and 7 post-infection, supernatants were collected for reverse transcriptase (RT) activity assays. Each plate contained infected (non-treated with drug) and uninfected (treated with culture medium alone) cell controls. All drug concentrations and controls were analysed in five parallels.
To measure HIV-1 replication, RT activity was determined by incubating 10 μl of infected sample media with a reaction mixture consisting of 0.05% Nonidet P-40 (Sigma–Aldrich) and [3H]-thymidine 5′-TP (2 Ci/mmol; Moravek Radiochemicals) in Tris-HCl buffer (pH 7.9) for 24 h at 37°C on days 5 and 7 post-infection [21]. Radiolabelled DNA was precipitated on paper filters in an automatic cell harvester (Skatron, Sterling, VA, USA) and incorporated activity was measured by liquid scintillation spectroscopy. The cytotoxicity of NRTIs and nano-NRTIs to MDMs was determined on day 7 using an MTT assay as previously described [22]. The mean values of optical density at 490 nm for non-treated controls was used as a normalization factor, and the other cytotoxicity data has been converted correspondingly. All observed HIV-1 RT activity values were divided by these normalization factors, and the adjusted HIV-1 RT activity expressed as DNA incorporated radioactivity (counts per min per ml) was plotted as a function of drug concentrations. Antiviral efficacy of NRTIs and nano-NRTIs was expressed as the effective drug concentration that inhibited HIV-1 RT activity by 90% (EC90) and was determined from concentration–effect curves generated using GraphPad Prism version 4.03 (GraphPad Software, San Diego, CA, USA).
Mitochondrial DNA analyses
The effect of NRTIs and nano-NRTIs on the content of mitochondrial DNA (mtDNA) was measured following the prolonged treatment of MDMs using an SYBR green dye real-time PCR method. Cells were treated with sterile solutions of AZT, AZT plus ddI, AZT-TP or AZT-TP plus ddI-TP-loaded nanogels at two drug concentrations (15 and 30 μg/ml) in full medium for 4 h at 37°C on days 1, 4, 7 and 10, and analysed on day 14. HepG2 cells (American Type Culture Collection, Manassas, VA, USA) were cultured as previously described [18]. The medium was renewed after every treatment. After trypsinolysis and treatment with RNase, DNA samples were isolated using the FujiFilm Quickgene DNA Tissue Kit S and Mini80 extraction cartridges (Autogen, Holliston, MA, USA). The obtained DNA samples were quantified and had absorbance ratios at 260 and 280 nm in the recommended range.
Human cytochrome b gene primers (mtDNA) 5′-CCAACATCTCCGCATGATGAAAC-3′ (forward) and 5′-GTGGGCGATTGATGAAAAGG-3′ (reverse), and β-actin gene primers nuclear DNA (nuDNA) 5′-AACACCCCAGCCATGTACGT-3′ (forward) and 5′-TCTCCTTAATGTCACGCACGA-3′ (reverse) were used in the PCR analysis [23]. All primers were synthesized and cartridge-purified in the Eppley Molecular Biology Core Facility, University of Nebraska Medical Center (Omaha, NE, USA). SYBR green dye real-time PCR reactions were performed in quadruplicates using the MiniOpticon Real-Time PCR detection system (BioRad, Hercules, CA, USA). The PCR reaction mixtures contained an equal amount of DNA (10–20 ng in 5 μl), 5 μl of 2 μM primer mix and 10 μl of SsoFast Evagreen Supermix (BioRad). PCR conditions were as follows: an initial denaturation at 98°C for 2 min, followed by amplification at 95°C for 15 s, and 60°C for 1 min (40 cycles) and a melting/annealing cycle. The quality of primers and conditions of amplification were checked at different dilutions of isolated DNA, and the efficacy of amplification was close to 0.9. The results were calculated as the content of mtDNA normalized to the content of nuDNA: m=2−C(t)mt/2−C(t)nu, where C(t)mt and C(t)nu are the quantification cycles observed for mtDNA and nuDNA in each sample. The normalized mtDNA content (m) in non-treated cells was taken as 100%, and all obtained data were expressed as the percentage change of the mtDNA content compared with non-treated cells.
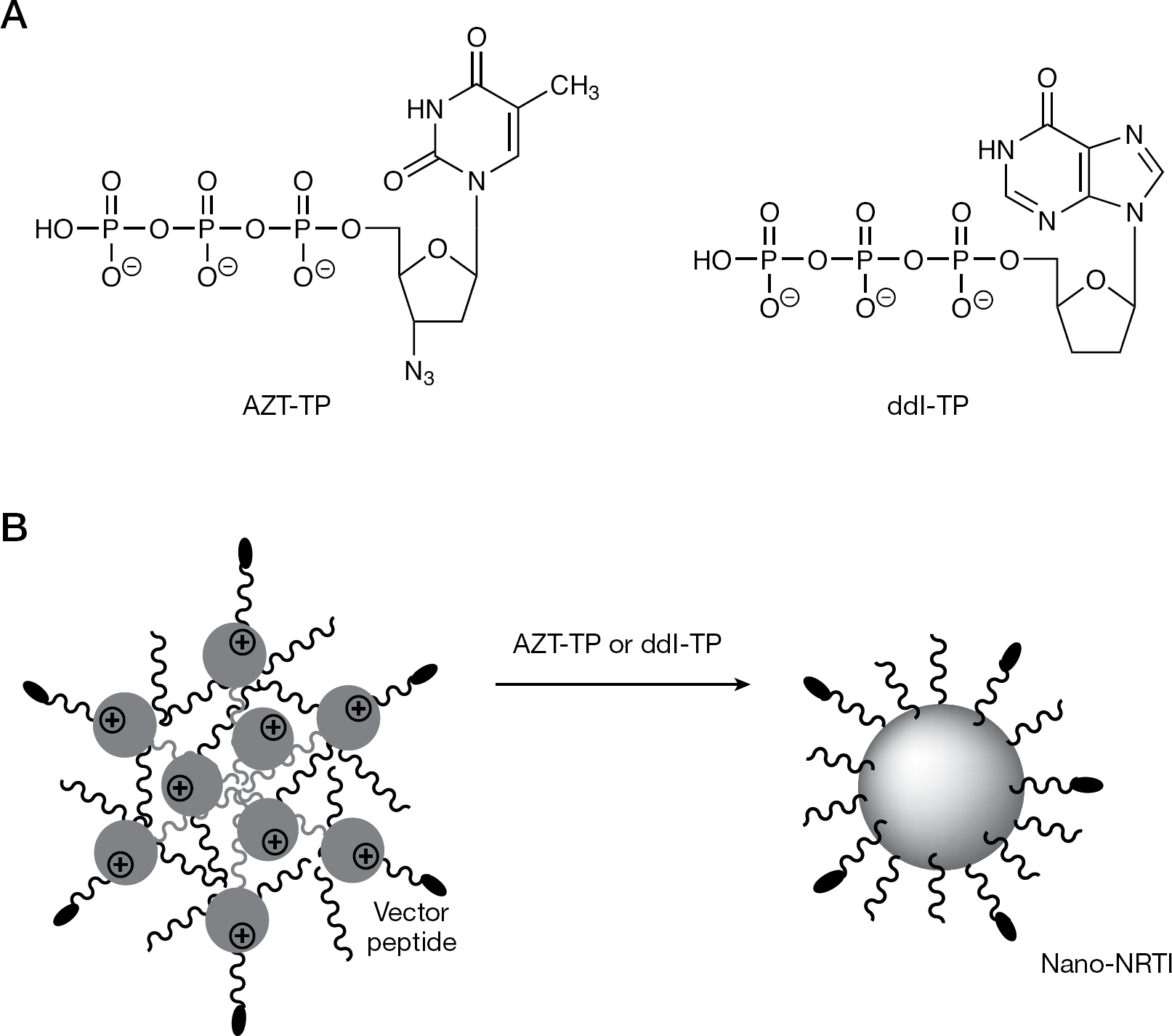
Formulation of nano-NRTIs
Statistical analyses
All experiments were performed at least 2–3x in duplicates. Data are presented as mean ± standard error of the mean. The experimental data were analysed using unpaired Student's t-tests. Results were considered significant at P<0.05, with a two-tailed test.
Results
Preparation of nano-NRTIs
Nanogel networks with an even spatial distribution of cationic and neutral polymeric molecules (NG1), a layered structure and a cationic polymer in the outer layer (NG2 and NG3), and a cationic core-neutral shell structure (NG4) were prepared in order to compare their efficacy in the delivery of 5′-TPs of antiviral nucleoside analogues to macrophages (Figure 1A). All these networks had a high ratio of neutral to cationic polymers to ensure lower cytotoxicity of nanocarriers, and the mean total nitrogen content measured by elemental analysis was 2.2 μmol/mg nanogels (Table 1). Biodegradable segmented PEI with disulfide bridges was used in the synthesis of nanogels NG1 and NG2 [18]. Following the reduction of disulfide bridges inside the cells, these nanogels are capable of quick degradation to non-toxic PEG/Pluronic-g-PEI conjugates (Mw<30 kDa), which can be effectively removed by the kidneys. For modification with a brain-specific peptide vector, AP [24], nanogel was initially treated with a bifunctional PEG linker (Figure 1B). Efficiency of the AP-coupling reaction was usually >70%. The maleimide-thiol linkage between PEG and peptide is stable in vivo, as well as the protected linear peptide (C-amide) attached to the PEG molecule. The peptide-modified nanogels were previously shown to efficiently bind brain capillary endothelial cells in culture and accumulate in the brain in vivo [25].
All nanogels were readily swollen in water, forming stable transparent dispersions after ultrasonication. Antiviral drugs AZT and ddI in active phosphorylated forms, AZT-TP and ddI-TP, have been used for preparation of nano-NRTIs in the study (Figure 2A). Nucleoside 5′-TP molecules easily penetrated into nanogel pores and spontaneously bound to the cationic PEI chains in the network after simple mixing of both aqueous solutions, forming compact nano-NRTI particles with a dense drug-loaded core and PEG molecules in the protective outer layer (Figure 2B). The hydrodynamic diameter of all nanogels in water was <200 nm. The dispersed nanogel networks demonstrated a very low light scattering efficacy in aqueous solution prior to association with 5′-TP. Upon association with 5′-TP, the nanogel networks formed compacted drug-loaded core-shell particles with a mean 1.5–2-fold smaller diameter (Table 1). Usually, a condensed core of 30–50 nm in diameter formed, therefore a protective PEG layer could be twice as thick [13]. Nano-NRTIs were separated from an unbound 5′-TP by gel filtration; drug loading capacity was calculated based on ultraviolet absorbance and molar extinction coefficients of each nucleoside (Table 1). Theoretical drug binding in nanogels calculated on the basis of their total nitrogen analysis was very close to the observed average value (13% or 0.28 μmol/mg).
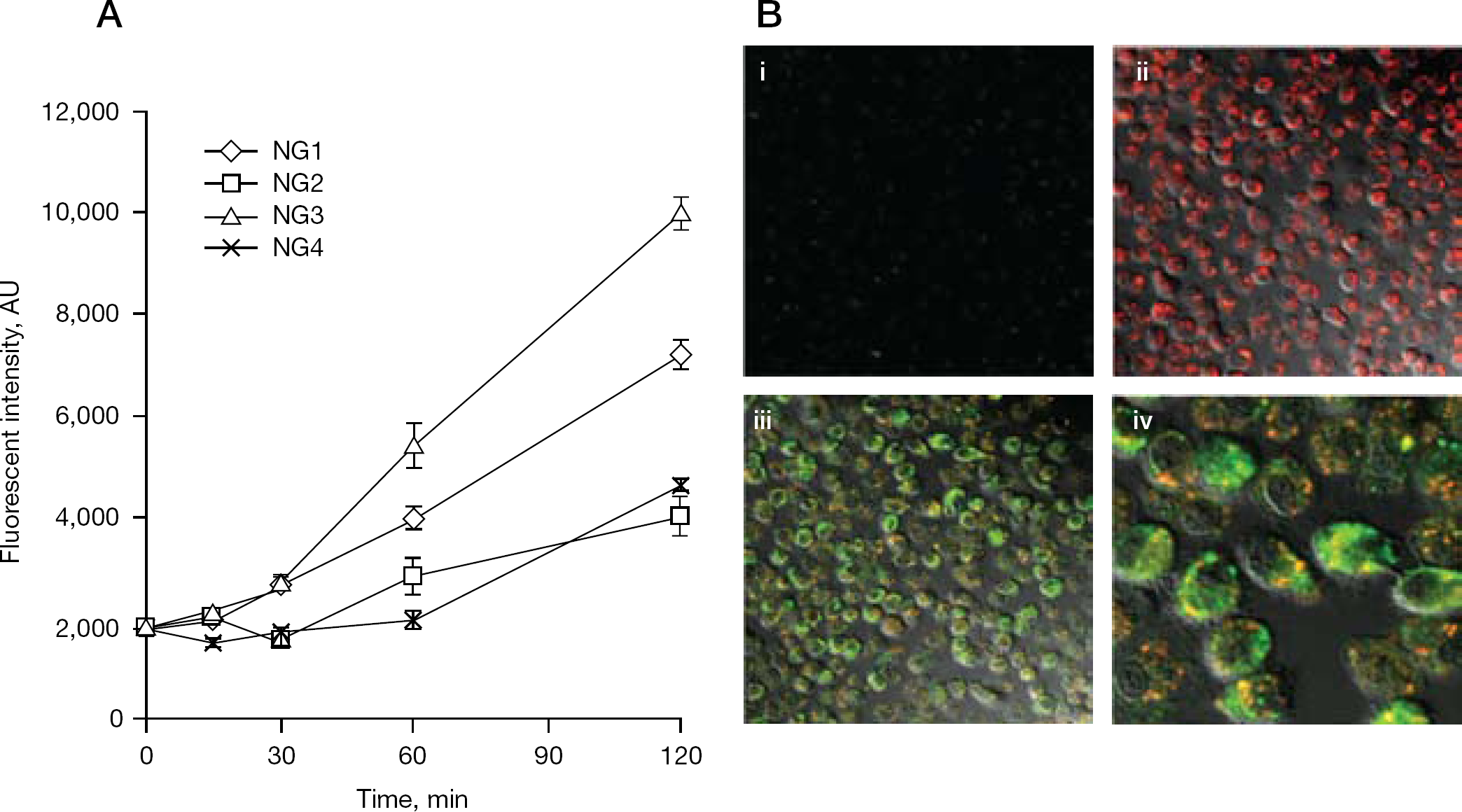
Cellular accumulation of nanogels in MDMs
Cellular accumulation
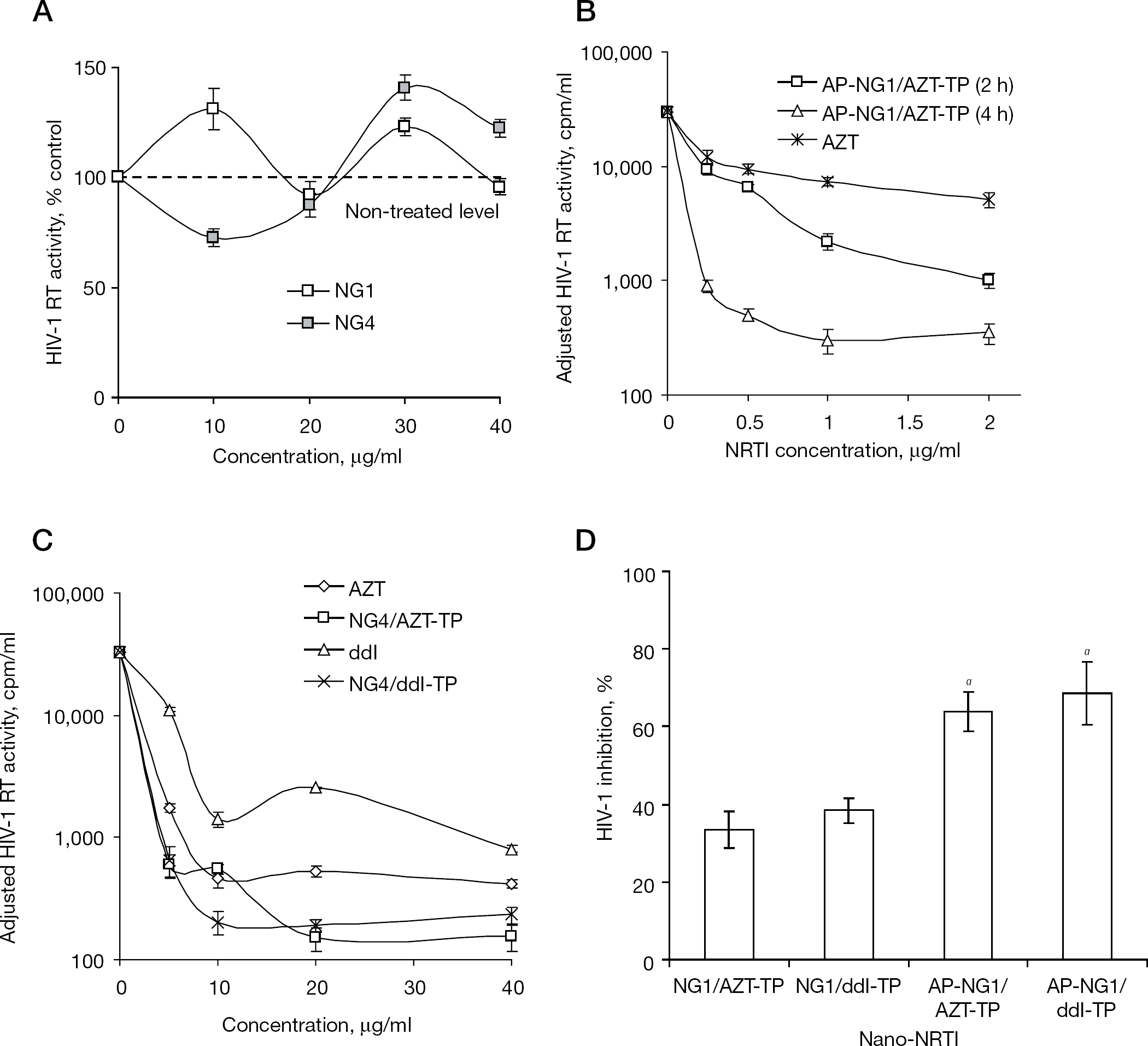
Capture of Rho-labelled nanogels was studied in cultured macrophages (Figure 3). Initially, Rho-NG1 was loaded with a fluorescent 5′-TP, ATP BODIPY FL, and the obtained double-labelled nanoformulation (100 μg/ml) was used to treat MDMs for 2 h at 37°C. No cytotoxicity was evident during the treatment of MDMs (the 50% inhibitory concentration [IC50] of the nanogel NG1 was 0.3 mg/ml). Fast and efficient capture of free and ATP BODIPY FL-loaded fluorescent nanogel particles was observed by confocal microscopy, showing their prevalent cytoplasmic accumulation. In contrast to the Rho-labelled nanogel, the accumulation level of free ATP BODIPY FL in macrophages was noticeably lower (Figure 3B, panels i and ii). Superposition pictures of both labels after incubation of ATP BODIPY FL-loaded fluorescent nanogel with MDMs demonstrated an intensive process of intracellular release of the 5′-TP from nanogels in the cytoplasm, which is visible as a diffuse green fluorescence (Figure 3B, panels iii and iv). Colocalized 5′-TP and nanogel are visible in yellow dotted areas, evidently encompassed in endosomes. These results directly confirm that nanogel-delivered 5′-TP can be available in free form for virus inhibition in macrophages very quickly after the beginning of the treatment.
The capture of all types of nanogels by MDMs has been investigated by flow cytometry. Here, we observed a significant difference in the cell accumulation rate among the various Rho-labelled nanogels (10 μg/ml). The fastest capture was observed for nanogel NG3, which was followed by NG1, whereas NG2 and NG4 showed equally lower capture efficacy (Figure 3A). These differences can be explained by various PEG densities and charge shielding in these nanogels. The first pair (NG1 and NG3) seems to have formed networks with a positive charge on their surface and a less uniform and dense outer PEG layer, whereas the second pair (NG2 and NG4) formed more dense particles with the cationic core and an expanded outer PEG layer. The first type of nanogel architecture should simplify binding with serum proteins (opsonization) and the consequent recognition by macrophages, but the second type of nanogel architecture has more protection against protein coating and the corresponding capture by macrophages. It is worth noting, that although the macrophage capture of NG3 is approximately twofold higher and NG1 50% higher, compared with NG2 and NG4, the overall antiviral activity might depend not only on the nanogel capture, but also on efficacies of both nanogel uptake and drug release processes, as shown in the following experiments on the antiviral efficacy of nano-NRTIs in HIV-1-infected MDMs.
Antiviral efficacy
Nano-NRTIs were examined for their ability to inhibit HIV-1 replication in an in vitro model system using infected MDMs. The cells were pretreated for 2–4 h with either free drug (AZT or ddI) or nano-NRTIs containing AZT, ddI or AZT plus ddI at concentrations from 0.25 to 16 μg/ml (0.5–30 μg/ml or 1–60 μM by 5′-TP), washed and inoculated with HIV-1ADA at an MOI of 0.01. Culture supernatants were monitored for viral replication by measuring RT activity by inclusion of 3H-thymidine 5′-TP on days 5 and 7 post-infection using a micro-RT assay. The RT data were normalized for cytotoxicity of free- and nano-NRTIs determined using an MTT assay. In preliminary experiments, we found that preincubation time had a visible effect on antiviral efficacy of nano-NRTIs; the strongest antiviral effect was obtained at a longer incubation time. It is evident from Figure 3 that cellular uptake of nanogels did not reach a plateau after 2 h of incubation; a longer time should increase the internalization of nanogels and also the release of activated drug molecules from nanogels into the cytoplasm. Because of the different cytotoxicities of nanogels, we compared the antiviral effect of nano-NRTIs, mostly at a 2 h preincubation time, in order to reduce any effects associated with cytotoxicity of nanocarriers. The nanogels, by themselves, displayed no visible antiviral activity (Figure 4A). Most of the studied nano-NRTIs were relatively non-cytotoxic to MDMs and showed no signs of cytotoxicity up to concentrations of 100–200 μg/ml. Both nano-AZT-TPs and nano-ddI-TPs demonstrated more efficient suppression of the RT activity than corresponding NRTIs (AZT and ddI) at equivalent drug concentrations (0.25, 0.5, 1 and 2 μg/ml) by comparison of the EC90 values calculated from these dose–effect curves adjusted for cytotoxicity (Figure 4B and 4C). The EC90 data presented in Table 2 has been calculated from three separate experiments where these dose–effect curves were obtained.
Antiviral effect of the treatment of HIV-1-infected MDMs with nano-NRTIs
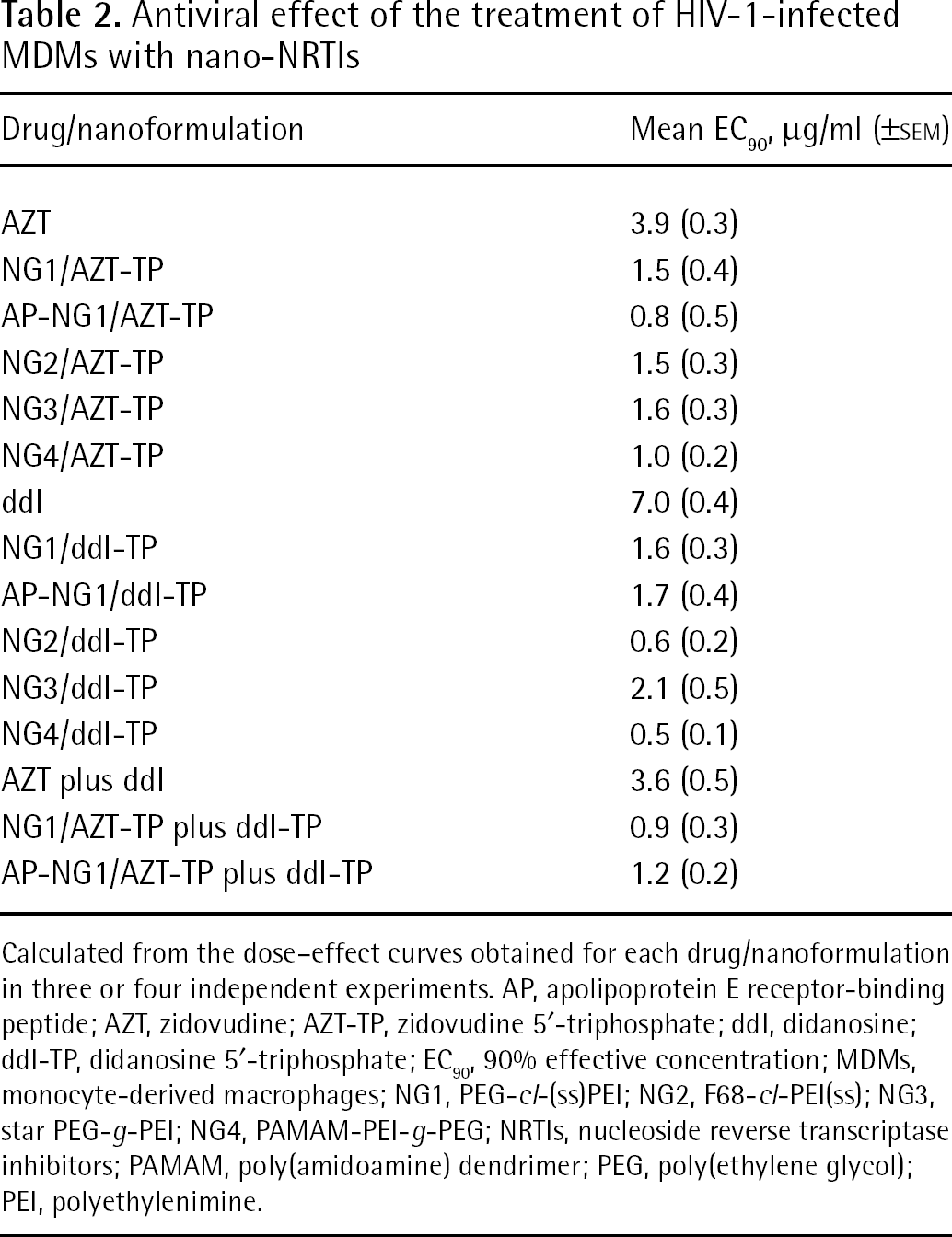
Calculated from the dose–effect curves obtained for each drug/nanoformulation in three or four independent experiments. AP, apolipoprotein E receptor-binding peptide; AZT, zidovudine; AZT-TP, zidovudine 5′-triphosphate; ddI, didanosine; ddI-TP, didanosine 5′-triphosphate; EC90, 90% effective concentration; MDMs, monocyte-derived macrophages; NG1, PEG-cl-(ss)PEI; NG2, F68-cl-PEI(ss); NG3, star PEG-g-PEI; NG4, PAMAM-PEI-g-PEG; NRTIs, nucleoside reverse transcriptase inhibitors; PAMAM, poly(amidoamine) dendrimer; PEG, poly(ethylene glycol); PEI, polyethylenimine.
All investigated nano-NRTIs showed strong antiviral activity at an EC90 as low as 0.5 μg/ml (Table 2). These EC90 values were calculated from data obtained in three or four independent experiments with HIV-1-infected MDMs. Corresponding free drugs showed similar activity at concentrations from 3.9 (AZT) to 7 μg/ml (ddI). For nano-AZT-TPs, the enhancement was 2.6- to 4.9-fold, whereas for nano-ddI-TPs, it reached 3.4- to 14-fold. Nearly complete eradication of virus by selected nano-NRTIs, when <1% viral activity was detected, could be reached at drug concentrations of only 2–4 μg/ml, although treatment with free NRTIs at these concentrations was ineffective (Figure 4C). Modification of nanogels with the brain-specific AP had an additional effect on the observed antiviral efficacy, and both AP-NG1/AZT-TP and AP-NG1/ddI-TP at the lowest studied drug concentrations (0.25 μg/ml or 1 μmol/l) demonstrated nearly twofold higher antiviral efficacy (Figure 4D). The antiviral activity of nano-AZT-TPs was similar for NG1, NG2 and NG3 (1.5–1.6 μg/ml), and the best EC90 values have been observed for NG4 (1 μg/ml) and AP-NG1 (0.8 μg/ml or 3 μmol/l; Table 2). Nano-ddI-TPs demonstrated the best results for NG2 (0.6 μg/ml) and NG4 (0.5 μg/ml or 2 μmol/l), whereas NG1, AP-NG1 and NG3 showed EC90 values between 1.6 and 2.1 μg/ml. The two-drug combination, AZT plus ddI (1:1), was slightly more efficient (3.6 μg/ml) than AZT or ddI alone (3.9 and 7 μg/ml, respectively), as well as the corresponding nano-NRTIs (EC90 0.9 versus 1.5 and 1.6 μg/ml, respectively). However, the vectorized AP-NG1/AZT-TP plus ddI-TP had an EC90 value that was an additive of EC90 values for single-drug AP-NG1/AZT-TP and AP-NG1/ddI-TP (1.2 versus 0.8 and 1.7 μg/ml, respectively; Table 2). In summary, it is evident that nano-NRTIs with a core-shell structure, such as nanogel NG4, or peptide-decorated AP-NG1, displayed the highest antiviral efficacy (EC90<1 μg/ml or 4 μmol/l, respectively).
Mitochondrial toxicity
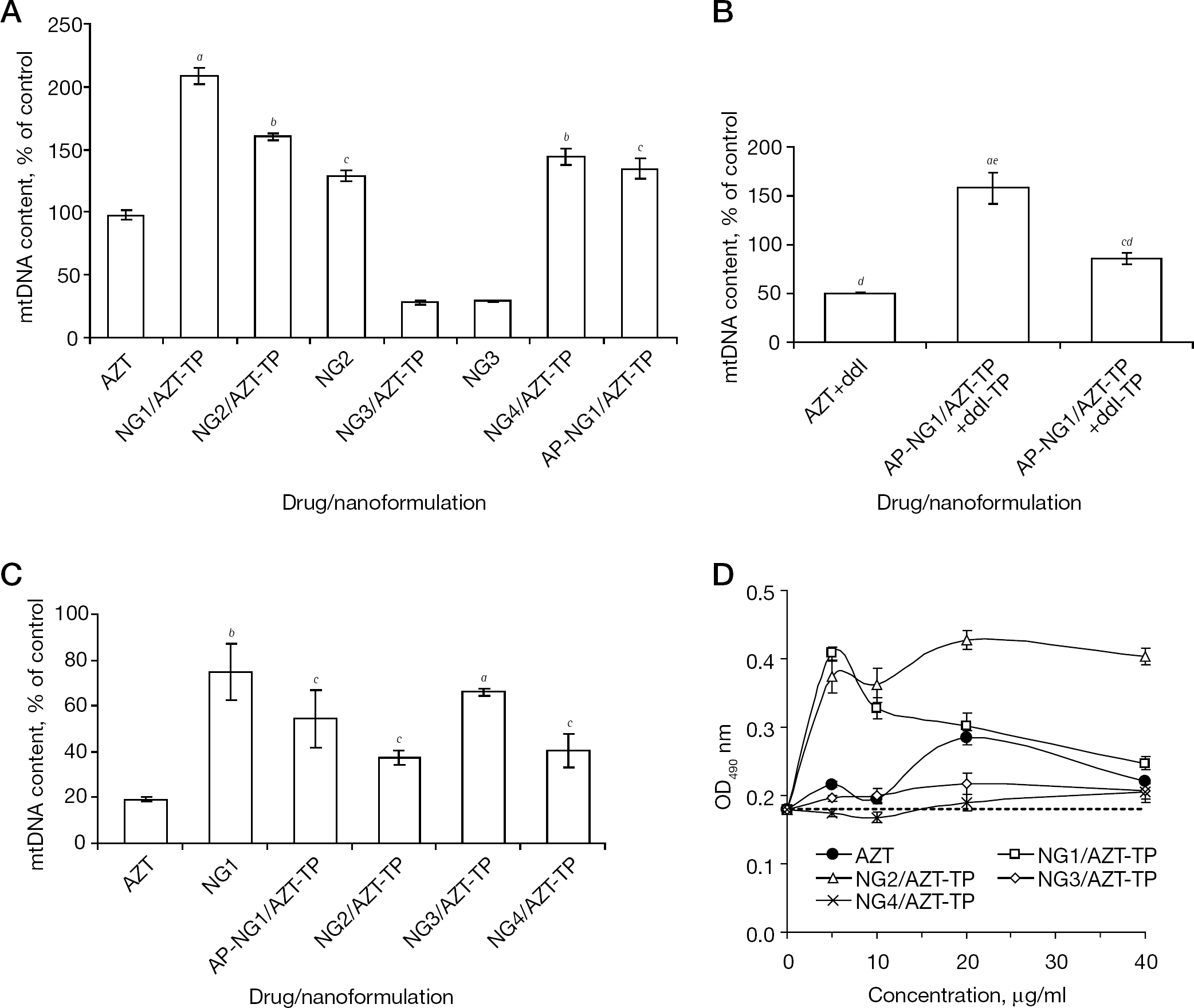
Mitochondrial toxicity caused by NRTIs results from the depletion of mtDNA, which occurs because of mtDNA chain growth termination or inhibition of DNA polymerase γ [26]. In order to compare NRTIs and nano-NRTIs, and evaluate their influence on mitochondrial toxicity, we measured the relative amount of mtDNA in macrophages using real-time PCR after a 2-week-long treatment (Figure 5). Additionally, mitochondrial toxicity was analysed under similar conditions in human hepatocyte HepG2 cells, a widely-accepted cellular model for drug toxicity studies [23]. Cells were treated with drugs and drug-loaded nanogels (15 or 30 μg/ml of AZT or AZT plus ddI, 1:1, w/w) for 4 h at 37°C every 3 days over a 2-week period (four doses). The total DNA was isolated from treated samples, and then an SYBR green dye quantitative real-time PCR analysis of mtDNA and nuDNA was performed in quadruplicates. In MDMs, the effect of AZT treatment was nearly zero at a concentration of 15 μg/ml, whereas the corresponding AZT plus ddI combination resulted in a 50% reduction of the mtDNA level (Figure 5A and 5B). Unexpectedly, the NG3/AZT-TP nanoformulation revealed significant 75% depletion of mtDNA in MDMs. Additionally, when a control NG3 nanoformulation without drug was tested, we obtained the same level of mtDNA depletion. Evidently, it suggests that the structure of nanogel NG3 has a specific adverse effect on the mitochondria function in MDMs. All other nano-NRTIs demonstrated no mtDNA depletion as a result of the treatment; conversely, they produced a 25–100% increase in the mtDNA content over non-treated MDMs. One plausible explanation could be the macrophage activation accompanied by a rise in the number of mitochondria. Previously, we also observed an unexplained increase in the formazan level during an MTT cytotoxicity assay of MDMs treated with selected nano-AZT-TPs or nano-ddI-TPs (Figure 5D). At that point, the strongest increase was detected for NG1/AZT-TP and NG2/AZT-TP. Similarly, we obtained the highest activation of MDMs and greatest increase in the mtDNA content after the treatment with NG1/AZT-TP (105% increase) and NG2/AZT-TP (60% increase) nanoformulations (Figure 5A). AP-NG1/AZT-TP and NG4/AZT-TP demonstrated a moderate MDM activation (30–40%). All observed data were statistically significant (P<0.05, compared with AZT).
Antiviral activity of nano-NRTIs in HIV-1-infected MDMs
We then tested the effect of nano-NRTIs containing a 1:1 mixture of AZT-TP and ddI-TP on the mtDNA content in MDMs at the drug doses of 15–30 μg/ml (Figure 5B). Peptide-modified AP-NG1/AZT-TP plus ddI-TP showed a low mitochondrial toxicity after 14 days of treatment, affecting the mtDNA content in a dose-dependent manner by producing a 60% increase at the dose of 15 μg/ml and by producing a 14% decrease at the dose of 30 μg/ml compared with non-treated MDMs. In general, the effect of AP-NG1/AZT-TP plus ddI-TP was similar to the effect of AP-NG1/AZT-TP on MDM activation (increases of 50% and 30%, respectively).
Conversely, we did not observe any increase in the mtDNA content after the treatment of HepG2 cells with free AZT and nano-AZT-TPs at drug concentrations of 15 μg/ml (Figure 5C). The 2-week treatment by AZT resulted in significant mtDNA depletion (80% reduction), whereas selected nano-AZT-TPs reduced mtDNA depletion by only 25–33%, with NG1, AP-NG1 and NG3 being the most effective nanocarriers. The mtDNA content was nearly identical after treatment with NG1 (75%, P<0.01), AP-NG1/AZT-TP (52%, P<0.05) and NG3/AZT-TP (64%, P<0.001) because differences between them were not significant (P= not significant). Both NG2/AZT-TP and NG4/AZT-TP demonstrated higher mitochondrial toxicities (57–62% depletion; P<0.05). Evidently, the mtDNA depletion might be, at least in part, nanocarrier-specific. The observed protective effect of selected nano-AZT-TPs versus AZT on mitochondria can be explained by a low efficacy of the released 5′-TP transport from cytoplasm through the mitochondrial membrane. The general conclusion from these results is that an efficient shielding of the positive charge in nano-NRTIs might play a decisive role in the mitochondrial toxicity of nanocarriers and activation of MDMs; thus, peptide-modified AP-NG1 and the core-shell NG4 showed the best characteristics in general, whereas unmodified nanogels NG1, NG2 and NG3, having a high density of positively charged molecules in the outer layer, demonstrated the highest mitochondrial toxicity and MDM activation.
Discussion
HAART has greatly improved the survival and the quality of life of AIDS patients, but has not reduced the recurrence of the disease because of an HIV-1 reservoir in the brain. Treatment of latent HIV-1 harbouring in the brain, mostly in macrophages, has proven to be a challenge with regard to HAART. Many of the current antiviral drugs cannot penetrate the BBB because of the activity of various drug efflux transporter proteins in the brain capillary endothelial cells forming the barrier. Development of novel, less toxic drugs and effective drug nanoformulations that are able to deliver antiviral drugs to the latent HIV-1 reservoirs is now considered the first priority in anti-HIV drug research [27]. Moreover, chronic exposure of peripheral neurons to NRTIs during treatment often results in gradual neurotoxic degradation in patients [28]. Chronic neurotoxicity, liver damage, lipodistrophy and myopathy are now recognized as major HAART-associated complications. Mitochondrial toxicity of many antiviral NRTIs, which affect the activity of DNA polymerase γ and mitochondria function, and incorporate into mtDNA, resulting in mtDNA depletion, is now considered a major cause of HAART-associated complications [29–31].
Mitochondrial toxicity of nano-NRTIs in MDMs and HepG2 cells
Development of nanosized drug delivery systems has shown tremendous potential for enhancement of drug penetration across the BBB [32,33]. Recent progress in the design of hydrophilic microgels and nanogels have made them promising drug carriers, together with liposomes, nanoparticles and micellar formulations [34]. Cationic nanogels demonstrated a strong ability to enhance drug penetration across the BBB and could be efficiently vectorized to the brain through receptormediated binding [17]. In this report, we evaluated the anti-HIV efficacy of nanogel formulations of AZT-TP and ddI-TP, active 5′-TP species of antiviral NRTIs, AZT and ddI, in an in vitro system of HIV-1-infected macrophages. Delivery of active NRTIs offers serious advantage because of their fast antiviral effect through bypassing the intracellular phosphorylation steps of drug activation. Recently, several groups have reported application of nanocarriers for delivery of antiviral nucleoside 5′-TPs. Hillaireau et al. [15] formulated the PEI-associated AZT-TP into hollow polyisobutylcyanoacrylate nanocapsules and demonstrated a 30-fold increase in cellular uptake of the encapsulated drug. Kohli et al. [18] demonstrated a 30-fold enhancement of antiviral activity and 10-fold increase in the selectivity index of nanogel-encapsulated ribavirin 5′-TP compared with free ribavirin in influenza-virus-infected Madin–Darby canine kidney cells. Saiyed et al. [35,36] demonstrated an efficient AZT-TP delivery in liposomes and suppression of HIV-1 replication in peripheral blood mononuclear cells, as well as a CNS delivery of magnetic liposomes encapsulating AZT-TP. Here, we observed a fast capture of nanogels by MDMs; 5′-TP-loaded nanogels exhibited a sufficient drug release in the cytosol 60 min after the capture, so that therapeutic levels of activated NRTIs could be reached in MDMs very quickly and at subtoxic concentrations of nano-NRTIs.
Nanogels represent the hydrophilic type of nanocarriers, which have advantages over liposomes for systemic administration and drug delivery to the brain [17]. The hydrophilic nature and high dispersion stability of drug-loaded nanogels, together with their ability to cross the BBB, make nanogels a convenient platform for the development of brain-targeted drug nanoformulations. The second feature of cationic nanogels is their ability to encapsulate 5′-TPs of nucleoside analogues and efficiently carry them inside various types of cells [14]. The active phosphorylated form of AZT, AZT-TP, was shown to induce a lower mitochondrial toxicity than free AZT when delivered in HepG2 cells in the form of an NG1/AZT-TP formulation [18].
The ability of nanogel formulations to reduce mitochondrial toxicity of NRTIs during prolonged treatment was confirmed in cultured MDMs and human hepatocyte HepG2 cells. According to Azzam et al. [37], treatment with AZT (≤25 μg/ml or 0.1 mM) did not affect the mtDNA content in MDMs, but could result in potent mitochondrial toxicity in other tissues/cells (for example, muscles, heart or liver). Among NRTIs, ddI had one of the highest potency of the inhibition of polymerase γ in mitochondria. Although we observed no effect of AZT and a moderate effect of the AZT plus ddI mixture on the mtDNA content in MDMs, a substantial increase in mtDNA content was detected after prolonged treatment with many of the nano-NRTIs (Figure 5A and 5B). We hypothesize that treatment with selected nanocarriers might provoke an activation of macrophages. Phagocytosis is an energy-dependent process, and macrophages derive most of this energy from glucose metabolism and oxidative phosphorylation in mitochondria. Activation of MDMs can result in increased mitochondrial numbers, a fact noted by some researchers [38]. One detrimental effect of higher doses of NRTIs is the impairment of phagocytic activity of MDMs; however, this might be compensated for by an increase in mitochondrial numbers. We also observed a significant twofold increase in the formation of formazan, the product of MTT conversion by mitochondrial enzymatic activities, during the cytotoxicity assay of HIV-1-infected MDMs treated with NG1/AZT-TP and NG2/AZT-TP (Figure 5D). Taken together, both results on mtDNA content and MTT conversion activity suggest a significant twofold increase in the amount of mitochondria following treatment of MDMs with the above-mentioned nano-NRTIs. Another cellular model for drug cytotoxicity studies, human HepG2 cells, did not show this effect and all nano-AZT-TPs demonstrated a 2- to 3.2-fold lower depletion in mtDNA compared with free AZT at the equivalent drug dose. The lower hepatotoxicity of nano-NRTIs is additionally enhanced by nanogel in vivo biodistribution properties. Although the intravenously injected nanogels were capable of liver accumulation to some extent, this accumulation was usually lower compared with many other nanocarriers, such as liposomes [17]. Generally, the surface pegylation reduced any interaction of nanocarriers with plasma proteins and increased circulation times [34]. Here, we observed the highest antiviral activity of nanogels with a core-shell structure, and nanocarriers with peptide modifications. These types of nano-NRTIs are worth further development to obtain targeted nanocarriers for systemic applications. Core-shell architecture is one of the most promising approaches to diminish the non-specific organ accumulation and toxicities of nano-NRTIs.
In order to develop an effective antiviral therapy against HIV-1 reservoirs in the brain, investigators face major problems associated with systemically administered nanocarriers: these are a highly polarized in vivo biodistribution, inefficient brain targeting, non-specific cellular damage and neurotoxicities. Only initial steps have been made in the area of design and evaluation of brain-targeted drug nanoformulations. Most of the currently available nanocarriers in the non-targeted form are effectively captured by peripheral macrophages of the lung, spleen and liver; therefore, their CNS delivery is far from optimal. Recently, several applications of nanocarriers modified with AP were described, which have demonstrated an increased brain accumulation compared with non-modified carriers [39–41]. Here, we suggested that placement of multiple copies of short AP molecules on the surface of nanogels could result in more specific binding of nanogels with the BBB receptors and lower non-specific biodistribution. As a first step, we demonstrated that decoration of nanogels with AP does not diminish the antiviral efficacy of nano-NRTIs in HIV-1-infected macrophages. This result makes us optimistic about the ensuing in vivo applications of vectorized nano-NRTIs. Further evaluation of selected brain-targeted nano-NRTIs in vivo is currently underway in an established mouse model of NeuroAIDS. Finally, because brain reservoirs serve as major hiding places for the virus to avoid contact with lethal drug levels, they might serve as a constant source of drug-resistant virus strains going back into circulation. Nano-NRTIs carrying activated phosphorylated drugs have the unique ability to produce more efficient inhibition of drug-resistant HIV-1 strains, especially those where drug activation is hampered by low activity of viral kinases.
Footnotes
Acknowledgements
This work was supported by the National Institutes of Health grants NS063879 and CA136921 to SVV. The authors are grateful to the very helpful output of the University of Nebraska Medical Center Flow Cytometry, Confocal Microscopy and Oligonucleotide Synthesis Core facilities.
The authors declare no competing interests.