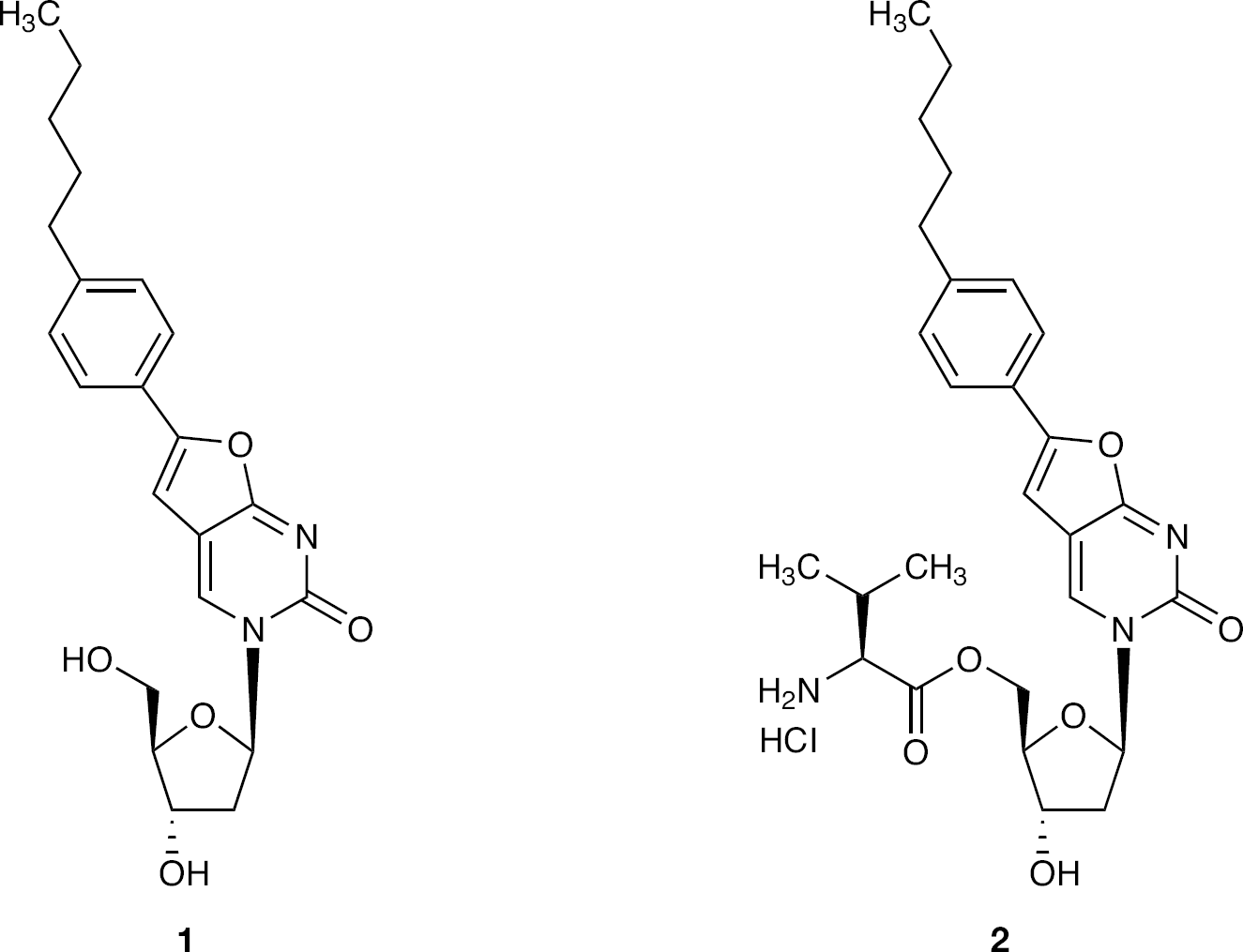
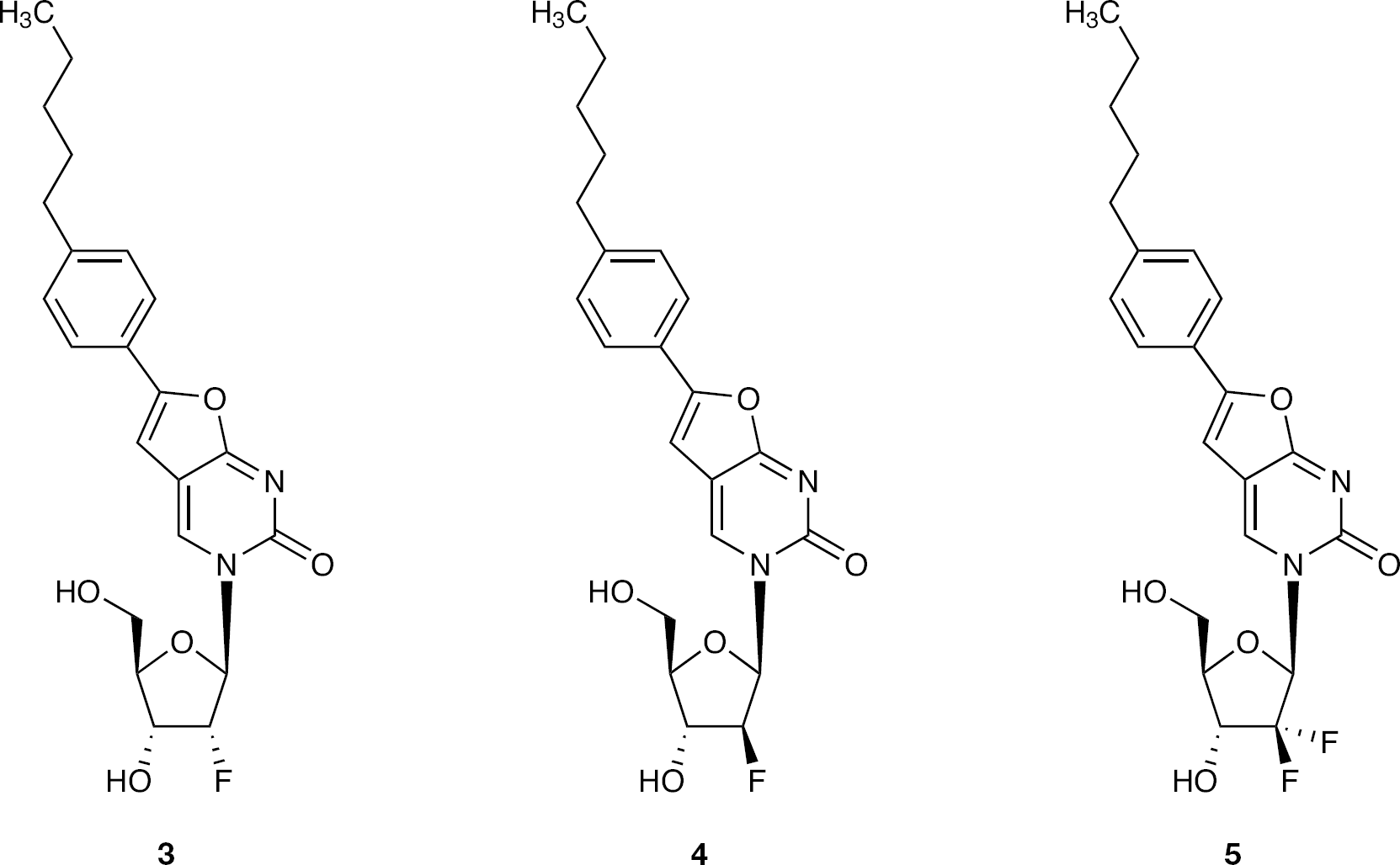
Methods
Solvents and reagents
The following anhydrous solvents were bought from Sigma–Aldrich Co. (St Louis, MO, USA) with a sure seal stopper: dichloromethane (DCM), diethyl ether, N-methylimidazole (NMI), pyridine, tetrahydrofuran (THF) and triethylamine (TEA). All reagents that were commercially available were used without further purification.
Thin-layer chromatography
Pre-coated aluminium-backed plates (60 F254, 0.2 mm thickness; Merck, Darmsted, Germany) were visualized under both short- and long-wave ultraviolet light (254 nm and 366 nm, respectively). Preparative thin-layer chromatography (TLC) plates (20×20 cm, 500–2,000 μm) were purchased from Merck.
Column chromatography
Column chromatography (CC) processes were carried out using silica gel supplied by Fisher Scientific (Hampton, NH, USA; 60 A, 35–70 μm). Glass columns were slurry packed using the appropriate eluent and samples were applied either as a concentrated solution in the same eluent or preadsorbed on silica gel.
HPLC
Analytical and semi-preparative HPLC were conducted using Varian ProStar (LC Work Station, Varian ProStar 335 Diode Array Detector, Varian 701 HPLC Fraction Collector and ProStar 210 Solvent Delivery Systems; Varian, Inc., Palo Alto, CA, USA), with Varian Polaris C18-A (10 μm) as an analytic column and Varian Polaris C18-A (10 μm) as a semi-preparative column. The software used was the Galaxie Chromatography Data System.
Nuclear magnetic resonance
1H-nuclear magnetic resonance (NMR; 500 MHz), 13C-NMR (125 MHz), 31P-NMR (202 MHz) and 19F-NMR (471 MHz) were recorded on a Bruker Avance 500 MHz Spectrometer (Bruker Instruments, Inc., Billerica, MA, USA) at 25°C. Spectra were calibrated to the residual signal of the deuterated solvent used. Chemical shifts are given in parts per million and coupling constants (J) in Hertz.
The following abbreviations are used in the assignment of NMR signals: singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), broad singlet (bs), doublet of doublet (dd) and doublet of triplet (dt).
Mass spectroscopy
High-resolution mass spectroscopy (MS) was performed as a service by Cardiff University (Cardiff, UK) using electrospray.
Elemental analyses
Elemental analysis (CHN) microanalyses were performed as a service by Medac, Ltd (Surrey, UK).
Standard procedure A: Synthesis of phosphorochloridates
To a stirred solution of the appropriate aryl dichlorophosphate (1.00 mol/eq) and the appropriate amino acid ester salt (1.00 mol/eq) in anhydrous DCM, anhydrous TEA (2.00 mol/eq) was added dropwise at −78°C under an argon atmosphere. Following the addition of TEA, the reaction mixture was stirred at −78°C for 0.5–1 h, then at room temperature (rt) for 2–3.5 h. Formation of the desired compound was monitored by 31P-NMR. After this period, the solvent was removed under reduced pressure and the residue triturated with anhydrous diethyl ether. The precipitate was filtered under nitrogen and the solution was concentrated to produce an oil. Most of the aryl phosphorochloridates synthesized were purified by flash CC (eluting with ethyl acetate/petroleum ether =60/40).
Standard procedure B: Synthesis of phosphoramidates
To a stirring solution of the appropriate nucleoside (1.00 mol/eq) and the appropriate phosphorochloridate (8.00–10.00 mol/eq) in anhydrous THF, NMI (8.00–13.00 mol/eq) was added dropwise and the reaction was stirred at rt for 1 day. After this period, the solvent was removed under reduced pressure. The residue was dissolved in DCM, washed with water (twice) and with 0.5 N HCl (twice). The organic phase was dried over MgSO4, filtered, reduced to dryness, and the crude product was purified by column chromatography eluting with DCM/MeOH in different proportions.
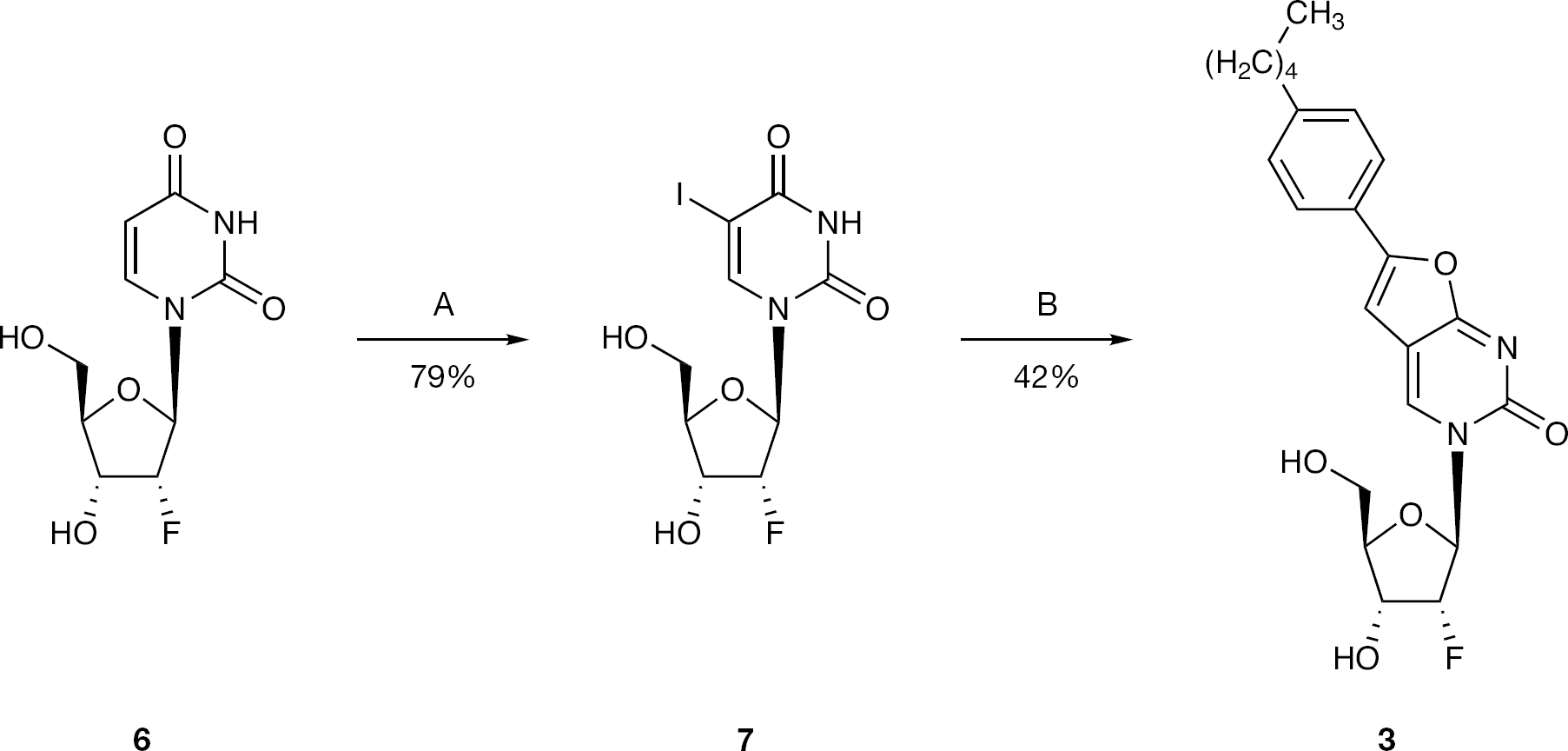
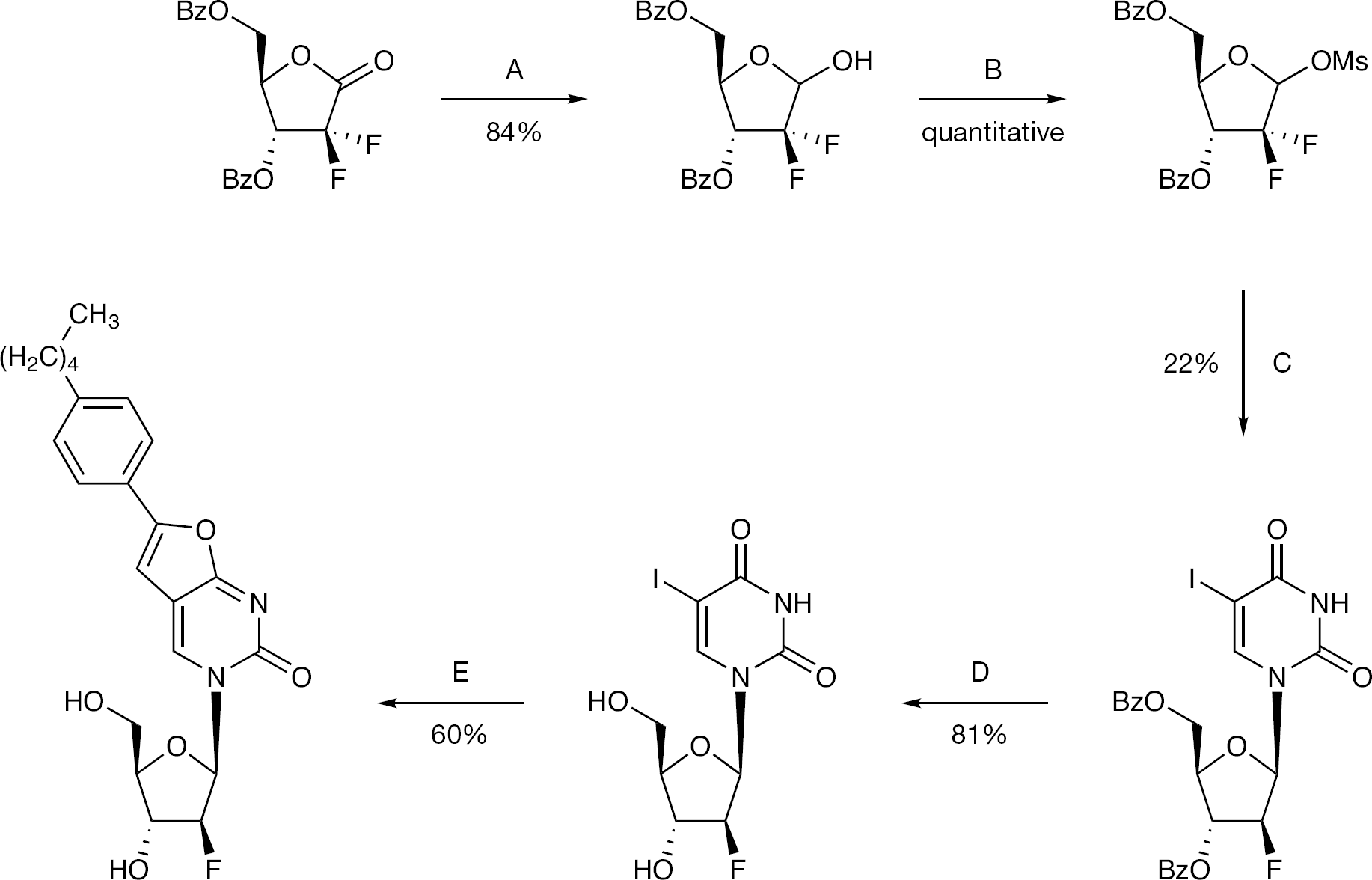
Synthesis of 5-iodo-2′-deoxy-2′-α-fluorouridine (7)
To a solution of 2′-α-fluoro-2′deoxyuridine (6; 2.00 g, 8.12 mmol) in anhydrous acetonitrile (50 ml), iodine (1.24 g, 4.87 mmol) and ceric ammonium nitrate (CAN; 2.23 g, 4.06 mmol) were added, and the reaction mixture was stirred under reflux for 1 h. After this period, the reaction mixture was quenched with a saturated solution of Na2S2O3 and then concentrated. The residue was dissolved in ethyl acetate and washed with brine (twice). The organic phase was dried over MgSO4, concentrated to give a pale yellow solid (79%, 2.40 g). 19F-NMR (DMSO, 471 MHz): δ −202.14. 1H-NMR (DMSO, 500 MHz): δ 11.71 (1H, s, NH), 8.52 (1H, s, H-6), 5.86 (1H, d, J=16.9, H-1′), 5.58 (1H, d, J=6.3, 3′-OH), 5.36 (1H, s, 5′-OH), 5.08 (0.5H, s, H-2′), 4.98 (0.5H, s, H-2′), 4.17 (1H, d, H-3′), 3.89 (1H, d, J=7.5, H-4′), 3.81 (1H, d, J=10.5, H-5′), 3.60 (1H, d, J=10.5, H-5′). 13C-NMR (DMSO, 126 MHz): δ 58.50 (C-5′), 66.73 (d, JC–F=16.3, C-3′), 69.04 (C-5), 83.00 (C-4′), 87.39 (d, JC–F=34.1, C-1′), 95.12 (d, JC–F=184.3, C-2′), 144.66 (C-6), 149.94 (C-2), 160.53 (C-4).
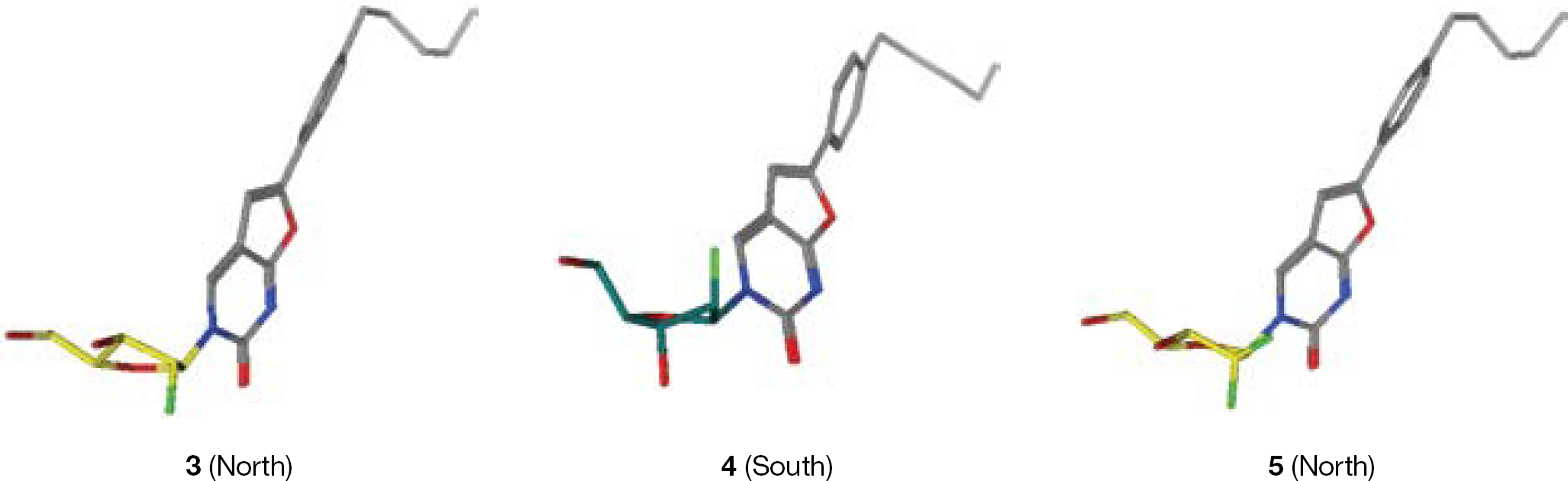
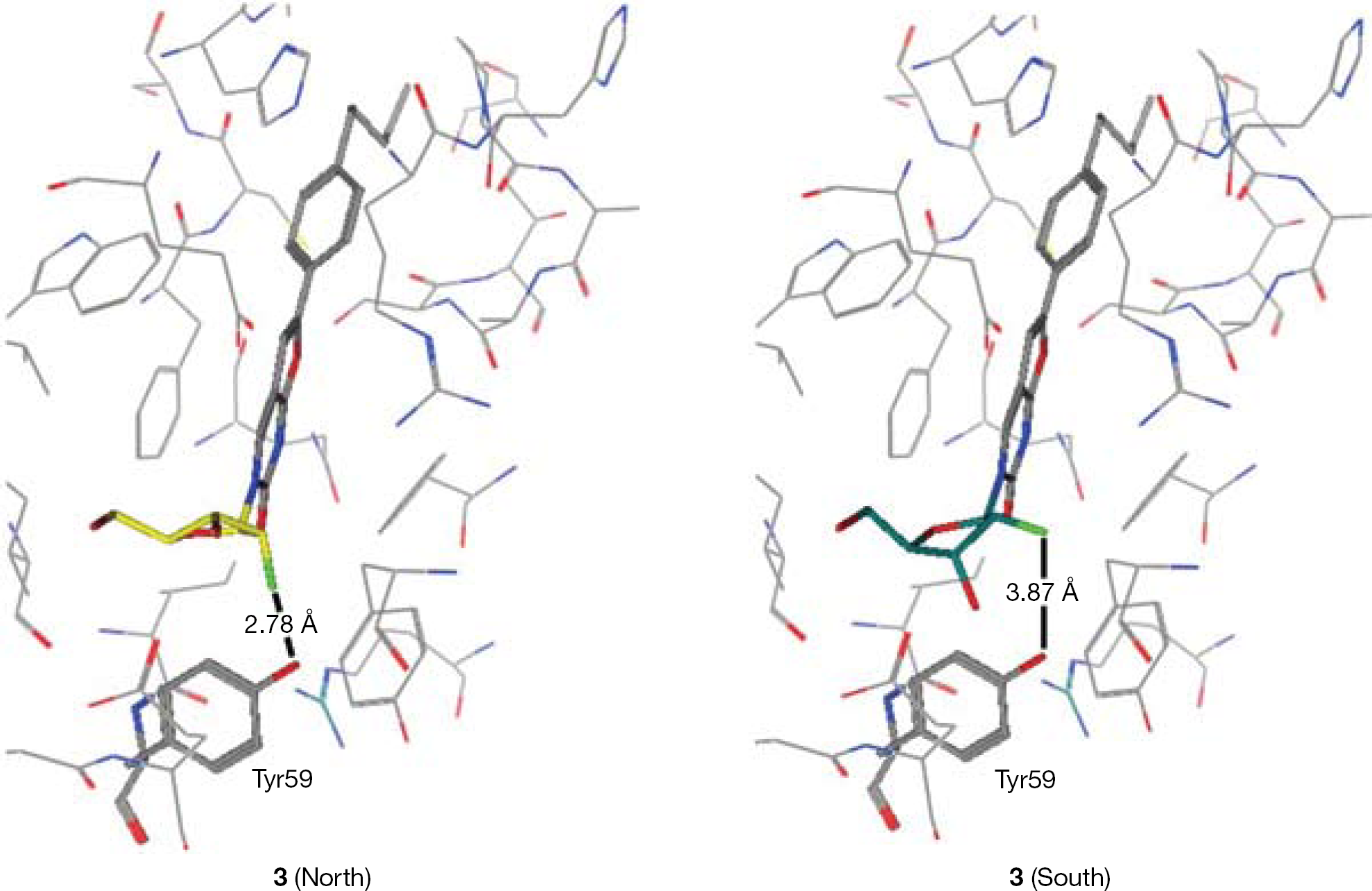
Synthesis of 3-(2′-α-fluoro-2′-deoxy-β-D-ribofuranosyl)-6-(4-n-pentylphenyl)-2,3-dihydrofuro-[2,3-d]pyrimidin-2-one (3)
To a solution of 7 (1.50 g, 4.03 mmol) in anhydrous dimethyl formamide (DMF; 20 ml) 4-n-pentylphenylacetylene (2.36 ml, 12.09 mmol), tetrakis (0.47 g, 0.40 mmol), copper (I) iodide (0.15 g, 0.81 mmol) and DIPEA (1.40 ml, 8.06 mmol) were added, and the reaction mixture was stirred at rt under an argon atmosphere overnight. After this period, copper (I) iodide (0.15 g, 0.81 mmol) and anhydrous TEA (20 ml) were added and the reaction mixture was stirred at 85°C for 7.5 h. The solvent was then removed in vacuo, and the resulting residue was triturated with methanol, filtered and washed with methanol and DCM to obtain a black solid (pure by NMR). The organic phase was concentrated and the residue purified by CC, eluting with DCM/MeOH=96/4. The compound was obtained as a dark solid, which was combined with the previous precipitate and filtered through a silica gel column, eluting with DCM/MeOH=96/4, to give a black solid. The solid obtained was washed with acetone, filtered and the solid was further washed with petroleum ether to give a white-grey solid (42%, 0.70 g). 19F-NMR (DMSO, 471 MHz): δ −201.19. 1H-NMR (DMSO, 500 MHz): δ 8.94 (1H, s, H-4), 7.75 (2H, d, J=8.1, Ha), 7.32 (2H, d, J=8.1, Hb), 7.22 (1H, s, H-5), 6.02 (1H, d, J=17.0, H-1′), 5.60 (1H, d, J=6.6, 3′-OH), 5.42 (1H, t, J=4.9, 5′-OH), 5.00 (1H, dd, J=3.7, JH-F=52.6, H-2′), 4.26–4.20 (1H, m, H-3′), 3.95 (1H, dd, J=12.4, 2.9, H-4′), 3.77–3.67 (2H, m, H-5′), 2.61 (2H, t, J=7.6, α-CH2), 1.65–1.61 (2H, m, β-CH2), 1.42–1.18 (4H, m, γ-CH2, δ-CH2), 0.86 (3H, t, J=6.9, ω-CH3). 13C-NMR (DMSO, 126 MHz): δ 13.84 (CH3), 21.88, 30.33, 30.79, 34.88 (C4H8), 58.42 (C-5′), 66.31 (d, JC–F=16.3, C-3′), 83.04 (C-4′), 89.54 (d, JC–F=34.0, C-1′), 94.16 (d, JC–F=185.7, C-2′), 98.56 (C-5), 107.19 (C-4a), 124.58 (C-Hb), 125.77 (ipso-C), 128.96 (C-Ha), 137.77 (C-4), 144.14 (para-C), 153.69 (C-6), 154.12 (C-2), 171.27 (C-7a). EI MS=416.1738 (M+). Anal. Calcd for C22H25FN2O5·0.5H2O: C, 62.11; H, 6.16; N, 6.58. Found: C, 61.73; H, 6.15; N, 6.41.
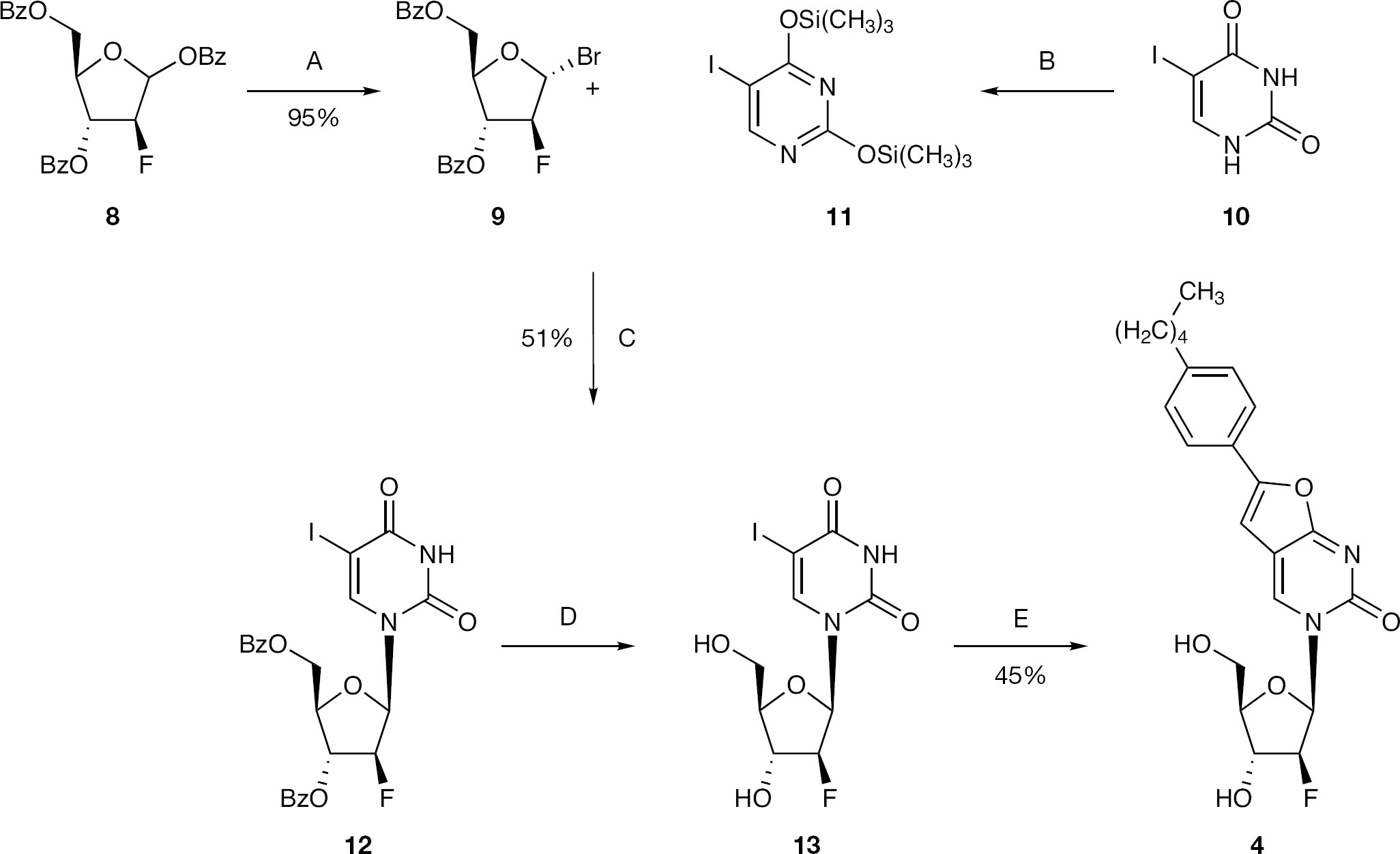
Synthesis of 2-deoxy-1-α-bromo-2-β-fluoro-3,5-di-O-benzoyl-d-ribofuranose (9)
To a solution of 2-β-fluoro-2-deoxy-1,3,5-tribenzoylribose (8; 2.00 g, 4.31 mmol) in anhydrous DCM (9 ml), a 33% by weight solution of HBr in acetic acid (1.54 ml, 9.04 mmol) was added dropwise under an argon atmosphere, and the reaction mixture was stirred at rt overnight. After this period, a 33% by weight solution of HBr in acetic acid (0.77 ml, 4.50 mmol) was added and the reaction mixture was stirred at rt for a further 7 h. Next, the reaction mixture was quenched with saturated solution of NaHCO3. The organic phase was washed with a saturated solution of NaHCO3 (twice), dried over MgSO4, then concentrated to give a colourless oil (95%, 1.73g). 19F-NMR (CDCl3, 471 MHz): δ −165.93. 1H-NMR (CDCl3, 500 MHz): δ 8.18–8.04 (4H, m, Bz), 7.69–7.43 (6H, m, Bz), 6.66 (1H, d, J=12.2, H-1′), 5.57 (1H, dd, J=3.2, JH-F=22.0, H-2′), 4.87–4.72 (4H, m, H-3′, H-4′, H-5′).
Synthesis of 2,4-bis-O-(trimethylsilyl)-5-iodouracil (11)
To a suspension of 10 (0.49 g, 2.04 mmol) in anhydrous acetonitrile (10 ml), ammonium sulphate (0.027 g, 0.2 mmol) and hexamethyldisilazane (0.48 ml, 2.30 mmol) were added, and the reaction mixture was stirred at reflux for 22 h. After this period, hexamethyldisilazane (0.96 ml, 4.08 mmol) was added, and the reaction mixture was stirred at reflux for a further 4 h and then concentrated. The crude product was used in the next step without purification.
Synthesis of 1-(2-deoxy-2-fluoro-3,5-di-O-benzoyl-D-arabino-furanosyl)-5-iodouracil (12)
To a solution of 9 (3.50 g, 8.27 mmol) in DCM (65 ml) and 11 (8.27 mmol) in acetonitrile (15 ml), NaI (0.99 g, 6.62 mmol) was added, and the reaction mixture was stirred at rt for 6 days. After this period, the suspension was filtered and the solid was washed with water and DCM to give the desired compound as a white solid (51%, 2.45 g). 19F-NMR (DMSO, 471 MHz): δ −199.13. 1H-NMR (DMSO, 500 MHz): δ 8.08–7.99 (5H, m, H-6, Bz), 7.74–7.66 (2H, m, Bz), 7.60–7.52 (4H, m, Bz), 6.31 (1H, dd, J=3.7, JH-F=19.4, H-1′), 5.71 (1H, ddd, J=4.8, 1.3, JH-F=20.3, H-3′), 5.54 (1H, ddd, J=3.8, 1.4, JH-F=50.7, H-2′), 4.81–4.71 (2H, m, H-5′), 4.63 (1H, dd, J=8.3, 4.3, H-4′).
Synthesis of 5-iodo-2′-β-fluoro-2′-deoxyuridine (13)
To a stirring solution of 12 (2.40 g, 4.14 mmol) in anhydrous methanol (60 ml), sodium methoxide (0.49 g, 9.70 mmol) was added, and the reaction mixture was stirred at rt for 1 h. After this period, the reaction was neutralized with amberlite, filtered and concentrated to give the desired product, which was used in the following step without further purification. 19F-NMR (DMSO, 471 MHz): δ −198.65. 1H-NMR (DMSO, 500 MHz): δ 8.21 (1H, s, H-6), 6.08 (1H, dd, J=4.5, JH-F=13.9, H-1′), 5.93 (1H, bs, 3′-OH), 5.27–5.25 (1H, m, 5′-OH), 5.14–5.11 (1H, m, H-2′), 5.04–4.99 (1H, m, H-2′), 4.29–4.19 (1H, m, H-3′), 3.81–3.78 (1H, m, H-4′), 3.71–3.65 (1H, m, H-5′), 3.61–3.56 (1H, m, H-5′).
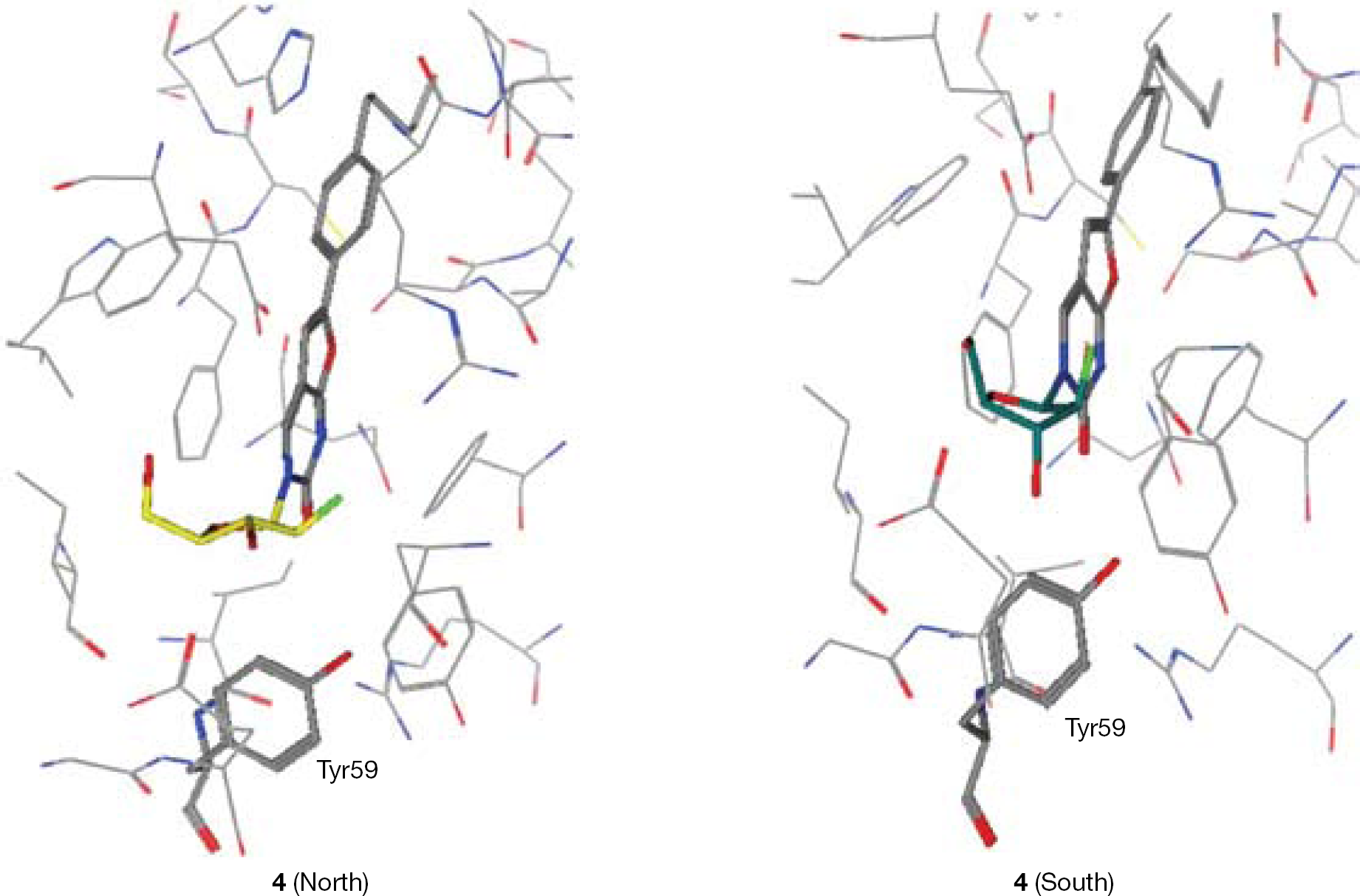
Synthesis of 3-(2′-α-fluoro-2′-deoxy-β-D-ribofuranosyl)-6-(4-n-pentylphenyl)-2,3-dihydrofuro-[2,3-d]pyrimidin-2-one (4)
To a solution of 13 (1.54 g, 4.14 mmol) in anhydrous DMF (20 ml), 4-n-pentylphenylacetylene (2.40 ml, 12.41 mmol), tetrakis (0.48 g, 0.41 mmol), copper (I) iodide (0.16 g, 0.83 mmol) and anhydrous DIPEA (1.44 ml, 8.27 mmol) were added, and the reaction mixture was stirred at rt under an argon atmosphere overnight. After this period, copper (I) iodide (0.16 g, 0.83 mmol) and anhydrous TEA (20ml) were added, and the reaction mixture was stirred at 85°C for 8 h. The solvent was then removed in vacuo and the residue was triturated with DCM, stirred at rt for 2 h, and then filtered and washed with DCM to give a white solid (45%, 0.77 g). A sample was filtered through silica gel for biological testing. 19F-NMR (DMSO, 471 MHz): δ −197.85. 1H-NMR (DMSO, 500 MHz): δ 8.73 (1H, s, H-4), 7.74 (2H, d, J=8.3, Ph), 7.33 (2H, d, J=8.3, Ph), 7.22 (1H, s, H-5), 6.25, 6.22 (1H, 2d, J=3.7, JH-F=17.1, H-1′), 6.07 (1H, d, J=4.6, 3′-OH), 5.28 (1H, t, J=5.8, 5′-OH), 5.24, 5.14 (1H, 2dd, J=3.6, 2.3, JH-F=52.0, H-2′), 4.29, 4.26 (1H, 2dd, J=6.1, 4.3, JH-F=18.2, H-3′), 3.99 (1H, q, H-4′), 3.71–3.63 (2H, m, H-5′), 2.62 (2H, t, α-CH2), 1.59 (2H, quintet, β-CH2), 1.35–1.24 (4H, m, γ-CH2, δ-CH2), 0.86 (3H, t, CH3). 13C-NMR (DMSO, 126 MHz): δ 13.85 (CH3), 21.87 (CH2), 30.32 (β-CH2), 30.78 (CH2), 34.87 (α-CH2), 60.13 (C-5′), 73.08 (d, JC-F=24.5 Hz, C-3′), 85.22 (C-4′), 86.15 (d, JC-F=16.6 Hz, C-1′), 94.66 (d, JC-F=191.5 Hz, C-2′) 98.60 (C-5), 107.14 (C-4a), 124.61 (Ph), 125.70 (ipso-C), 128.99 (Ph), 138.70 (C-4), 144.20 (para-C), 153.47 (C-6), 154.20 (C-2), 171.30 (C-7a). EI MS=416.1749 (M+). HPLC=H2O/AcCN from 100/0 to 0/100 in 30 min = retention time 23.81 min.
Synthesis of 2-deoxy-D-erythro-2,2-difluoro-ribofuranose-3,5-dibenzoate (15)
A solution of 14 (5.0 g, 0.013 mmol) in anhydrous THF (40 ml) and anhydrous diethyl ether (10 ml) was cooled to −78°C under an argon atmosphere and lithium tri(tert-butoxy)aluminium hydride (14.58 ml, 1.0 M in THF) was added dropwise. The reaction mixture was stirred for 1 h at −78°C and quenched by the slow addition of methanol (3.2 ml). The reaction mixture was allowed to warm to rt and then ethyl acetate (162 ml) was added. The organic phase was washed with equal volumes of saturated NaHCO3 solution and brine, and the organic layer was dried over Na2SO4 and concentrated to give a thick oil as a mixture of anomers (84%, 4.14 g). 19F-NMR (CDCl3, 471 MHz): δ −108.97, −109.50, −123.19, −123.70, −124.93, −125.46. 1H-NMR of major (approximately 55%) anomer (CDCl3, 500 MHZ): δ 8.14–7.99 (4H, m, Bz), 7.66–7.55 (2H, m, Bz), 7.51–7.39 (4H, m, Bz), 5.81–5.73 (1H, m, H-3), 5.52–5.49 (1H, m, H-1), 4.82–4.58 (3H, m, H-4, H-5), 3.66 (1H, s, OH). 1H-NMR of minor (approximately 45%) anomer (CDCl3, 500 MHZ): δ 8.14–7.99 (4H, m, Bz), 7.66–7.55 (2H, m, Bz), 7.51–7.39 (4H, m, Bz), 5.55–5.49 (1H, m, H-3), 5.40–5.35 (1H, m, H-1), 4.77–4.60 (2H, m, H-5), 4.51–4.45 (1H, m, H-4), 3.93 (1H, s, OH).
Synthesis of 2-deoxy-D-erythro-2,2-difluoro-ribofuranose-3,5-dibenzoate-1-methanesulfonate (16)
A solution of 15 (4.14 g, 10.9 mmol) in anhydrous DCM (52 ml) and anhydrous TEA (2.4 ml) was cooled to 0°C and methane sulfonylchloride (1.23 ml, 15.8 mmol) was added dropwise. The reaction was stirred at rt for 18 h. After this period, the reaction mixture was partitioned between DCM (140 ml) and a saturated solution of NaHCO3 (56 ml). The organic phase was dried over Na2SO4 and concentrated to give an oil as a mixture of anomers (quantitative, 5.03 g). 19F-NMR (CDCl3, 471 MHz): δ −107.70, −108.22, −120.65, −121.17, −122.21, −122.73, −123.76, −124.45. 1H-NMR of major (approximately 60%) anomer (CDCl3, 500 MHZ): δ 8.13–8.04 (4H, m, Bz), 7.65–7.54 (2H, m, Bz), 7.50–7.41 (4H, m, Bz), 6.20–6.15 (1H, d, J=5.62 Hz, H-1), 5.65–5.58 (1H, dd, J1=4.25 Hz, J2=16.45 Hz, H-3), 4.93–4.89 (1H, m, H-4), 4.81–4.61 (2H, m, H-5), 3.17 (3H, s, CH3). 1H-NMR of minor (approximately 40%) anomer (CDCl3, 500 MHZ): δ 8.13–8.04 (4H, m, Bz), 7.65–7.54 (2H, m, Bz), 7.50–7.41 (4H, m, Bz), 6.11–6.07 (1H, d, J=6.37 Hz, H-1), 6.03–5.94 (1H, m, H-3), 4.81–4.61 (3H, m, H-4, H-5), 3.03 (3H, s, CH3).
Synthesis of 1-(3,5-Di-O-benzoyl-2-deoxy-2,2-difluoro-β-D-erythro-pentofuranos-1-yl)-5-iodouracil (17)
Compound 10 (3.61 g, 15.2 mmol) was treated with an excess of hexamethyldisilazane (100 ml) in the presence of (NH4)2SO4 (0.10 g, 0.76 mmol) under an argon atmosphere and refluxed at 125–130°C for 4 h. The solvent was removed under reduced pressure and the resulting syrup was dissolved in anhydrous dichloroethane (57 ml). A solution of 16 (3.46 g, 7.59 mmol) in anhydrous dichloroethane (86 ml) was added. The reaction mixture was stirred for 10 min, then trimethylsilyl trifluoromethanesulfonate (2.95 ml, 16.30 mmol) was added slowly and the reaction mixture was refluxed at 90–100°C for 10 h. The reaction mixture was cooled to rt and washed with equal volumes of saturated solution of NaHCO3 and brine. The β-anomer was obtained by precipitation from the organic solvent and washed with methanol to remove traces of α-anomer as a white solid (1.00 g, 22%). 19F-NMR (DMSO, 471 MHz): δ −111.37, −111.90, −114.17 (broad). 1H-NMR (DMSO, 500 MHZ): δ 12.03 (1H, s, NH), 8.17 (1H, s, H-6), 8.08–7.94 (4H, m, Bz), 7.77–7.63 (2H, m, Bz), 7.61–7.46 (4H, m, Bz), 6.36 (1H, t, J=9.06 Hz, H-1′), 5.93–5.84 (1H, m, H-3′), 4.83–4.73 (3H, m, H-4′, H-5′).
Synthesis of 1-(2-deoxy-2,2-difluoro-β-D-erythro-pentofuranos-1-yl)-5-iodouracil (18)
To a stirring solution of 17 (0.93 g, 1.56 mmol) in anhydrous methanol (30 ml), sodium methoxide (0.25 g, 4.67 mmol) was added, and the reaction mixture was stirred at rt overnight. The reaction was neutralized with amberlite, filtered and concentrated. The residue was purified by CC, eluting with chloroform/methanol 90/10 to give a white solid (0.56 g, 92%). 19F-NMR (DMSO, 471 MHz): δ −117.16. 1H-NMR (DMSO, 500 MHZ): δ 11.89 (1H, s, NH), 8.37 (1H, s, H-6), 6.30 (1H, d, J=6.51 Hz, 3′-OH), 6.02 (1H, t, J=6.78 Hz, H-1′), 5.45 (1H, m, 5′-OH), 4.29–4.17 (1H, m, H-3′), 3.91–3.84 (1H, m, H-4′), 3.84–3.60 (2H, 2m, H-5′).
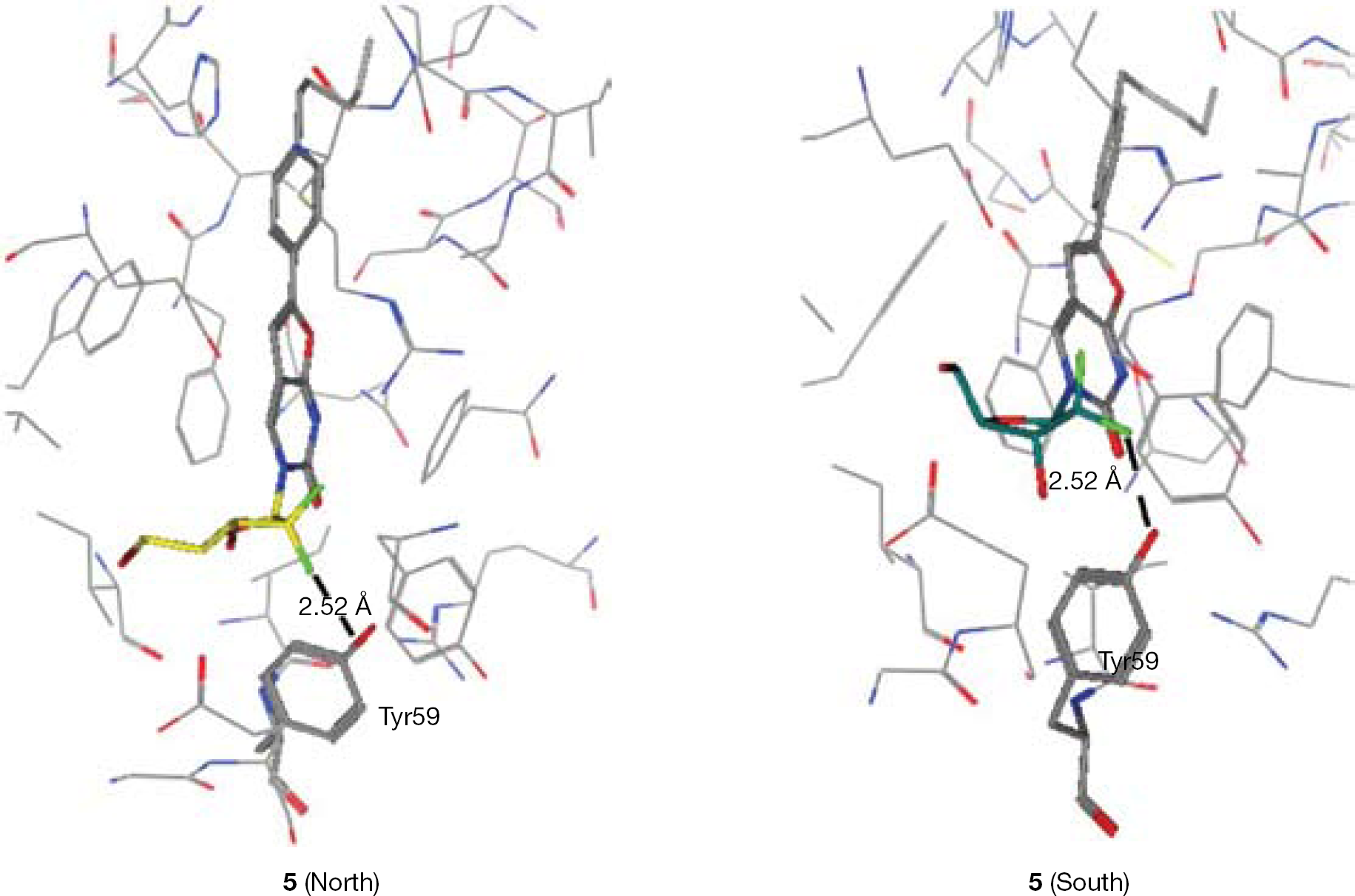
Synthesis of 3-(2′-difluoro-2′-deoxy-β-D-ribofuranosyl)-6-(4-n-pentylphenyl)-2,3-dihydrofuro-[2,3-d]pyrimidin-2-one (5)
To a solution of 18 (0.18 g, 0.47 mmol) in anhydrous DMF (5 ml), 4-n-pentylphenylacetylene (0.27 ml, 1.41 mmol), tetrakis (0.055 g, 0.047 mmol), copper (I) iodide (0.018 g, 0.047 mmol) and DIPEA (0.16 ml, 0.94 mmol) were added, and the reaction mixture was stirred at rt under an argon atmosphere overnight. After this period, copper (I) iodide (0.018 g, 0.047 mmol) and anhydrous TEA (5 ml) were added and the reaction mixture was stirred at 85°C for 7.5 h. The solvent was then removed in vacuo and the residue was purified by CC, with gradient elution of DCM, then DCM/MeOH=98/2, then 96/4. The compound was obtained as a brown-dark solid, which was triturated with acetone and petroleum ether, then filtered. The solid was washed with acetone and petroleum ether to give a white-pale-yellow solid (60%, 0.075 g). 19F-NMR (DMSO, 471 MHz): δ −116.84. 1H-NMR (DMSO, 500 MHz): δ 8.75 (1H, s, H-4), 7.76 (2H, d, J=8.2, Ha), 7.34 (2H, d, J=8.2, Hb), 7.23 (1H, s, H-5), 6.35 (1H, d, J=6.5, H-1′), 6.33–6.30 (1H, m, 3′-OH), 5.43 (1H, t, J=5.3, 5′-OH), 4.36–4.18 (1H, m, H-3′), 3.99–3.95 (1H, m, H-4′), 3.91–3.85 (1H, m, H-5′), 3.75–3.69 (1H, m, H-5′), 2.63 (2H, t, J=7.6, α-CH2), 1.68–1.52 (2H, m, β-CH2), 1.40–1.21 (4H, m, γ-CH2, δ-CH2), 0.87 (3H, t, J=7.0, CH3). 13C-NMR (DMSO, 126 MHz): δ 13.85 (CH3), 21.88, 30.32, 30.79, 34.90 (C4H8), 58.62 (C-5′), 68.14 (t, JC–F=22.4, C-3′), 81.11 (C-4′), 85.14 (t, JC–F=31.2, C-1′), 98.37 (C-5), 107.94 (C-4a), 120.84, 122.91 (C-2′), 124.75 (C-Hb), 125.56 (ipso-C), 129.01 (C-Ha), 137.26 (C-4), 144.44 (para-C), 153.68 (C-6), 154.87 (C-2), 171.53 (C-7a). EI MS=435.1731 (M+H). Anal. Calcd for C22H24F2N2O5·0.5H2O: C, 59.59; H, 5.68; N, 6.32. Found: C, 59.38; H, 5.59; N, 6.25.
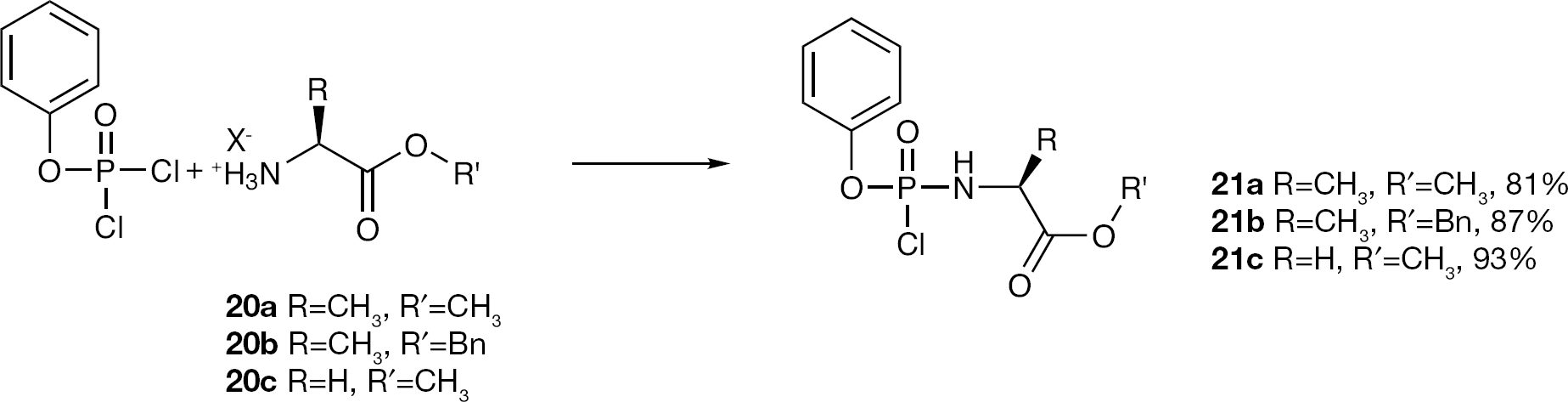
Synthesis of phenyl-(methoxy-L-alaninyl)-phosphorochloridate (21a)
Compound 21a was prepared according to standard procedure A, from phenyldichlorophosphate (19; 2.24 ml, 15.00 mmol), L-alanine methyl ester HCl (20a; 2.09 g, 15.00 mmol), anhydrous TEA (4.20 ml, 30.00 mmol) and anhydrous DCM (80 ml). The reaction mixture was stirred at −78°C for 30 min, then at rt for 2.5 h. The crude product was purified by CC, eluting with ethyl acetate/hexane =6/4, to give an oil (81%, 3.35 g). 31P-NMR (CDCl3, 202 MHz): δ 7.95, 7.66. 1H-NMR (CDCl3, 500 MHz): δ 7.32–7.15 (5H, m, PhO), 4.42–4.34 (1H, m, NH), 4.17–4.08 (H, m, CHNH), 3.72, 3.70 (3H, 2s, CH3O), 1.45–1.43 (3H, m, CHCH3).
Synthesis of phenyl-(benzoxy-L-alaninyl)-phosphorochloridate (21b)
Compound 21b was prepared according to standard procedure A, using 19 (0.30 ml, 2.00 mmol), L-alanine benzyl ester tosylate (20b; 0.43 g, 2.00 mmol) and anhydrous TEA (0.56 ml, 4.00 mmol) in anhydrous DCM (15 ml). The reaction mixture was stirred at −78°C for 30 min, then at rt for 3.5 h. The crude product was obtained as an oil (87%, 0.62 g). 31P-NMR (CDCl3, 202 MHz): δ 7.86, 7.52. 1H-NMR (CDCl3, 500 MHz): δ 7.33–7.28 (10H, m, PhO, OCH2Ph), 5.15–5.13 (2H, m, OCH2Ph), 4.18–4.13 (1H, m, CHNH), 1.46–1.44 (3H, m, CH3).
Synthesis of phenyl-(methoxy-glycinyl)-phosphorochloridate (21c)
Compound 21c was prepared according to standard procedure A, from 19 (2.24 ml, 15.00 mmol), glycine methyl ester HCl (20c; 1.88 g, 15.00 mmol), anhydrous TEA (4.20 ml, 30.00 mmol) and anhydrous DCM (80 ml). The reaction mixture was stirred at −78°C for 30 min, then at rt for 2 h. The crude product was obtained as an oil (93%, 3.70 g) and used in the next step without further purification. 31P-NMR (CDCl3, 202 MHz): δ 9.03. 1H-NMR (CDCl3, 500 MHz): δ 7.28–7.12 (5H, m, PhO), 4.43 (1H, bs, NHCH2), 3.84 (2H, d, NHCH2), 3.72 (3H, s, OCH3).
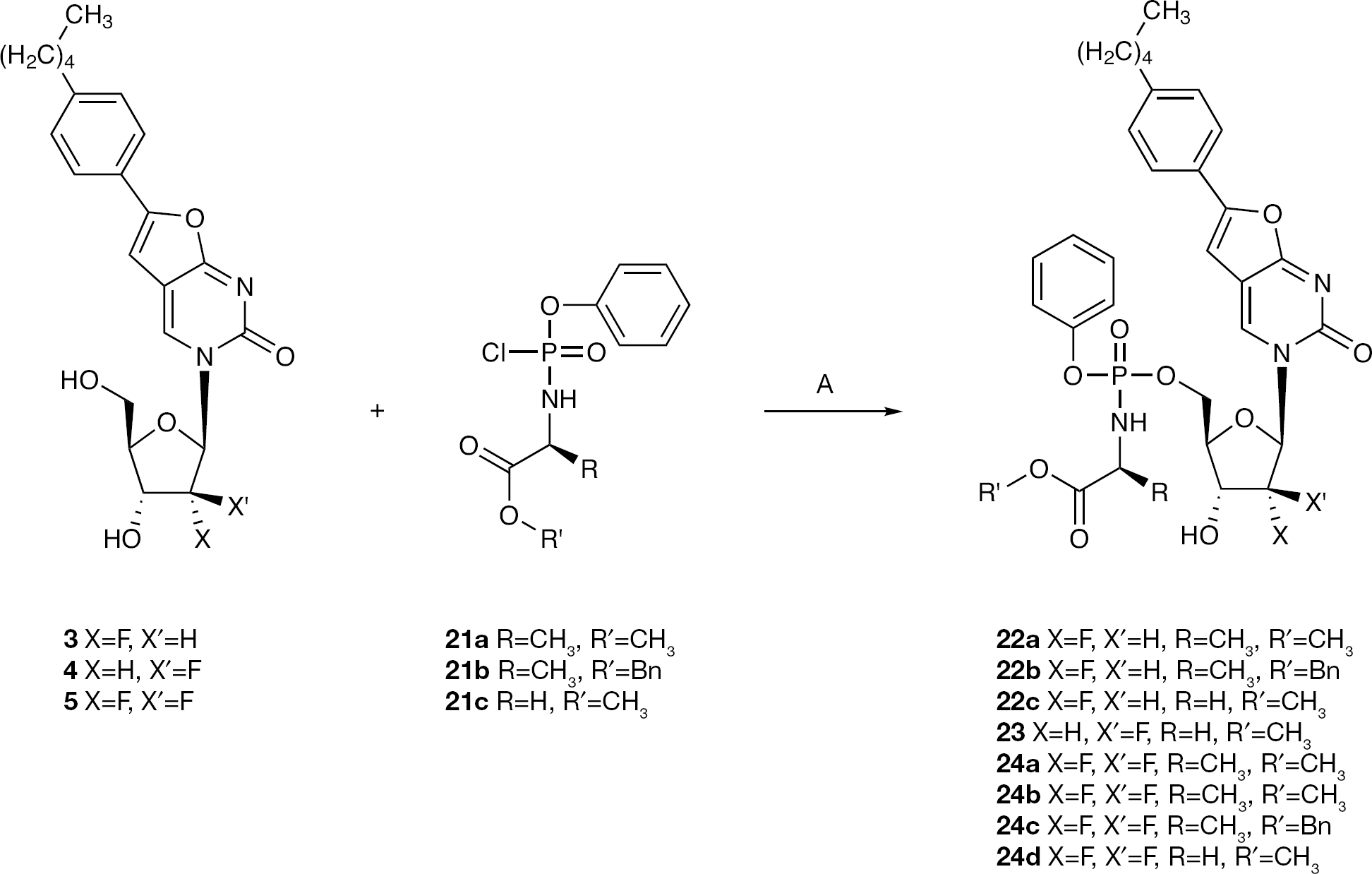
Synthesis of 3-(2′-α-fluoro-2′-deoxy-β-D-ribofuranosyl-5-[phenyl-(methoxy-l-alaninyl)]-phosphate)-6-(4-n-pentylphenyl)-2,3-dihydrofuro-[2,3-d]pyrimidin-2-one (22a)
Compound 22a was prepared according to standard procedure B, from 4 (0.20 g, 0.48 mmol) in anhydrous THF (5 ml) and anhydrous pyridine (5 ml), 21a (0.67 g, 2.40 mmol) in anhydrous THF (2 ml)and NMI (0.19 ml, 2.40 mmol), and the reaction mixture was stirred at rt overnight. After this period, 21a (0.40 g, 1.44 mmol) in anhydrous THF (2 ml) and NMI (0.11 ml, 1.44 mmol) were added, and the reaction mixture was stirred at rt for a further 7 h. After this period, the solvent was removed and the residue dissolved in DCM. The organic phase was washed with a 0.5 M aqueous solution of citric acid (twice) and water (twice), dried over MgSO4 and concentrated. The residue was purified by CC gradient elution of DCM/MeOH=98/2, then 97/3, to give a white solid that was further purified by preparative TLC (gradient elution of DCM/MeOH=99/1, then 98/2, then 96/4) to give a white solid (5%, 0.015 g). 31P-NMR (MeOD, 202 MHz): δ 3.90, 3.83. 19F-NMR (DMSO, 471 MHz): δ −203.40, −203.72. 1H-NMR (MeOD, 500 MHz): δ 8.72, 8.71 (1H, 2s, H-4), 7.66–7.59 (2H, m, Ph), 7.43–7.17 (7H, m, Ph), 6.87, 6.75 (1H, 2s, H-5), 6.16 (0.5H, d, J=8.3, H-1′), 6.13 (0.5H, d, J=8.5, H-1′), 5.09 (1H, dd, J=3.1, JH-F=52.1, H-2′), 4.79–4.64 (1H, m, H-5′), 4.57–4.42 (1H, m, H-4′), 4.40–4.27 (2H, m, H-3′, H-5′), 4.08–3.99 (1H, m, CHCH3), 3.68 (3H, s, OCH3), 2.66 (2H, t, J=7.7, α-CH2), 1.73–1.60 (2H, m, β-CH2), 1.44–1.28 (7H, m, γ-CH2, δ-CH2, CHCH3), 0.93 (3H, t, J=7.0, CH3). 13C-NMR (MeOD, 126 MHz): δ 14.35 (CH3), 20.34 (d, JC–P=7.4, CHCH3), 20.53 (d, JC–P=6.4, CHCH3), 23.56, 32.14, 32.59, 36.74 (C4H8), 51.55, 51.77 (CHCH3), 52.81, 52.85 (2s, OCH3), 65.64 (d, JC–P=2.8, C-5′), 65.68 (d, JC–P=2.0, C-5′), 68.80 (d, JC–P=16.5, C-4′), 68.93 (d, JC–P=16.2, C-4′), 82.56, (d, JC–P=8.3, C-3′) 82.63 (d, JC–P=8.3, C-3′), 92.26, 92.54 (2d, JC–F=34.8, C-1′), 94.30, 95.80 (2d, JC–F=187.7, C-2′), 98.96, 98.99 (2s, C-5), 110.45 (C-4a), 121.42, 121.50, 121.51, 121.53, 121.55, 123.94, 125.90, 126.02, 126.40, 127.12, 127.14, 130.09, 130.17, 130.20, 130.59, 130.72, 130.93, 131.00, (PhO, Ph), 138.52, 138.54 (C-4), 146.49, 146.50 (2s, para-C), 152.00, 152.05 (2s, ‘ipso’ PhO), 156.59 (C-6), 157.45, 157.49 (C-2), 173.36 (C-7a), 175.31 (d, JC–P=5.2, COOCH3), 175.58 (d, JC–P=4.5, COOCH3). EI MS=658.2330 (M+H). HPLC = H2O/AcCN from 100/0 to 0/100 in 30 min = retention time 20.43 min.
Synthesis of 3-(2′-α-fluoro-2′-deoxy-β-D-ribofuranosyl-5-[phenyl-(benzoxy-L-alaninyl)]-phosphate)-6-(4-n-pentylphenyl)-2,3-dihydrofuro-[2,3-d]pyrimidin-2-one (22b)
Compound 22b was prepared according to standard procedure B, from 4 (0.17 g, 0.40 mmol) in anhydrous THF (5 ml) and anhydrous pyridine (5 ml), 21b (0.70 g, 2.00 mmol) in anhydrous THF (2 ml) and NMI (0.16 ml, 2.00 mmol), and the reaction mixture was stirred at rt overnight. After this period, 21b (0.42 g, 1.20 mmol) in anhydrous THF (2 ml) and NMI (0.09 ml, 1.20 mmol) were added, and the reaction mixture was stirred at rt for a further 24 h. NMI (0.09 ml, 1.20 mmol) was then added and the reaction mixture stirred at rt for a further 7 h. After this period, the solvent was removed and the residue dissolved in DCM. The organic phase was washed with a 0.5 M aqueous solution of citric acid (twice) and water (twice), dried over MgSO4 and concentrated. The residue was purified by CC gradient elution of DCM/MeOH=98/2, then 97/3, to give a white solid that was further purified by preparative TLC (gradient elution of DCM/MeOH=99/1, then 98/2, then 96/4) to give another white solid (7%, 0.020 g). 31P-NMR (MeOD, 202 MHz): δ 3.94, 3.71. 19F-NMR (DMSO, 471 MHz): δ −200.55, −200.94. 1H-NMR (MeOD, 500 MHz): δ 8.70, 8.67 (1H, 2s, H-4), 7.70–7.60 (2H, m, Ph), 7.47–7.18 (12H, m, Ph, PhO, CH2Ph), 6.86, 6.79 (1H, 2s, H-5), 6.14 (0.5H, d, J=9.3, H-1′), 6.11 (0.5H, d, J=10.0, H-1′), 5.18–5.06 (2H, m, CH2Ph), 5.00–4.93 (1H, m, H-2′), 4.71–4.51 (1H, m, H-5′), 4.50–4.41 (1H, m, H-5′), 4.36–4.23 (2H, m, H-3′, H-4′), 4.16–4.02 (1H, m, CHCH3), 2.68 (2H, t, J=7.4, α-CH2), 1.73–1.55 (2H, m, β-CH2), 1.48–1.27 (7H, m, γ-CH2, δ-CH2, CHCH3), 0.93 (3H, t, J=6.6, CH3). 13C-NMR (MeOD, 126 MHz): δ 14.35 (CH3), 20.28 (d, JC–P=7.7, CHCH3), 20.51 (d, JC–P=6.3, CHCH3), 23.56, 32.15, 32.58, 36.74 (C4H8), 51.68, 51.94 (CHCH3), 65.59 (d, JC–P=5.0, C-5′), 65.80 (d, JC–P=5.4, C-5′), 68.03 (CH2Ph), 68.71, 68.85 (C-4′), 82.52, 82.58 (2s, C-3′), 92.23, 92.51 (2d, JC–F=24.4, C-1′), 95.27, 95.77 (2d, JC–F=188.7, C-2′), 99.00, 99.02 (2s, C-5), 110.46 (C-4a), 121.45, 121.49, 121.53, 121.56, 125.93, 126.37, 126.42, 127.14, 129.14, 129.18, 129.32, 129.57, 130.19, 130.93, 130.98, (PhO, CH2Ph, Ph), 137.17 (‘ipso’ OCH2Ph), 138.53 (C-4), 146.50 (para-C), 152.03 (‘ipso’ PhO), 156.58 (C-6), 157.45 (C-2), 173.34 (C-7a), 174.95 (COOCH3). EI MS=734.2616 (M+H) 756.2448 (M+Na). HPLC=H2O/MeOH 20/80 isocratic = retention time 28.08, 30.03 min.
Synthesis of 3-(2′-α-fluoro-2′-deoxy-β-D-ribofuranosyl-5-[phenyl-(methoxy-glycinyl)]-phosphate)-6-(4-n-pentylphenyl)-2,3-dihydrofuro-[2,3-d]pyrimidin-2-one (22c)
Compound 22c was prepared according to standard procedure B, from 4 (0.20 g, 0.48 mmol) in anhydrous THF (10 ml), 21c (0.25 g, 0.96 mmol) in anhydrous THF (2 ml) and NMI (0.08 ml, 0.96 mmol), and the reaction mixture was stirred at rt overnight. After this period were added anhydrous pyridine (2 ml), 21c (0.25 g, 0.96 mmol) in anhydrous THF (2 ml) and NMI (0.08 ml, 0.96 mmol), and the reaction mixture was stirred at rt for a further 7 h. Next, 21c (0.38 g, 1.44 mmol) in anhydrous THF (2 ml) and NMI (0.11 ml, 1.44 mmol) were added, and the reaction mixture was stirred at rt for 16 h. After this period, the solvent was removed and the residue dissolved in DCM. The organic phase was washed with a 0.5 M aqueous solution of citric acid (twice) and water (twice), dried over MgSO4 and concentrated. The residue was purified by CC gradient elution of DCM/MeOH=98/2, then 97/3, to give a white solid (15%, 0.046 g). 31P-NMR (MeOD, 202 MHz): δ 5.24, 4.95. 19F-NMR (MeOD, 471 MHz): δ −203.47, −203.56. 1H-NMR (MeOD, 500 MHz): δ 8.71, 8.69 (1H, 2s, H-4), 7.61–7.56 (2H, m, Ph), 7.49–7.34 (2H, m, Ph), 7.34–7.17 (5H, m, Ph), 6.84, 6.73 (1H, 2s, H-5), 6.13 (1H, d, J=17.0, H-1′), 5.14, 5.04 (1H, 2t, J=3.8, JH-F=52.0, H-2′), 4.78–4.68 (1H, m, H-5′), 4.59–4.47 (1H, m, H-4′), 4.41–4.28 (2H, m, H-3′, H-5′), 3.89–3.80 (2H, m, NHCH2), 3.68 (3H, 2s, OCH3), 2.64 (2H, t, J=7.7, α-CH2), 1.71–1.59 (2H, m, β-CH2), 1.46–1.28 (4H, m, γ-CH2, δ-CH2), 0.93 (3H, t, J=7.0, CH3). 13C-NMR (MeOD, 126 MHz): δ 14.38 (CH3), 23.57, 32.11, 32.62, 36.75 (C4H8), 43.76 (CH2NH), 52.69, 52.71 (2s, OCH3), 65.63 (d, JC–P=5.0, C-5′), 65.72 (d, JC–P=4.8, C-5′), 68.75 (d, JC–P=11.7, C-4′), 68.89 (d, JC–P=11.7, C-4′), 82.57, 82.64 (2s, C-3′), 92.17, 92.44 (2d, JC–F=34.8, C-1′), 95.09 (d, JC–F=187.8, C-2′), 98.98 (C-5), 110.42 (C-4a), 121.51, 121.55, 125.87, 125.90, 126.43, 127.08, 130.14, 130.97, 131.02 (PhO, Ph), 138.50, 138.56 (C-4), 146.41 (para-C), 152.00, 152.06 (2s, ‘ipso’ PhO), 156.77 (C-6), 157.35, 157.42 (C-2), 172.97 (d, JC–P=4.5, COOCH3), 173.03 (C-7a), 173.27 (d, JC–P=1.8, COOCH3). EI MS=644.2202 (M+H) and 667.2043 (M+Na). HPLC=H2O/AcCN from 100/0 to 0/100 in 30 min = retention time 27.59 min, H2O/MeOH 20/80 isocratic = retention time 5.88 min.
Synthesis of 3-(2′-β-2′-deoxy-β-D-ribofuranosyl-5-[phenyl-(methoxy-glycinyl)]-phosphate)-6-(4-n-pentylphenyl)-2,3-dihydrofuro-[2,3-d]pyrimidin-2-one (23)
Compound 23 was prepared according to standard procedure B, from 5 (0.20 g, 0.48 mmol) in anhydrous THF (10 ml) and anhydrous pyridine (2 ml), 21c (0.63 g, 2.40 mmol) in anhydrous THF (5 ml) and NMI (0.20 ml, 2.40 mmol), and the reaction mixture was stirred at rt overnight. After this period, 21c (0.63 g, 2.40 mmol) in anhydrous THF (5 ml) and NMI (0.20 ml, 2.40 mmol) were added, and the reaction mixture was stirred at rt for a further 24 h. After this period, the solvent was removed and the residue dissolved in DCM. The organic phase was washed with a 0.5 M aqueous solution of HCl (twice) and water (twice), dried over MgSO4 and concentrated. The residue was purified by CC gradient elution of DCM/MeOH=98/2, then 97/3, to give another white solid, which was further purified by preparative TLC (gradient elution of DCM/MeOH=99/1, then 98/2, then 96/4) to give a white solid (4%, 0.012 g). 31P-NMR (MeOD, 202 MHz): δ 5.26, 5.15. 19F-NMR (DMSO, 471 MHz): δ −200.08. 1H-NMR (MeOD, 500 MHz): δ 8.66, 8.64 (1H, 2s, H-4), 7.70–7.16 (9H, m, Ph, PhO), 6.88, 6.86 (1H, 2s, H-5), 6.41, 6.37 (1H, 2t, J=3.5, JH-F=18.5, H-1′), 5.31–5.30 (0.5H, m, H-2′), 5.21–5.20 (0.5H, m, H-2′), 4.49–4.32 (4H, m, H-3′, H-4′, H-5′), 3.86–3.81 (2H, m, NHCH2), 3.68 (3H, 2s, OCH3), 2.67 (2H, t, J=8.3, α-CH2), 1.70–1.64 (2H, m, β-CH2), 1.42–1.28 (4H, m, γ-CH2, δ-CH2), 0.93 (3H, t, J=7.3, CH3). 13C-NMR (MeOD, 126 MHz): δ 14.36 (CH3), 23.56, 32.14, 32.59, 36.75 (C4H8), 43.70 (CH2NH), 52.57, 52.66 (2s, OCH3), 66.92 (C-5′), 75.25 (d, JC–P=10.9, C-4′), 75.46 (d, JC–P=10.6, C-4′), 85.24, 85.31 (2s, C-3′), 89.06 (d, JC–F=13.7, C-1′), 89.16 (d, JC–F=14.3, C-1′), 96.16 (d, JC–F=194.4, C-2′), 98.89, 98.91 (C-5), 110.12 (C-4a), 121.56, 121.60, 121.64, 121.67, 121.70, 121.74, 125.78, 125.96, 126.21, 126.35, 127.17, 130.19, 130.66, 130.89, 130.90 (PhO, Ph), 139.75, 139.78 (C-4), 146.47 (para-C), 152.18 (2s, ‘ipso’ PhO), 156.46 (C-6), 157.46 (C-2), 172.94 (COOCH3), 173.26 (C-7a), 173.28 (COOCH3). EI MS=666.2001 (M+Na). HPLC = H2O/AcCN from 95/5 to 0/100 in 30 min = retention time 28.60 min.
Synthesis of 3-(2′-di-fluoro-2′-deoxy-β-D-ribofuranosyl-5-[phenyl-(methoxy-L-alaninyl)]-phosphate)-6-(4-n-pentylphenyl)-2,3-dihydrofuro-[2,3-d]pyrimidin-2-one (24a) and (24b)
Compounds 24a and 24b were prepared according to standard procedure B from 6 (0.20 g, 0.46 mmol) in anhydrous THF (10 ml), 21a (0.64 g, 2.30 mmol) in anhydrous THF (5 ml) and NMI (0.18 ml, 2.30 mmol), and the reaction mixture was stirred at rt overnight. After this period, 21a (0.64 g, 2.30 mmol) in anhydrous THF (5 ml) and NMI (0.18 ml, 2.30 mmol) were added, and the reaction mixture was stirred at rt for a further 8 h. Next, NMI (0.09 ml, 1.20 mmol) was added and the reaction mixture stirred at rt for a further 24 h. After this period, the solvent was removed and the residue dissolved in DCM. The organic phase was washed with a 0.5 M aqueous solution of HCl (twice) and water (twice), dried over MgSO4 and concentrated. The residue was purified by CC gradient elution of DCM/MeOH=100/0, then 98/2, then 97/3, to give a white solid that was further purified by preparative TLC (gradient elution of DCM/MeOH=99/1, then 98/2, then 96/4) to give another white solid as a mix of two diastereoisomers (0.005 g) and as a single diastereoisomer (0.004 g).
24a 31P-NMR (MeOD, 202 MHz): δ 3.98, 3.96. 19F-NMR (DMSO, 471 zMHz): δ −118.46, −118.83, −118.93, −119.44, −120.23. 1H-NMR (MeOD, 500 MHz): δ 8.55, 8.51 (1H, 2s, H-4), 7.81–7.65 (2H, m, Ph), 7.48–7.16 (7H, m, Ph), 6.96, 6.88 (1H, 2s, H-5), 6.48–6.44 (1H, m, H-1′), 4.68–4.46 (2H, m, H-5′), 4.43–4.31 (1H, m, H-4′), 4.31–4.24 (1H, m, H-3′), 4.04–3.92 (1H, m, CHCH3), 3.68, 3.67 (3H, 2s, OCH3), 2.69 (2H, t, J=7.6, α-CH2), 1.71–1.65 (2H, m, β-CH2), 1.43–1.30 (7H, m, γ-CH2, δ-CH2, CHCH3), 0.94 (3H, t, J=6.9, CH3). 13C-NMR (MeOD, 126 MHz): δ 14.35 (CH3), 21.00 (d, JC–P=7.8, CHCH3), 21.06 (d, JC–P=7.0, CHCH3), 23.55, 32.15, 32.57, 36.76 (C4H8), 50.04 (CHCH3), 52.79 (OCH3), 65.76 (C-5′), 69.82, 70.04 (2s, C-4′), 82.86, 82.92 (2s, C-3′), 94.47 (C-1′), 98.86, 98.89 (2s, C-5), 110.87 (C-4a), 121.32, 121.52, 121.56, 126.06, 126.09, 126.22, 126.44, 126.48, 127.01, 129.59, 129.83, 130.23, 130.40, 130.94, 131.03, 132.51, 132.64 (C-2′, PhO, Ph), 138.38 (C-4), 145.11, 146.70 (2s, para-C), 150.97 (‘ipso’ PhO), 156.66 (C-6), 158.01 (C-2), 173.56 (C-7a), 174.50 (COOCH3). HPLC = H2O/AcCN from 95/5 to 0/100 in 40 min = retention time 29.72, 33.48 min.
24b 31P-NMR (MeOD, 202 MHz): δ 3.89. 19F-NMR (DMSO, 471 MHz): δ −118.54. 1H-NMR (MeOD, 500 MHz): δ 8.54 (1H, s, H-4), 7.71–7.69 (2H, m, Ph), 7.45–7.19 (7H, m, Ph), 6.83 (1H, 2s, H-5), 6.56–6.38 (1H, m, H-1′), 4.66–4.51 (1H, m, H-5′), 4.50–4.41 (1H, m, H-5′), 4.40–4.29 (1H, m, H-4′), 4.29–4.20 (1H, m, H-3′), 4.08–3.98 (1H, m, CHCH3), 3.69 (3H, s, OCH3), 2.69 (2H, t, J=7.7, α-CH2), 1.73–1.58 (2H, m, β-CH2), 1.46–1.26 (7H, m, γ-CH2, δ-CH2, CHCH3), 0.94 (3H, t, J=7.0, CH3). 13C-NMR (MeOD, 126 MHz): δ 14.35 (CH3), 20.45 (d, JC–P=6.6, CHCH3), 23.56, 32.15, 32.58, 36.75 (C4H8), 50.04 (CHCH3), 52.78 (OCH3), 65.56 (C-5′), 68.87 (C-4′), 81.04 (C-3′), 94.47 (C-1′), 98.72 (C-5), 110.81 (C-4a), 121.44, 121.48, 121.53, 121.57, 126.05, 126.24, 127.00, 129.55, 129.83, 130.23, 130.82, 130.98, 132.63 (C-2′, PhO, Ph), 138.38 (C-4), 146.72 (para-C), 156.66 (C-6), 158.02 (C-2), 173.56 (C-7a), 174.50 (COOCH3). HPLC=H2O/AcCN from 95/5 to 0/100 in 40 min = retention time 34.41 min; H2O/MeOH 20/80 isocratic =23.53.
Synthesis of 3-(2′-di-fluoro-2′-deoxy-β-D-ribofuranosyl-5-[phenyl-(benzoxy-L-alaninyl)]-phosphate)-6-(4-n-pentylphenyl)-2,3-dihydrofuro-[2,3-d]pyrimidin-2-one (24c)
Compound 24c was prepared according to standard procedure B from 6 (0.20 g, 0.46 mmol) in anhydrous THF (12 ml), 21b (0.81 g, 2.30 mmol) in anhydrous THF (5 ml) and NMI (0.18 ml, 2.30 mmol), and the reaction mixture was stirred at rt overnight. After this period, 21b (0.81 g, 2.30 mmol) in anhydrous THF (5 ml) and NMI (0.18 ml, 2.30 mmol) were added, and the reaction mixture was stirred at rt for a further 8 h. Next, NMI (0.09 ml, 1.20 mmol) was added and the reaction mixture was stirred at rt overnight. After this period, the solvent was removed and the residue dissolved in DCM. The organic phase was washed with a 0.5 M aqueous solution of citric acid (twice) and water (twice), dried over MgSO4 and concentrated. The residue was purified by CC gradient elution of DCM/MeOH=100/0, then 98/2, then 96/4, to give a white solid that was further purified by preparative HPLC (isocratic MeOH/H2O=80/20) to give another white solid (4%, 0.012 g). 31P-NMR (MeOD, 202 MHz): δ 3.99, 3.79. 19F-NMR (DMSO, 471 MHz): δ −117.93, −118.23, −118.44, −118.74, −119.61. 1H-NMR (MeOD, 500 MHz): δ 8.52, 8.49 (1H, 2s, H-4), 7.66 (2H, d, J=7.9, Ph), 7.40–7.17 (12H, m, Ph, PhO, CH2Ph), 6.85, 6.79 (1H, 2s, H-5), 6.44 (1H, dd, J=14.8, 7.4, H-1′), 5.21–5.10 (2H, m, CH2Ph), 4.65–4.52 (1H, m, H-5′), 4.51–4.38 (1H, m, H-5′), 4.39–4.29 (1H, m, H-4′), 4.27–4.16 (1H, m, H-3′), 4.16–4.04 (1H, m, CHCH3), 2.67 (2H, t, J=7.5, α-CH2), 1.72–1.60 (2H, m, β-CH2), 1.47–1.28 (7H, m, γ-CH2, δ-CH2, CHCH3), 0.93 (3H, t, J=7.0, CH3). 13C-NMR (MeOD, 126 MHz): δ 14.36 (CH3), 20.26 (d, JC–P=7.7, CHCH3), 20.43 (d, JC–P=6.9, CHCH3), 23.56, 32.14, 32.58, 36.75 (C4H8), 51.71, 51.93 (CHCH3), 65.55 (d, JC–P=6.1, C-5′), 65.62 (d, JC–P=5.8, C-5′), 68.02, 68.05 (2s, CH2Ph), 70.75, 70.91 (2s, C-4′), 81.03 (C-3′), 87.68 (C-1′), 98.75, 98.78 (2s, C-5), 110.80 (C-4a), 121.51, 121.54, 121.55, 121.59, 126.05, 126.41, 126.98, 129.12, 129.20, 129.35, 129.58, 130.21, 130.91, 130.97, (C-2′, PhO, CH2Ph, Ph), 137.17 (‘ipso’ OCH2Ph), 138.25, 138.35 (2s, C-4), 146.68 (para-C), 152.07 (‘ipso’ PhO), 156.62 (C-6), 157.94 (C-2), 173.49 (C-7a), 174.66, 174.96 (COOCH2Ph). EI MS=752.2555 (M+H), 774.2372 (M+Na). HPLC=H2O/AcCN from 95/5 to 0/100 in 40 min = retention time 38.96 min; H2O/MeOH 20/80 isocratic = retention time 33.48, 35.43 min.
Synthesis of 3-(2′-di-fluoro-2′-deoxy-β-D-ribofuranosyl-5-[phenyl-(methoxy-glycinyl)]-phosphate)-6-(4-n-pentylphenyl)-2,3-dihydrofuro-[2,3-d]pyrimidin-2-one (24d)
Compound 24d was prepared according to standard procedure B from 6 (0.20 g, 0.46 mmol) in anhydrous THF (10 ml), 21c (0.61 g, 2.30 mmol) in anhydrous THF (5 ml) and NMI (0.18 ml, 2.30 mmol), and the reaction mixture was stirred at rt overnight. After this period, 21c (0.61 g, 2.30 mmol) in anhydrous THF (5 ml) and NMI (0.18 ml, 2.30 mmol) were added, and the reaction mixture was stirred at rt for further 8 h. Next, NMI (0.09 ml, 1.20 mmol) was added, and the reaction mixture stirred at rt for a further 24 h. After this period, the solvent was removed and the residue dissolved in DCM. The organic phase was washed with a 0.5 M aqueous solution of HCl (twice) and water (twice), dried over MgSO4 and concentrated. The residue was purified by CC gradient elution of DCM/MeOH=98/2, then 97/3, to give a white solid that was further purified by preparative TLC (gradient elution of DCM/MeOH =99/1, then 98/2, then 96/4) to give another white solid (14%, 0.042 g). 31P-NMR (MeOD, 202 MHz): δ 5.42, 4.92. 19F-NMR (DMSO, 471 MHz): δ −118.13, −118.65. 1H-NMR (MeOD, 500 MHz): δ 8.55, 8.52 (1H, 2s, H-4), 7.70–7.59 (2H, m, Ph), 7.48–7.17 (7H, m, Ph, PhO), 6.88, 6.79 (1H, 2s, H-5), 6.45 (1H, d, J=7.0, H-1′), 4.73–4.63 (1H, m, H-5′), 4.60–4.48 (1H, m, H-5′), 4.47–4.30 (1H, m, H-4′), 4.30–4.23 (1H, m, H-3′), 3.89–3.82 (2H, m, NHCH2), 3.70 (3H, 2s, OCH3), 2.64 (2H, t, J=7.6, α-CH2), 1.69–1.59 (2H, m, β-CH2), 1.45–1.26 (4H, m, γ-CH2, δ-CH2), 0.92 (3H, t, J=7.0, CH3). 13C-NMR (MeOD, 126 MHz): δ 14.39 (CH3), 23.57, 32.10, 32.61, 36.76 (C4H8), 43.72 (CH2NH), 52.72 (OCH3), 65.55 (C-5′), 70.90 (C-4′), 80.97 (C-3′), 87.40, 87.52 (C-1′), 98.76, 98.79 (2s, C-5), 110.80, 110.83 (2s, C-4a), 121.53, 121.56, 121.60, 123.49, 126.01, 126.03, 126.44, 126.47, 126.92, 126.94, 129.57, 130.18, 130.86, 130.96, 131.00, 132.61 (C-2′, PhO, Ph), 138.32, 138.44 (C-4), 146.63 (para-C), 152.03, 152.09 (2s, ‘ipso’ PhO), 156.64 (C-6), 157.88, 157.94 (2s, C-2), 173.04, 173.08 (2s, C-7a), 173.47 (COOCH3). EI MS=662.2073 (M+H), 684.1885 (M+Na). HPLC = H2O/AcCN from 100/0 to 0/100 in 40 min = retention time 35.12 min; H2O/MeOH 20/80 isocratic = retention time 21.44 min.
Procedures for the anti-VZV experiments in HEL cell cultures
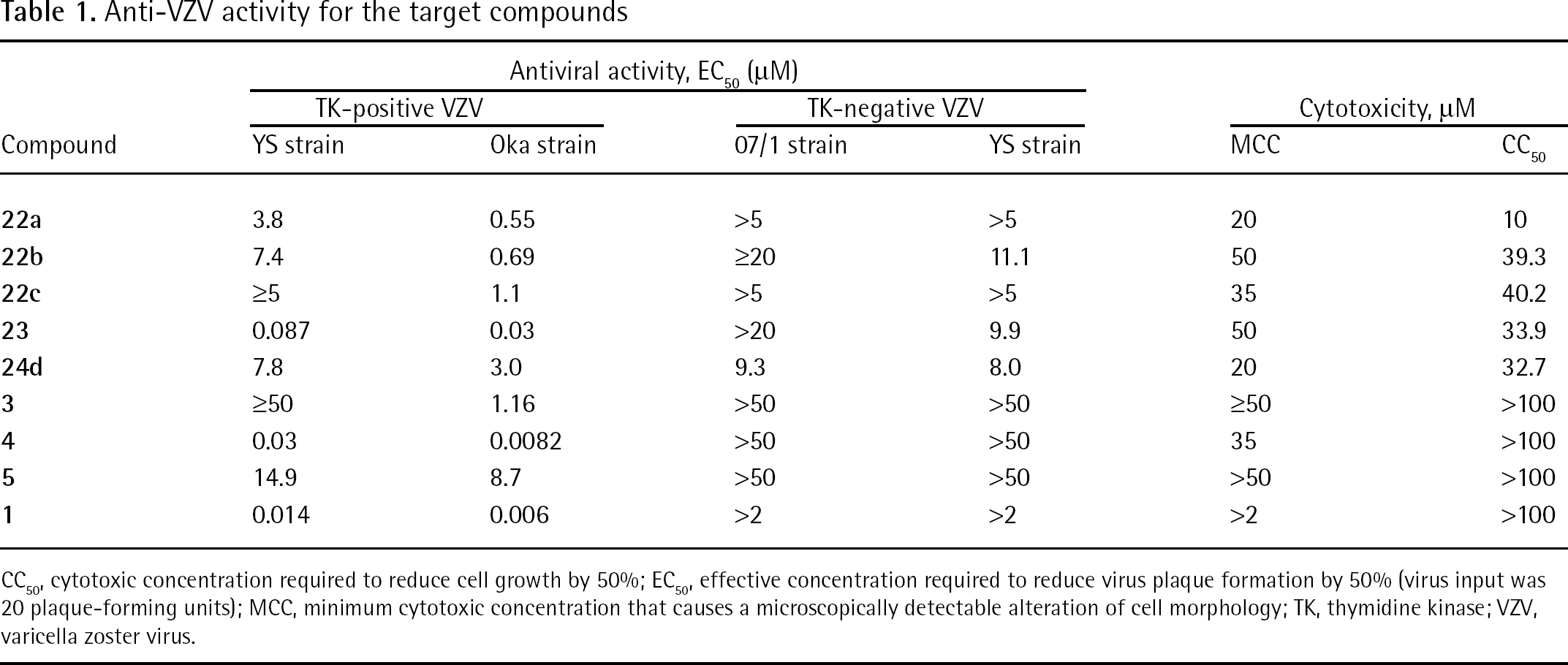
The laboratory wild-type VZV strains Oka and Davis and the TK-deficient VZV strains 07/1 and YS were used. Confluent HEL cell cultures grown in 96-well microtitre plates were inoculated with VZV at an input of 20 plaque-forming units per well. After a 2 h incubation period, residual virus was removed and varying concentrations of the test compounds were added (in duplicate). Antiviral activity was expressed as the effective concentration required to reduce viral plaque formation after 5 days by 50% (EC50) as compared with untreated controls. Cytotoxicity was expressed as the minimum cytotoxic concentration or the compound concentration that causes a microscopically detectable alteration of cell morphology.
Molecular modelling
All molecular modelling studies were performed on a MacPro dual 2.66 GHz Xeon running Ubuntu 8 using Molecular Operating Environment 2008.10 (MOE; Chemical Computing Group, Inc., Montreal, QC, Canada) and Plants 1.0 [25]. Hydrogen atoms were added to the crystal structure of proteins and minimised with MOE until a gradient of 0.05 kcal/mol/Å was reached, using the MMFF94x forcefield. The partial charges were automatically calculated. Docking experiments were carried out using Plants 1.0 with the default parameters. The results were examined with MOE.