
Abstract
Background:
The development of carbohydrate-binding agents as novel therapeutics for the inhibition of highly glycosylated enveloped viruses has generated much attention in recent literature. Possessing a potential dual mode of action by inhibiting virus entry and exposing the virion to neutralization by the host immune system upon the deletion of envelope glycans under drug pressure, these substances might provide a new direction in antiviral treatment. Phenylboronic acids are widely known to bind the cis-diol functionality of carbohydrate structures, thereby identifying themselves as potential lead structures. To date, few details have been disclosed of the structure–activity relationship of these substances in correlation to their antiviral activity.
Methods:
In this study, a compound library of a diverse range of ortho-, meta- and para- ring-substituted monophenylboronic acids and glutamine phenylboronic acid analogues was prepared, characterized and evaluated to probe antiviral activity versus a broad range of (enveloped) viruses.
Results:
The compounds described herein lack antiviral activity. They also did not show measurable binding to HIV type-1 (HIV-1) gp120, using surface plasmon resonance technology. However, of note is the general lack of toxicity, which suggests that further investigation of the compounds as potential therapeutics is needed.
Conclusions:
The monophenylboronic acids tested exhibited no antiviral activity as potential carbohydrate binders versus a broad range of enveloped and non-enveloped viruses. The compounds tested did not bind HIV-1 gp120, possibly because of their small size and lack of multivalency.
Introduction
The use of carbohydrate binding agents (CBAs) has recently been identified as a novel and potentially efficacious approach to antiviral chemotherapy [1,2]. CBAs are perceived to posses two distinct modes of antiviral action. Firstly, this action is demonstrated through their ability to covalently bind to cis-diol functionalities of carbohydrate moieties displayed on the glycans of certain viral envelopes (that is, HIV gp120), thereby blocking viral entry. Secondly, mutation of the virus to alleviate the drug pressure by progressive deletions in the carbohydrate surface of the glycan shield (resistance development) will engender the virus to the immune response of the host organism, targeting previously concealed immunogenic epitopes of the viral envelope [2].
Several peptidic and non-peptidic compounds acting with the CBA mode of action have been described [1–4]. These include the α(1,3)- and the α(1,6)-mannose-specific Hippeastrum hybrid agglutinin or the Galanthus nivalis agglutinin, the acetylglucosamine-specific Urtica dioica agglutinin plant lectins and the α(1,2)-mannose-specific non-peptidic antibiotics pradimicin A and S [4,5]. These CBAs have been demonstrated to posses antiviral activity against a range of viruses, including HIV [3,4], influenza virus [6], coronaviruses [7,8], Ebola virus [9], hepatitis C virus [10] and human T-lymphotropic virus type 1 [11].
Examination of the structure of the HIV gp120 glycan and other enveloped viruses shows an unusually high terminal mannose residue concentration (in particular, α-1,2-mannose, α-1,3-mannose and α-1,6-mannose oligomers) at the surface of the glycan. The majority of mammalian cells display carbohydrate termini of the glycans that are almost completely devoid of mannose residues; thus, selectivity for a high-mannose glycan surface should be a potential antiviral target. Most plant-derived CBAs, highlighted above, that show anti-HIV activity are specific for α-1,3-mannose and α-1,6-mannose oligomer binding.
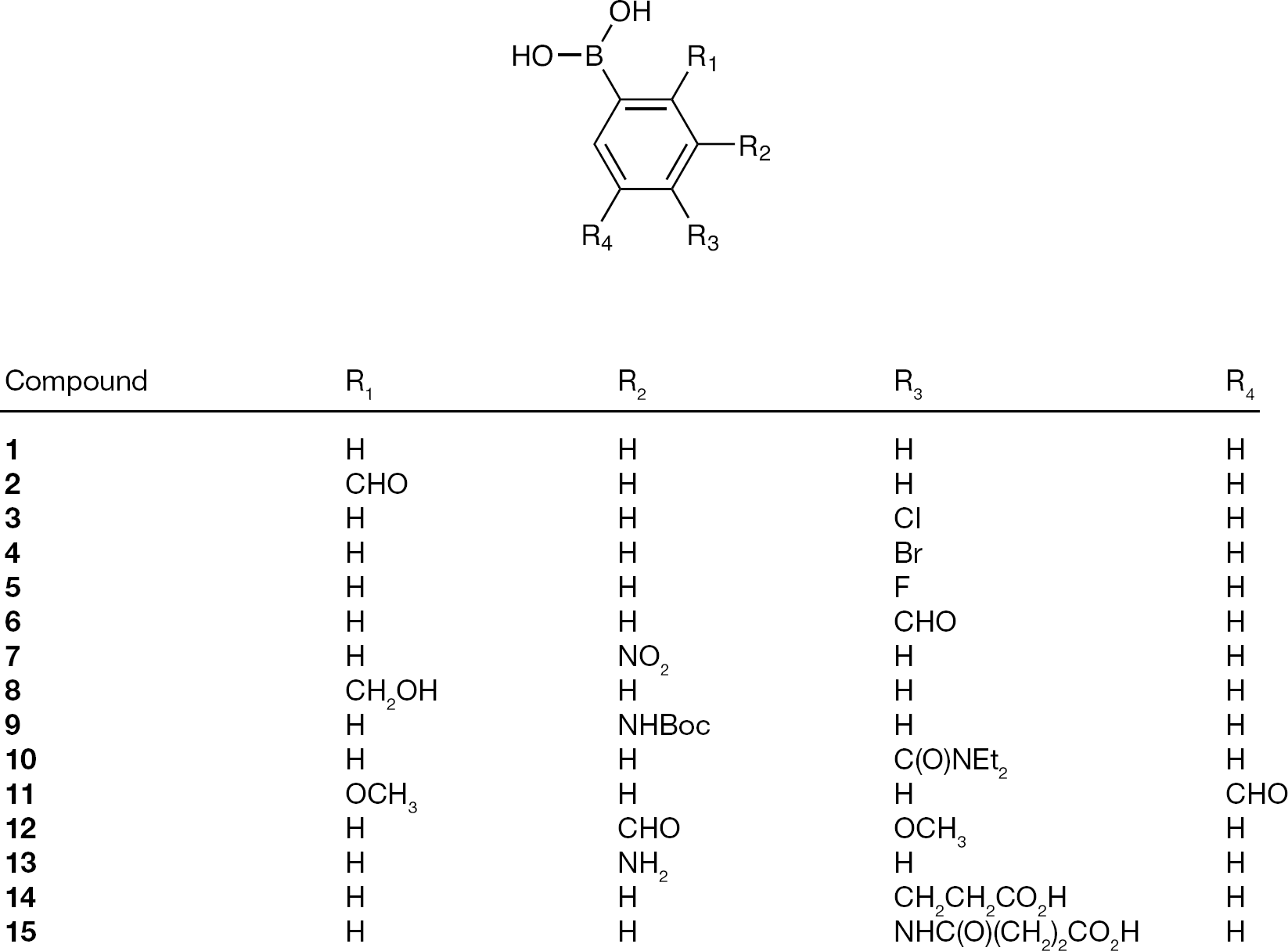
Substituted monophenylboronic acid compounds
Phenylboronic acids (PBAs) have also been shown [12] to demonstrate carbohydrate recognition via formation of reversible covalent bonds with the 1,2- or 1,3-diols commonly displayed on carbohydrates to provide five- or six-membered cyclic esters in aqueous media. The rigid nature of the saccharide cis-diols ensures a highly stable cyclic ester. Selectivity between saccharide structures was established for simple PBA [13]: D-fructose > D-galactose > D-mannose > D-glucose >> ethylene glycol. This pattern reportedly remains constant for all monoboronic acids; however, both the definition and method of measurement of the ‘binding constant’ provides some discrepancies in the literature (for a review of the subject and for design factors of boronic acid CBAs see references [14,15]).
Extensive studies have been conducted on the selective binding of D-glucose because the breakdown of this saccharide is an indicator in the pathology of several diseases, including diabetes [16]; therefore, several binder–indicator compounds have been synthesized. Studies to design mannose-selective PBA-based CBAs have, however, proved to be much more elusive.
Recently, the first synthesis of a boronic-acid-containing drug has been reported to specifically bind to HIV type-1 (HIV-1) gp120, exhibiting antiviral activity against HIV in cell culture [17].
Herein, we describe initial efforts in identifying mannose selective PBA-based CBAs. Structurally diverse monophenylboronic acids were obtained from both commercial sources and via synthesis using selected literature methods (Figures 1 and 2), and these were combined with novel glutamine analogue amino-acid-based monophenylboronic acids (Figure 3) to complete the compound library. These structures where exposed to in-house testing methods for binding affinity against HIV gp120 and for determining their inhibitory activity against a range of other enveloped (glycosylated) viruses.
Methods
Chemistry
1H NMR spectra were recorded on a Bruker AMX500 (500 MHz) spectrometer (Bruker Instruments, Inc., Billerica, MA, USA). Chemical shifts (δH) are reported in parts per million (ppm) and are referenced to the residual protonated solvent peak. Abbreviations used in the description of multiplicities are s (singlet), d (doublet), dd (double doublet), dt (double triplet), t (triplet), q (quartet), m (multiplet) and br (broad). Coupling constants (J) are quoted in Hertz (Hz) to the nearest 0.1 Hz.
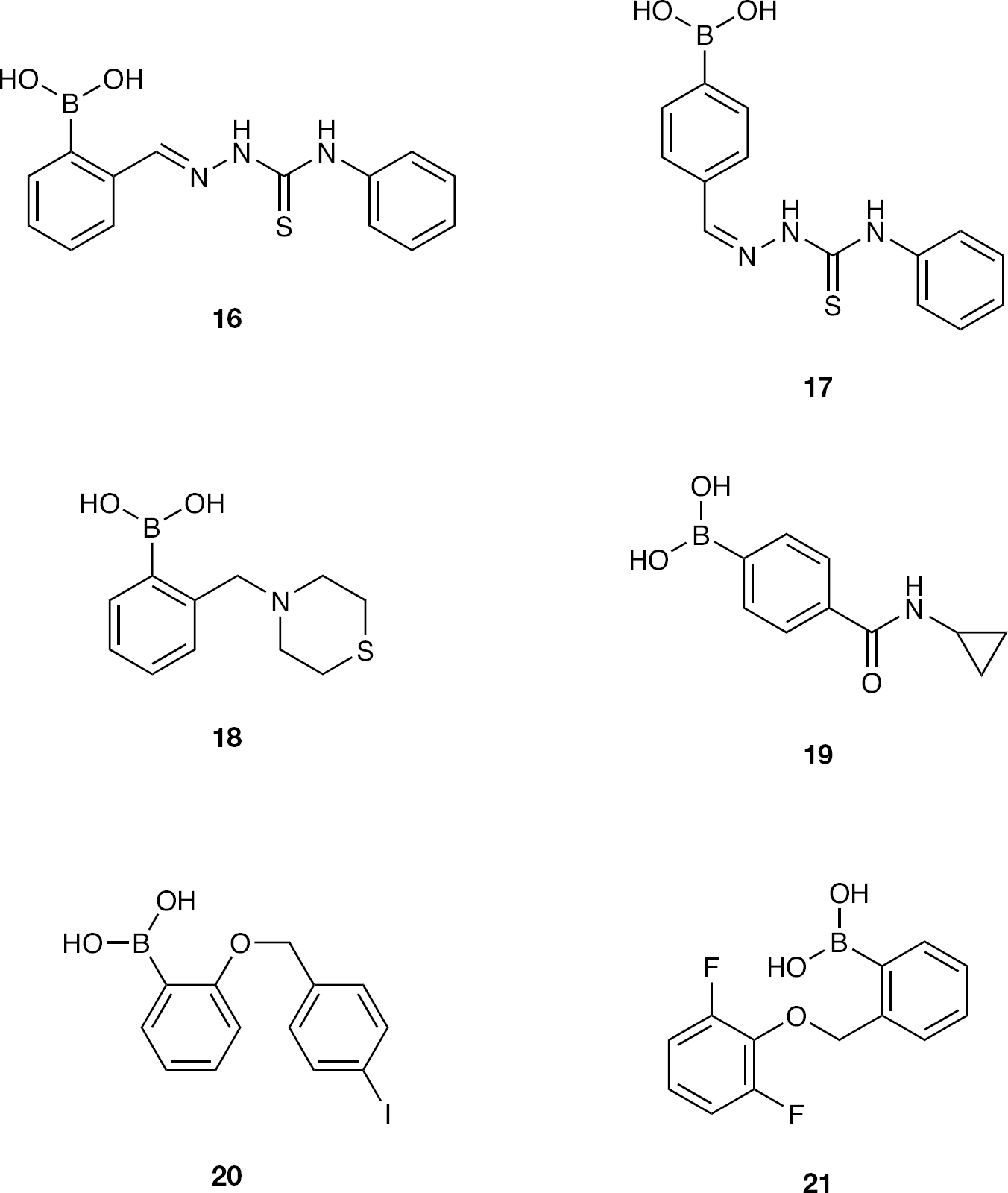
Further elaborated phenylboronic acids
13C NMR spectra were recorded on a Bruker AMX500 (125.8 MHz) spectrometer (Bruker Instruments, Inc.). Chemical shifts are quoted in ppm and referenced to the residual protonated solvent peak with proton decoupling, using DEPT, HMQC, and APT editing to aid assignment.
11B NMR spectra were recorded on a Bruker AMX500 (160.5 MHz) spectrometer (Bruker Instruments, Inc.). Chemical shifts are quoted in ppm and referenced to the residual protonated solvent peak with proton decoupling. Samples were performed in a pure quartz 5 mm NMR tube.
Low-resolution and high-resolution mass spectra (m/z) were recorded on a Waters GCT Premier spectrometer (Waters, Elstree, UK) using an electron ionization (EI) source. m/z values of major peaks are reported in Da, with intensities quoted as percentages of the base peak.
Thin-layer chromatography (TLC) was performed on Merck aluminium foil-backed sheets precoated with 0.2 mm Kieselgel 60 F-54 (Merck, Darmsted, Germany) with development by the ascending method. Product spots were visualized by the quenching of ultraviolet fluorescence (λmax 254 nm) or by staining with a solution of 5% (w/v) dodecamolybdophosphoric acid in ethanol.
Flash column chromatography was performed under pressure on silica gel, 60 A and 40–60 μm (Phase Separations Ltd, Deeside, Clwyd, UK). Samples were applied as a concentrated solution in the same eluent or pre-adsorbed on silica gel. Fractions containing the product were identified by TLC, pooled and the solvent removed in vaccuo.
All anhydrous solvents were used as obtained from Sigma–Aldrich Co. (St Louis, MO, USA). All reactions were performed under an inert argon atmosphere unless otherwise stated.
Compounds
Compounds
3-(4-boronophenyl)propanoic acid (14)
1,2-dibromoethane (catalyst) and (4-bromophenyl)propionic acid (500 mg, 2.18 mmol) were added to a stirred suspension of magnesium turnings (60 mg) in Et2O (100 ml), and stirring was continued at room temperature (RT) overnight. The mixture was cooled to −78°C and triisopropyl borate (0.5 ml, 2.18 mmol) in dry Et2O (1 ml) was added dropwise, the reaction was stirred for a further 1 h at −78°C and allowed to warm to RT overnight. The reaction was cooled to 0°C and 8% HCl (10 ml) was added slowly, the reaction was then diluted with water (10 ml) and stirred for 10 min at 0°C. The organic layer was extracted with Et2O (2×25 ml), the organic layers were combined, dried (MgSO4), filtered and concentrated in vaccuo to yield the title compound as a white powder (370 mg, 1.91 mmol, 87%): 1H NMR (500 MHz; dimethyl sulfoxide [DMSO]) δH: 2,49 (2H, t, J=7.5 Hz, CH2), 2.77 (2H, t, J=7.5 Hz, CH2CO2H), 7.17 (2H, d, J=8.0 Hz, ArH), 7.46 (2H, d, J=8.0 Hz, ArH), 12.08 (1H, br, s, CO2H); 13C NMR (125.8 MHz; CD3OD) δC: 31.33 (CH2), 36.35 (CH2), 120.84, 131.38, 132.49 (ArCH), 141.50 (Cipso-CH2), 176.36 (C(O)); 11B NMR (160.5 MHz; CD3CN) δB: 19.92 (B(OH)2).
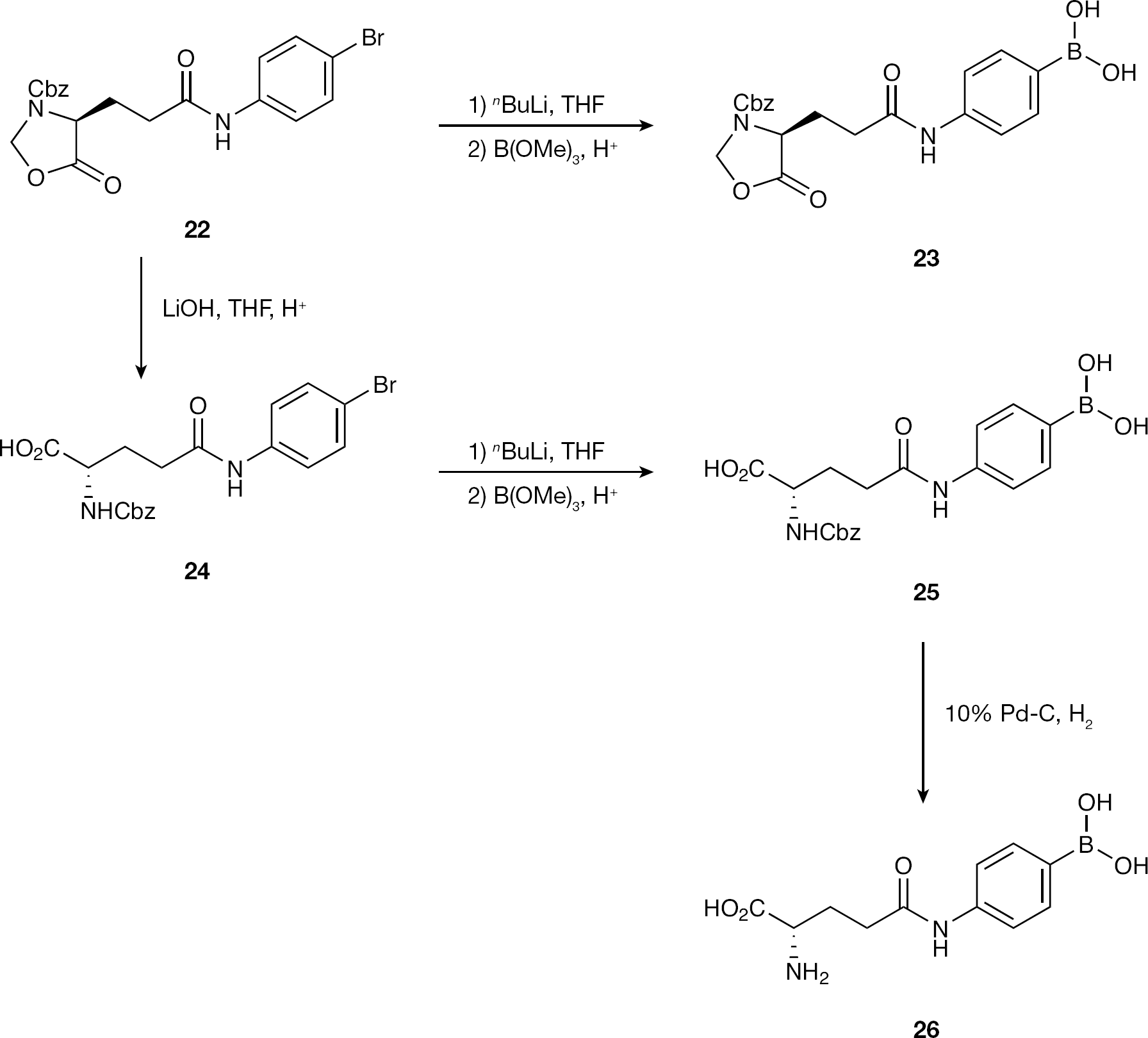
Synthesis of phenylboronic acid glutamine analogues
(S)-4-(3-(3-(benzyloxycarbonyl)-5-oxo-oxazolidin-4yl) propanamido)-phenylboronic acid (23)
Known bromide
(S)-2-(benzyloxycarbonylamino)-5-(4-boronophenylamino)-5-oxopentanoic acid (25)
Known bromide
(S)-2-amino-5-(4-boronophenylamino)-5-oxopentanoic acid (26)
Carbobenzyloxy (Cbz)-protected amide
Biology
Virological assays
The antiviral assays, other than the anti-HIV assays, were based on inhibition of virus-induced cytopathic effect in human lung fibroblast (herpes simplex virus type-1 [strain KOS], herpes simplex virus type-2 [strain G], vaccinia virus and vesicular stomatitis virus), African green monkey kidney (parainfluenza-3, reovirus-1, Sindbis, Coxsackie B4 and Punta Toro virus), human cervix carcinoma (vesicular stomatitis virus, Coxsackie virus B4 and respiratory syncytial virus) or Madin–Darby canine kidney (MDCK; influenza A [H1N1 and H3N2] and influenza B) cell cultures. Confluent cell cultures in microtitre 96-well plates were inoculated with 100 50% cell culture infective doses (CCID50; where 1 CCID50 is the virus dose to infect 50% of the cell cultures) of virus. After a 1 h virus adsorption period, residual virus was removed, and the cell cultures were incubated in the presence of varying concentrations (100, 40, 8, 1.6 and 0.32 μM) of the test compounds. Viral cytopathicity was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. For the human cytomegalovirus assays, confluent human embryonic lung (HEL) fibroblasts were grown in 96-well microtitre plates and infected with the human cytomegalovirus strains Davis and AD-169 at 100 plaque-forming units per well. After a 2 h incubation period, residual virus was removed and the infected cells were further incubated with medium containing different concentrations of the test compounds (in duplicate). After incubation for 7 days at 37°C, virus-induced cytopathogenicity was monitored microscopically after ethanol fixation and staining with Giemsa. Varicella zoster virus drug susceptibility tests were performed on confluent HEL cells in 96-well microtitre plates by the plaque reduction assay. Monolayers were infected with 20 plaque-forming units of cell-associated virus per well. For each assay, virus controls (infected untreated cells) were included. After a 2 h incubation period, the virus inoculum was removed and the media replaced by the different dilutions (in duplicate) of the tested molecules. Serial dilutions of test compounds were incubated with the infected monolayers for 5 days. After the 5-day incubation period, the cells were fixed and stained with Giemsa and the level of virus-induced cytopathic effect was determined by counting the number of plaques for each dilution. The methodology of the anti-HIV assays was as follows: human T-lymphocyte CEM cells (approximately 3×105 cells/cm3) were infected with 100 CCID50 of HIV-1(IIIB) or HIV type-2(ROD)/ml and seeded in 200 μl wells of a microtitre plate containing appropriate dilutions of the test compounds. After 4 days of incubation at 37°C, HIV-induced CEM giant cell formation was examined microscopically.
Determination of binding of the test compounds to HIV-1 gp120 through surface plasmon resonance analyses
Recombinant gp120 protein from HIV-1 strain IIIB (ImmunoDiagnostics, Inc., Woburn, MA, USA; produced by Chinese hamster ovary [CHO] cell cultures) was covalently immobilized on the carboxymethylated dextran matrix of a CM5 sensor chip (GE Healthcare, Uppsala, Sweden) in 10 mM sodium acetate (pH 4.0), using standard amine coupling chemistry to a final density of 122 RU (approximately 1 fmol of gp120). A reference flow cell was used as a control for non-specific binding and refractive index changes. All interaction studies were performed at 25°C on a Biacore T100 instrument (GE Healthcare). The compounds were diluted in HBS-P (10 mM 2-[4-(2-hydroxyethyl) piperazin-1-yl]ethanesulphonic acid, pH 7.0, 150 M NaCl and 0.05% P20 surfactant; pH 7.4) and with 10 mM CaCl2. Samples were injected for 2 min at a flow rate of 45 μl/min and the dissociation was followed for 6 min. Several buffer blanks were used as double referencing. The CM5 sensor chip surface was regenerated with one injection of 50 mM NaOH.
Results
The synthetic route to the generation of the desired PBA glutamine analogue involved hydrolysis of the lactone-protected bromide starting material, installation of the boronic acid and finally hydrogenation of the carbobenzoate to generate the free amino acid (Figure 3). The synthesis was chosen to allow the introduction of the desired boronic acid functionality at each point in the route to generate further compounds for evaluation.
Introduction of the boronic acid moiety to known bromides
Compounds
Anti-HIV-1 activity, anti-HIV-2 activity and cytotoxicity in human T-lymphocyte cells
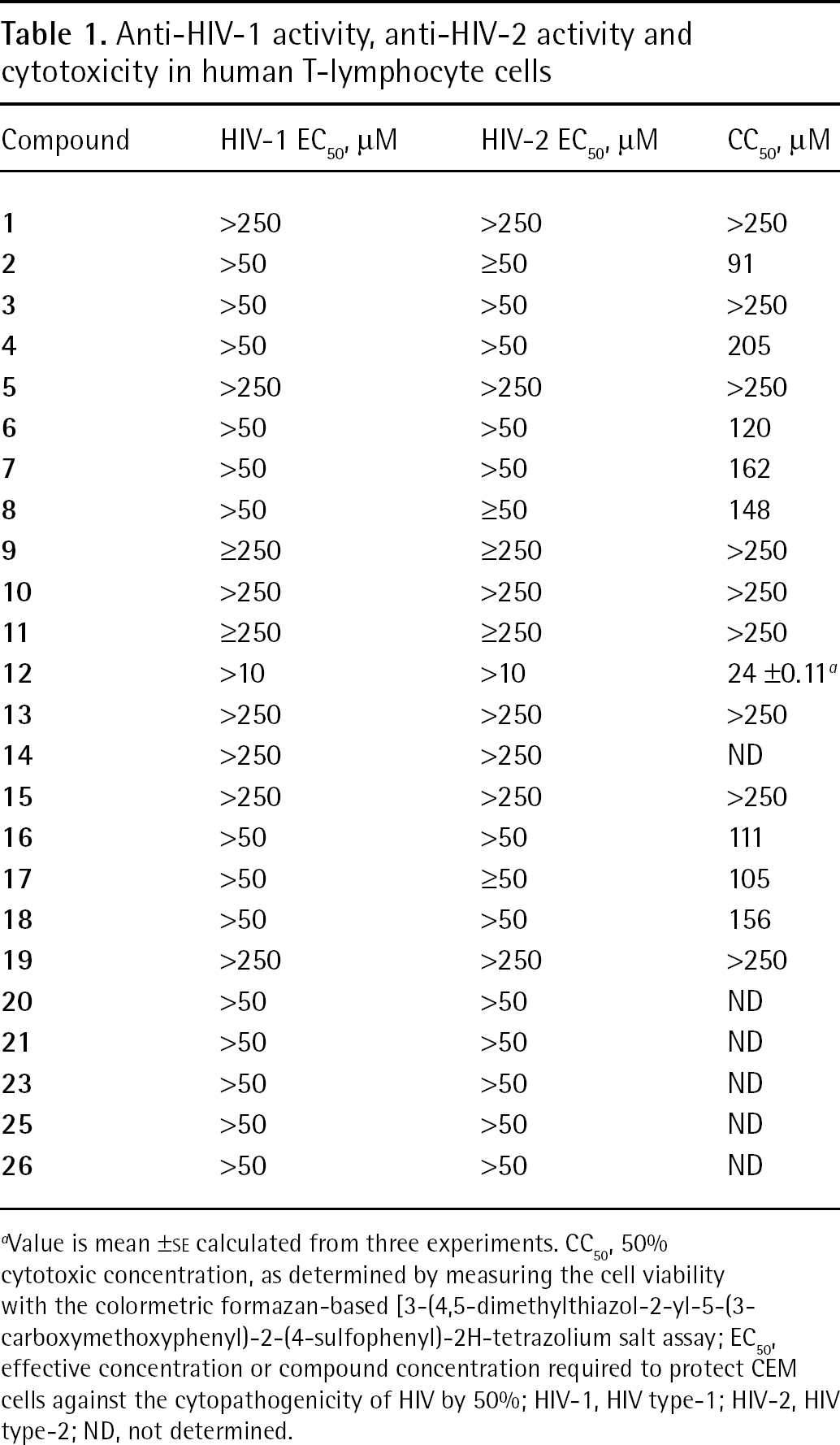
Value is mean ±SE calculated from three experiments. CC50, 50% cytotoxic concentration, as determined by measuring the cell viability with the colormetric formazan-based [3-(4,5-dimethylthiazol-2-yl-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt assay; EC50, effective concentration or compound concentration required to protect CEM cells against the cytopathogenicity of HIV by 50%; HIV-1, HIV type-1; HIV-2, HIV type-2; ND, not determined.
Anti-varicella zoster virus activity and cytostatic/cytotoxic activity in human embryonic lung cells
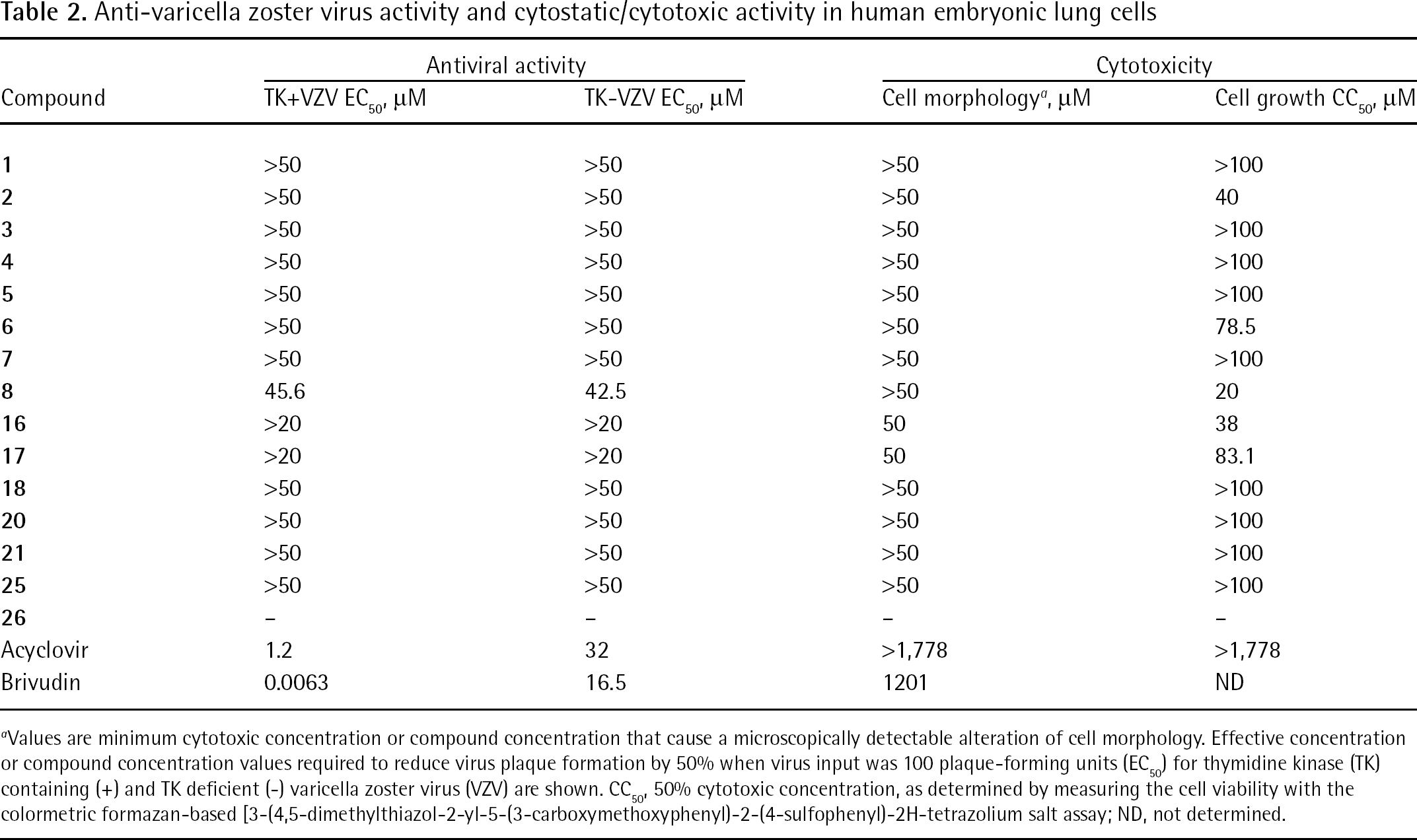
Values are minimum cytotoxic concentration or compound concentration that cause a microscopically detectable alteration of cell morphology. Effective concentration or compound concentration values required to reduce virus plaque formation by 50% when virus input was 100 plaque-forming units (EC50) for thymidine kinase (TK) containing (+) and TK deficient (−) varicella zoster virus (VZV) are shown. CC50, 50% cytotoxic concentration, as determined by measuring the cell viability with the colormetric formazan-based [3-(4,5-dimethylthiazol-2-yl-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt assay; ND, not determined.
Anti-influenza virus activity and cytotoxicity in Madin–Darby canine kidney cells
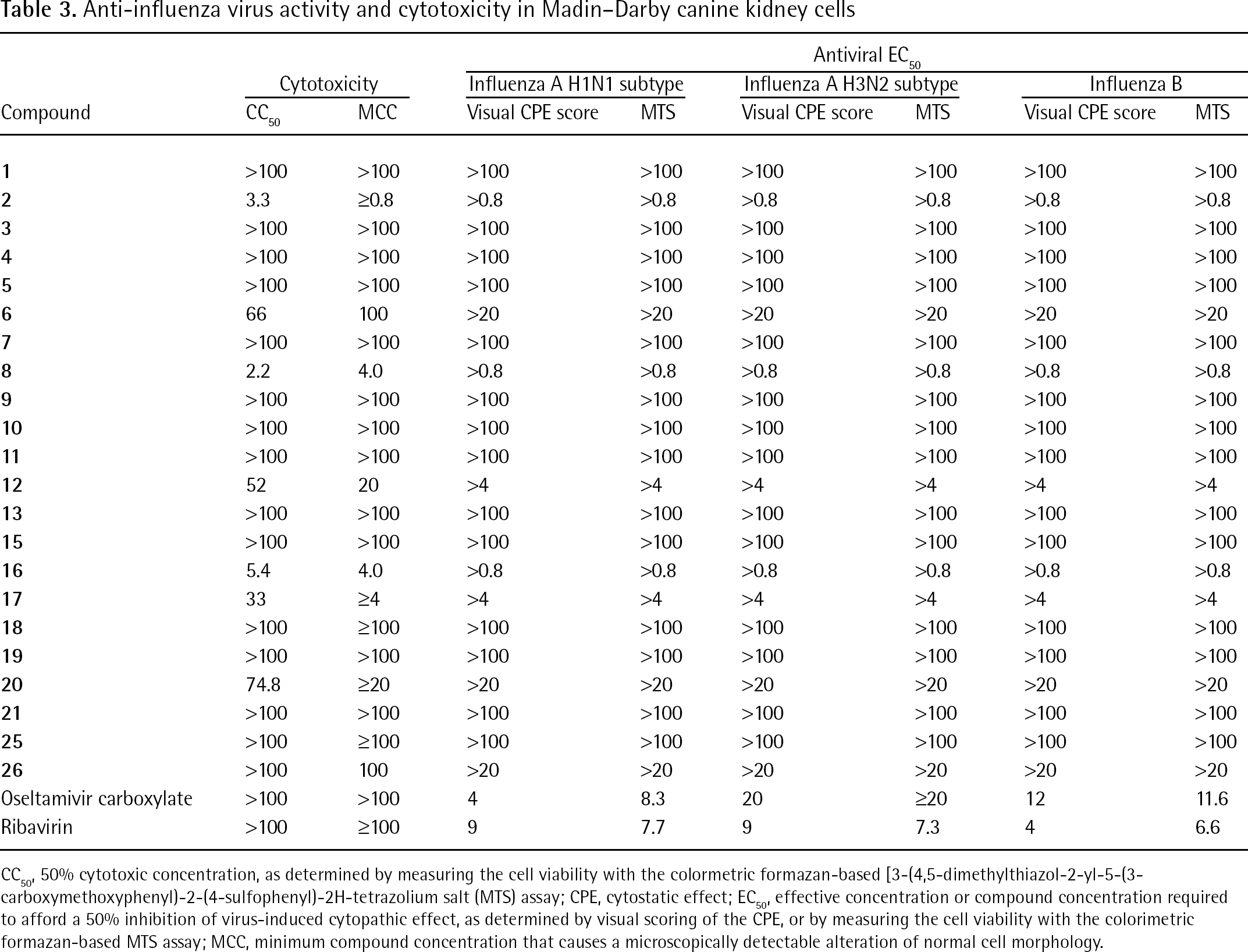
CC50, 50% cytotoxic concentration, as determined by measuring the cell viability with the colormetric formazan-based [3-(4,5-dimethylthiazol-2-yl-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTS) assay; CPE, cytostatic effect; EC50, effective concentration or compound concentration required to afford a 50% inhibition of virus-induced cytopathic effect, as determined by visual scoring of the CPE, or by measuring the cell viability with the colorimetric formazan-based MTS assay; MCC, minimum compound concentration that causes a microscopically detectable alteration of normal cell morphology.
Activity of compounds in human embryonic lung cell cultures
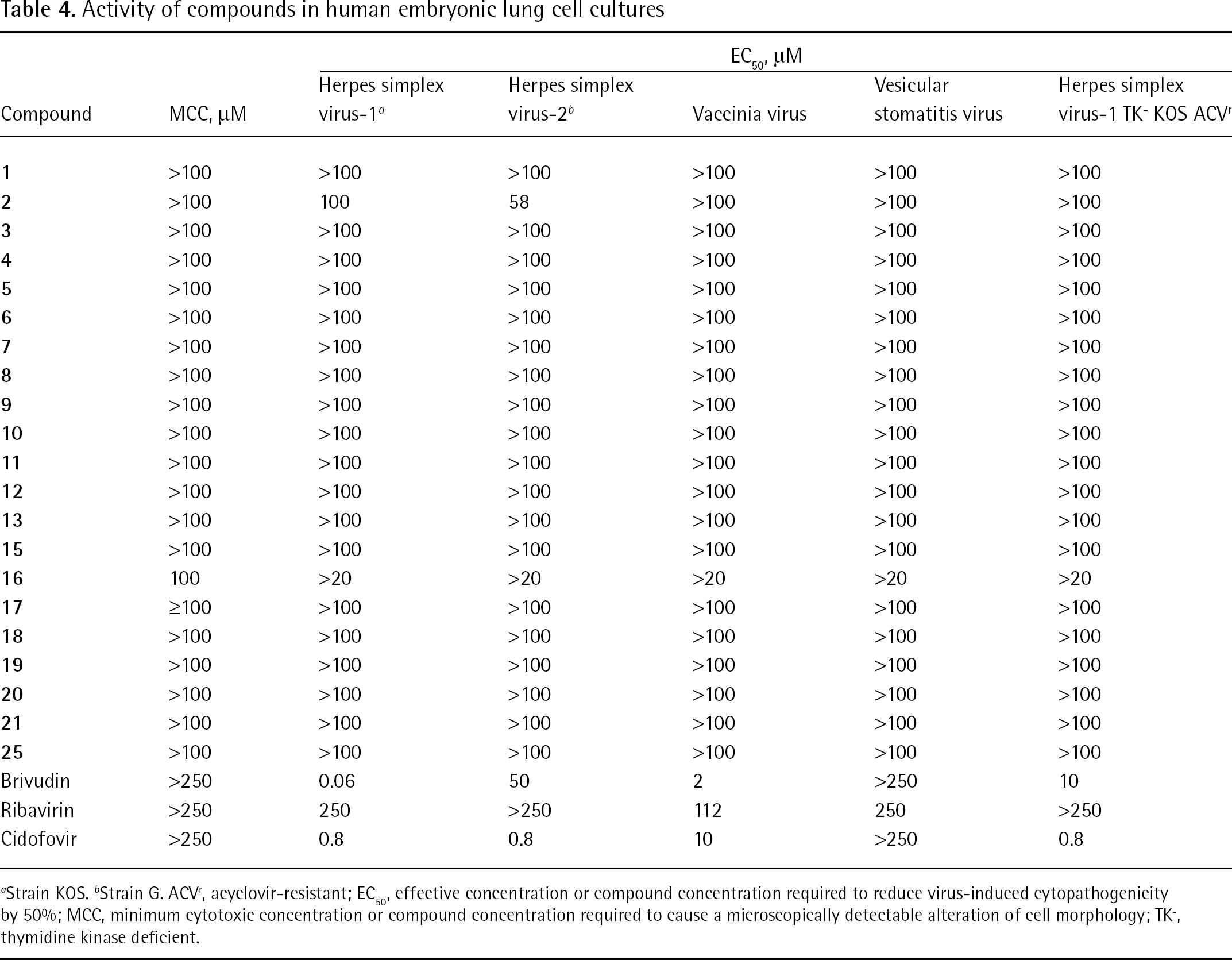
Strain KOS.
Strain G. ACVr, acyclovir-resistant; EC50, effective concentration or compound concentration required to reduce virus-induced cytopathogenicity by 50%; MCC, minimum cytotoxic concentration or compound concentration required to cause a microscopically detectable alteration of cell morphology; TK−, thymidine kinase deficient.
Discussion
None of the compounds tested were endowed with antiviral activity at subtoxic concentrations. Interestingly, a multivalent benzoboroxole functionalized polymer has been reported as an HIV-1 gp120 glycan-targeted entry inhibitor [17]. This high molecular weight polymer could neutralize both CXC chemokine receptor type 4-tropic and C–C chemokine receptor type 5-tropic HIV-1 by blocking virus entry in the nanomolar range; therefore, it might well be possible that the compounds described in this study are too small in size and lack multivalency to show antiviral potential. The surface plasmon resonance data (lack of significant affinity against HIV-1 gp120) support this view. Compound
Compound
In conclusion, the monophenylboronic acids tested herein exhibit no antiviral activity as potential carbohydrate binders versus a broad range of enveloped and non-enveloped viruses. The compounds tested did not bind HIV-1 gp120. It is, therefore, intended to design and prepare a series of diphenylboronic acids with linker lengths designed to mimic molecular modelling distances between the terminal mannose residues of the HIV-1 gp120 glycans. Such compounds would be expected to exhibit greater carbohydrate-binding activity. Efforts towards this goal are ongoing in our laboratories and will be reported forthwith.
Footnotes
Acknowledgements
We thank Rob Jenkins (Chemistry Department, Cardiff University, Cardiff, UK) for use of a 11B probe NMR spectrometer. The technical assistance of Leentje Persoons, Frieda De Meyer, Lies Van den Heurck, Steven Carmans and Anita Camps for the antiviral experiments and Marleen Renders for the SPR experiments is highly appreciated. The research was supported by a grant from the European Commission (CHAARM), the Concerted Actions of the K.U. Leuven (GOA number 10/014) and the Fund for Scientific Research (grant numbers G.485.08 and G.188.07).
The authors declare no competing interests.