HIV type-1 (HIV-1) non-nucleoside reverse transcriptase inhibitors (NNRTIs) are key drugs of highly active antiretroviral therapy (HAART) in the clinical management of AIDS/HIV infection. NNRTI-based HAART regimes effectively suppress viral reproduction, are not cytotoxic and show favourable pharmacokinetic properties. First-generation NNRTIs suffer the rapid selection of viral variants, hampering the binding of inhibitors into the reverse transcriptase (RT) non-nucleoside binding site (NNBS). Efforts to improve these first inhibitors led to the discovery of second-generation NNRTIs that proved to be effective against the drug-resistant mutant HIV-1 strains. The success of such agents launched a new season of NNRTI design and synthesis. This paper reviews the characteristics of second-generation NNRTIs, including etravirine, rilpivirine, RDEA-806, UK-453061, BIRL 355 BS, IDX 899, MK-4965 and HBY 097. In particular, the binding modes of these inhibitors into the NNBS of the HIV-1 RT and the most clinically relevant mutant RTs are analysed and discussed.
Introduction
HIV is the causative agent of AIDS and infection, characterized by loss of helper T-lymphocytes and heavy damage to lymphatic tissues. In 2007, people living with HIV were an estimated 33 million, and there were 2.7 million new HIV infections and 2 million HIV-related deaths at the same time [1].
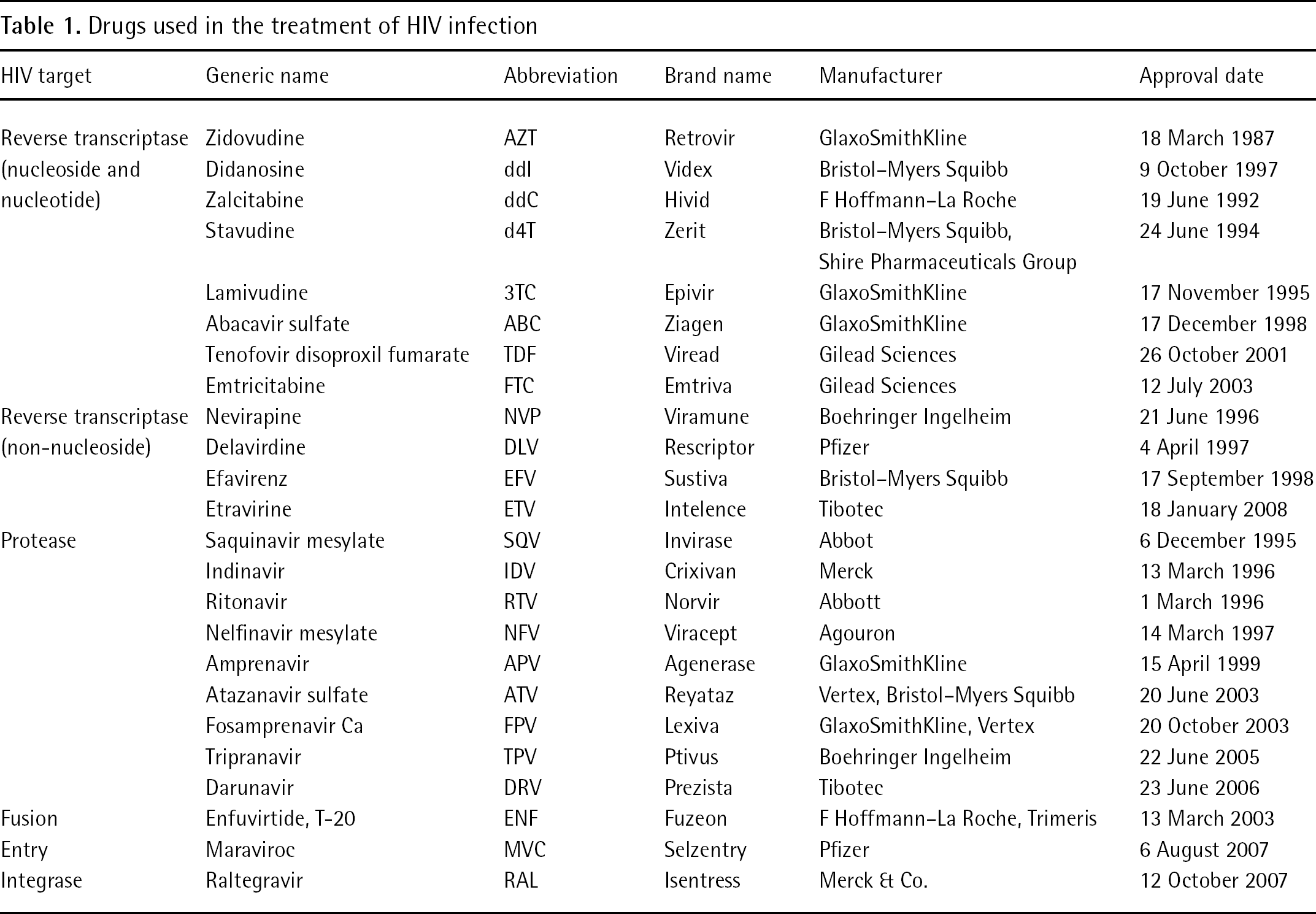
Clinical management of AIDS and HIV relies on the administration of chemotherapeutic agents because an effective vaccine has not yet been developed [2]. The US Food and Drug Adminstration (FDA)-approved HIV drugs fall into the following categories: nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs) [3,4], viral entry inhibitors [5], the new entry inhibitors (for example, the CCR5 coreceptor antagonist, maraviroc) and the integrase inhibitors (for example, raltegravir) [6] (Table 1).
Drugs used in the treatment of HIV infection
HIV target
Generic name
Abbreviation
Brand name
Manufacturer
Approval date
Reverse transcriptase (nucleoside and nucleotide)
Zidovudine
AZT
Retrovir
GlaxoSmithKline
18 March 1987
Didanosine
ddI
Videx
Bristol–Myers Squibb
9 October 1997
Zalcitabine
ddC
Hivid
F Hoffmann–La Roche
19 June 1992
Stavudine
d4T
Zerit
Bristol–Myers Squibb, Shire Pharmaceuticals Group
24 June 1994
Lamivudine
3TC
Epivir
GlaxoSmithKline
17 November 1995
Abacavir sulfate
ABC
Ziagen
GlaxoSmithKline
17 December 1998
Tenofovir disoproxil fumarate
TDF
Viread
Gilead Sciences
26 October 2001
Emtricitabine
FTC
Emtriva
Gilead Sciences
12 July 2003
Reverse transcriptase (non-nucleoside)
Nevirapine
NVP
Viramune
Boehringer Ingelheim
21 June 1996
Delavirdine
DLV
Rescriptor
Pfizer
4 April 1997
Efavirenz
EFV
Sustiva
Bristol–Myers Squibb
17 September 1998
Etravirine
ETV
Intelence
Tibotec
18 January 2008
Protease
Saquinavir mesylate
SQV
Invirase
Abbot
6 December 1995
Indinavir
IDV
Crixivan
Merck
13 March 1996
Ritonavir
RTV
Norvir
Abbott
1 March 1996
Nelfinavir mesylate
NFV
Viracept
Agouron
14 March 1997
Amprenavir
APV
Agenerase
GlaxoSmithKline
15 April 1999
Atazanavir sulfate
ATV
Reyataz
Vertex, Bristol–Myers Squibb
20 June 2003
Fosamprenavir Ca
FPV
Lexiva
GlaxoSmithKline, Vertex
20 October 2003
Tripranavir
TPV
Ptivus
Boehringer Ingelheim
22 June 2005
Darunavir
DRV
Prezista
Tibotec
23 June 2006
Fusion
Enfuvirtide, T-20
ENF
Fuzeon
F Hoffmann–La Roche, Trimeris
13 March 2003
Entry
Maraviroc
MVC
Selzentry
Pfizer
6 August 2007
Integrase
Raltegravir
RAL
Isentress
Merck & Co.
12 October 2007
The NRTI zidovudine (AZT) became the first drug to be approved for the treatment of AIDS and HIV infection in 1987 [7]; however, it was soon evident that the NRTIs, even in combination, could produce only transient viral suppression. In 1995, an effective reduction of viral infection was observed by combining a PI with two NRTIs into the highly active antiretroviral therapy (HAART) [8]. Later, NNRTI-based HAART showed similar effectiveness. Over the past decade, HAART has gradually improved its efficacy by combining drugs from different antiretroviral classes [9]. In 1997, the fixed-dose combination of two NRTIs became the backbone of HAART (the first HIV regimens required taking as many as 18 pills daily). During 2006, the first cross-class combination containing an NNRTI and two NRTIs (one pill per day) was approved in the US [6]. HAART effectively reduces symptoms and prolongs survival of AIDS patients [10,11]; however, HAART fails in eradicating the massive viral multiplication (>109 virions daily) and long-term drug administration favours the emergence of both drug-resistant mutant HIV type-1 (HIV-1) strains and unwanted side effects [12].
Reverse transcriptase
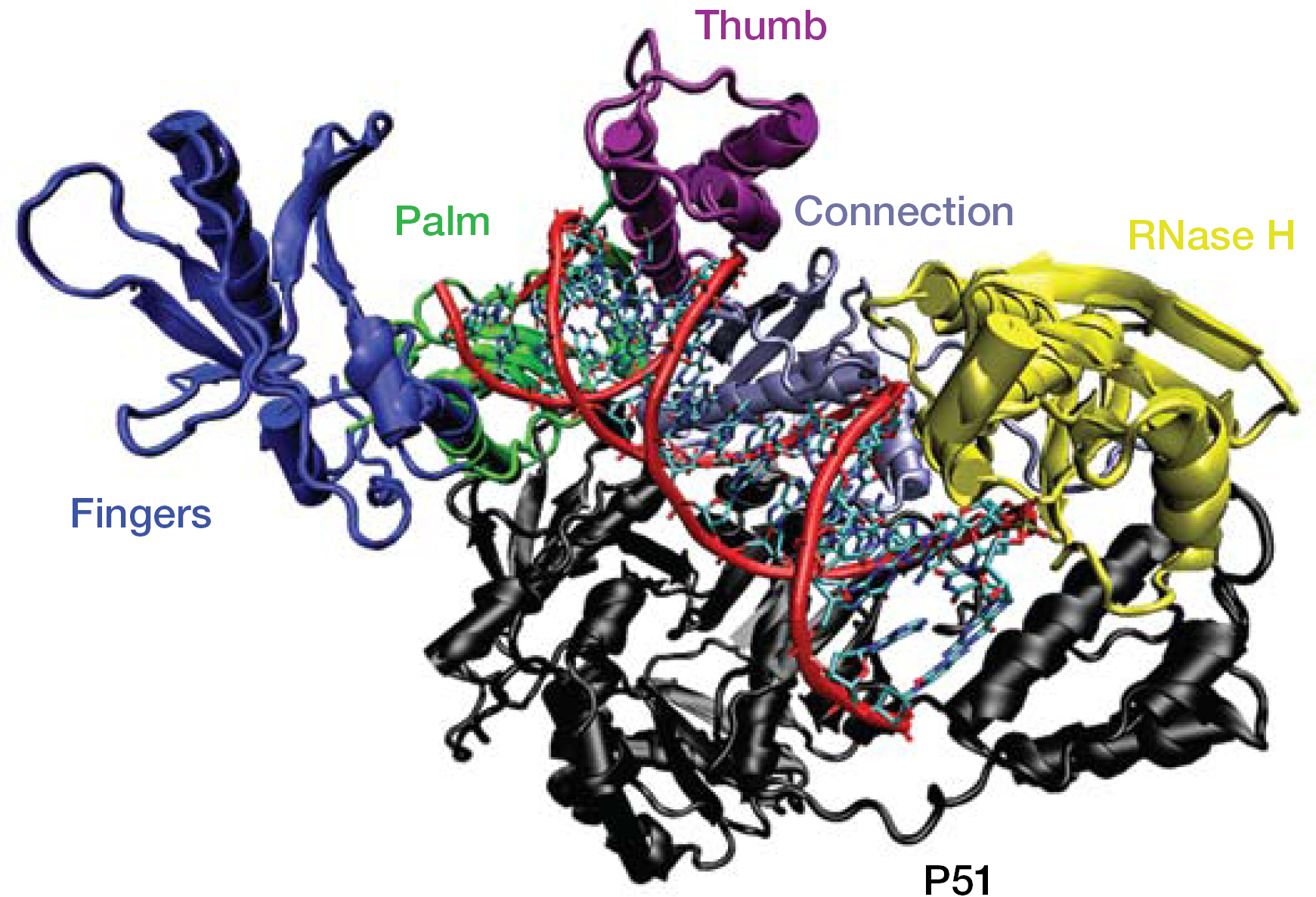
The enzyme reverse transcriptase (RT) catalyses the synthesis of proviral DNA from RNA retroviral genome [13] using three distinct activities: RNA-dependent DNA polymerization (RNA reverse transcription) by means of a cellular lysine transfer RNA primer, which generates an RNA–DNA hybrid; RNase H degradation of the original template strand leaving a single-strand DNA; and DNA-dependent DNA polymerization to form double-stranded DNA. The RT is a heterodimeric macromolecule formed by the subunits p66 and p51 [14,15]. The p66 subunit contains the RT catalytic core and resembles a right hand with fingers, palm, thumb and connection subdomains [15,16] (Figure 1). The p51 subunit shows the same p66 domains, but it plays only a structural role and is deprived of any catalytic activity. During catalysis, the p66 thumb [15] moves onto the finger subdomain [17], allowing the nucleic acid shift and elongation by binding a dNTP unit. The NNRTIs act as non-competitive allosteric inhibitors binding to a hydrophobic pocket, the non-nucleoside binding site (NNBS) located in the p66 palm subdomain close to the catalytic site [18,19].
Non-nucleoside reverse transcriptase inhibitors
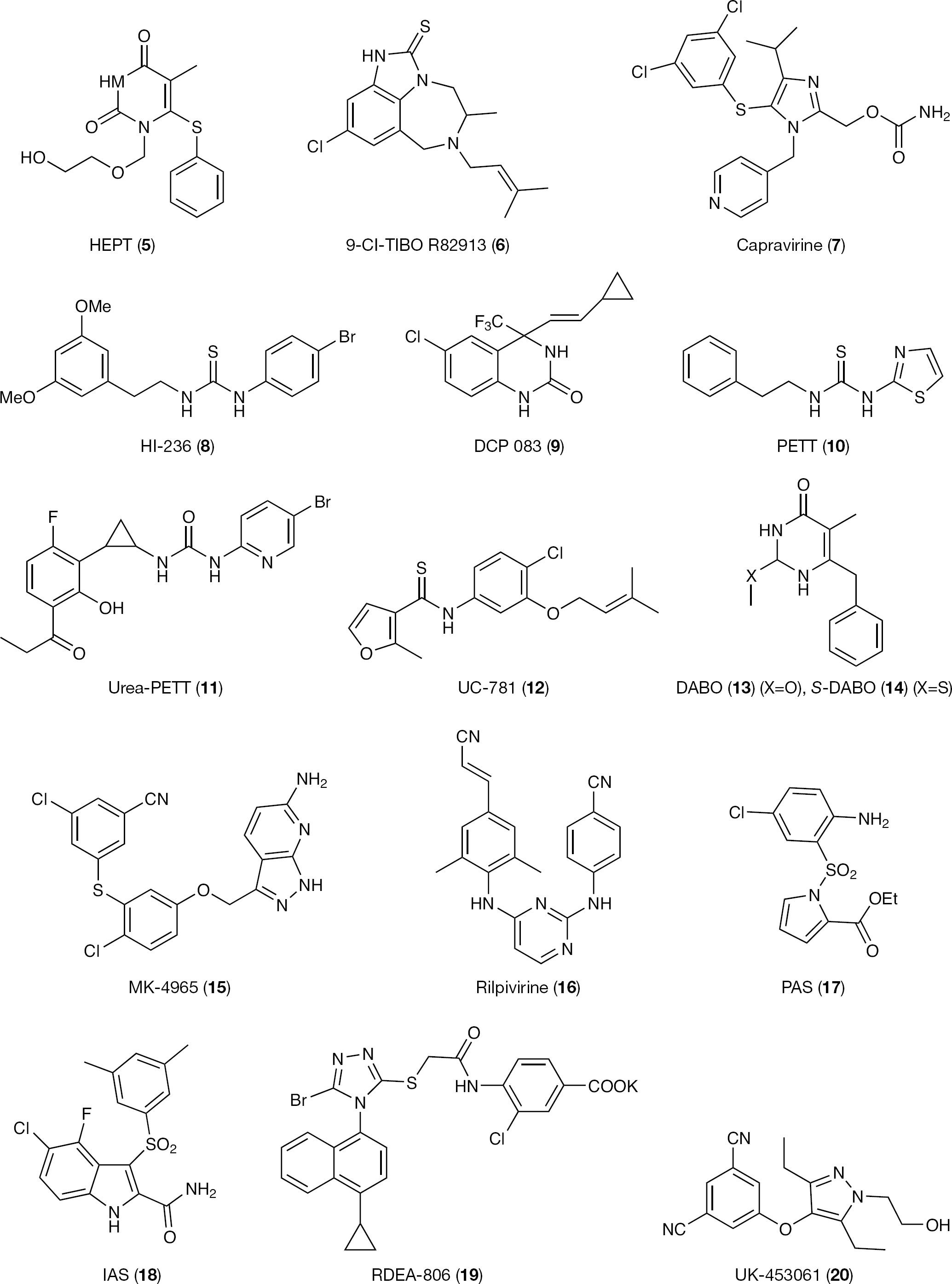
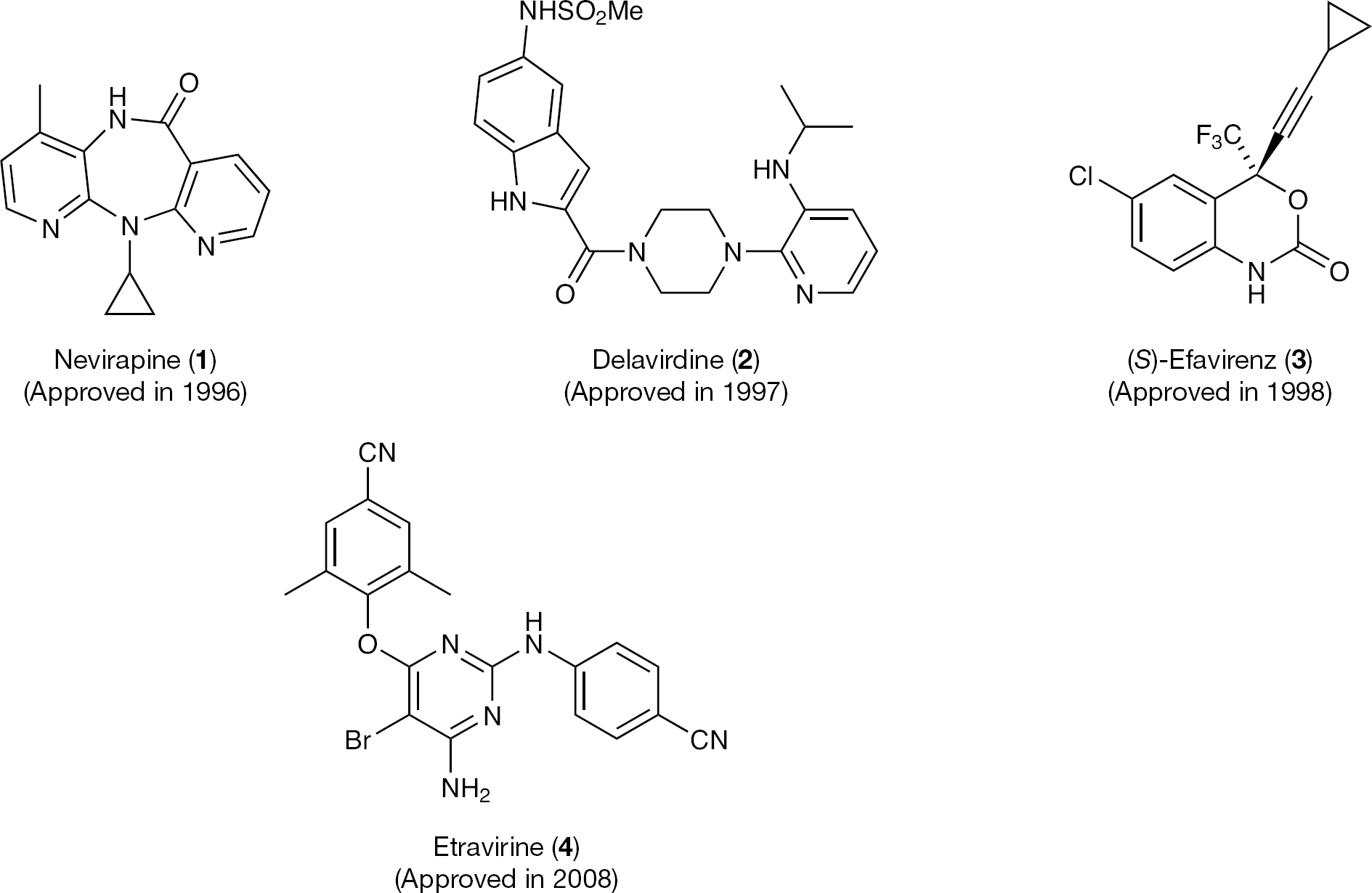
Nevirapine (1; Viramune; Boehringer Ingelheim) was approved by the FDA in 1996 for use in combination with NRTIs in adults with HIV infection. Nevirapine was followed by delavirdine mesylate (2; Rescriptor; Pharmacia & Upjohn) and efavirenz (3; Sustiva; DuPont) in 1997 and 1998, respectively. In the same period of time, 1-[(2-hydroxyethoxy)methyl]-6-(phenylsulfanyl)thymine (HEPT; 5) [20,21] and 4,5,6,7-tetrahydroimidazo[4,5,1-jk] [1,4]benzodiazepin-2(1H)-one and -thione (TIBO; 6) [22,23] were also discovered, introducing the era of NNRTIs (Figure 2). Following HEPT and TIBO [24–26] derivatives, dipyridodiazepinone (1) [27–30], bis(heteroaryl)piperazine (BHAP; 2) [31,–35] and pyridinone [36,37] derivatives were soon discovered. Etravirine (ETV; 4; TMC-125, Intelence; Tibotec) is the newest NNRTI approved by the FDA in January 2008 for treatment in drug combination of HIV-1-infected individuals for whom NNRTI-based therapies have failed (Figure 3).
The research into new NNRTIs rapidly boomed [38,–44] and led to the identification of approximately 30 classes of structurally unrelated HIV-1 inhibitors. Representative NNRTI classes are imidazole (capravirine; 7) [45], thiourea (HI-236; 8) [46], pyrido[1,2-a]indole [47], 3,4-dihydroquinazolin-2-(1H)-ones (3 [48,–52]; and DPC082 and DPC083 [10] analogues [53,54]), phenethylthiazolylthiourea (PETT; 10) [55,56], cyclopropyl urea-PETT (11) [57], thiocarboxanilide (UC 781; 12) [58,59], 3,4-dihydro-2-alkoxy-6-benzyl-4-oxopyrimidines (DABO; 13) [60–63], S-DABO (14) [64–66], pyrazolo[3,4-b]pyridine (MK-4965; 15) [67], diaryltriazino (DATA)/diamidino-2-phenylindole (DAPY; 4 [68] and 16 [69]), pyrrylarylsulfone (PAS; 17) [70–72] and indolylarylsolfone (IAS; 18) [73,,–80].
Non-nucleoside reverse transcriptase inhibitors in highly active antiretroviral therapy
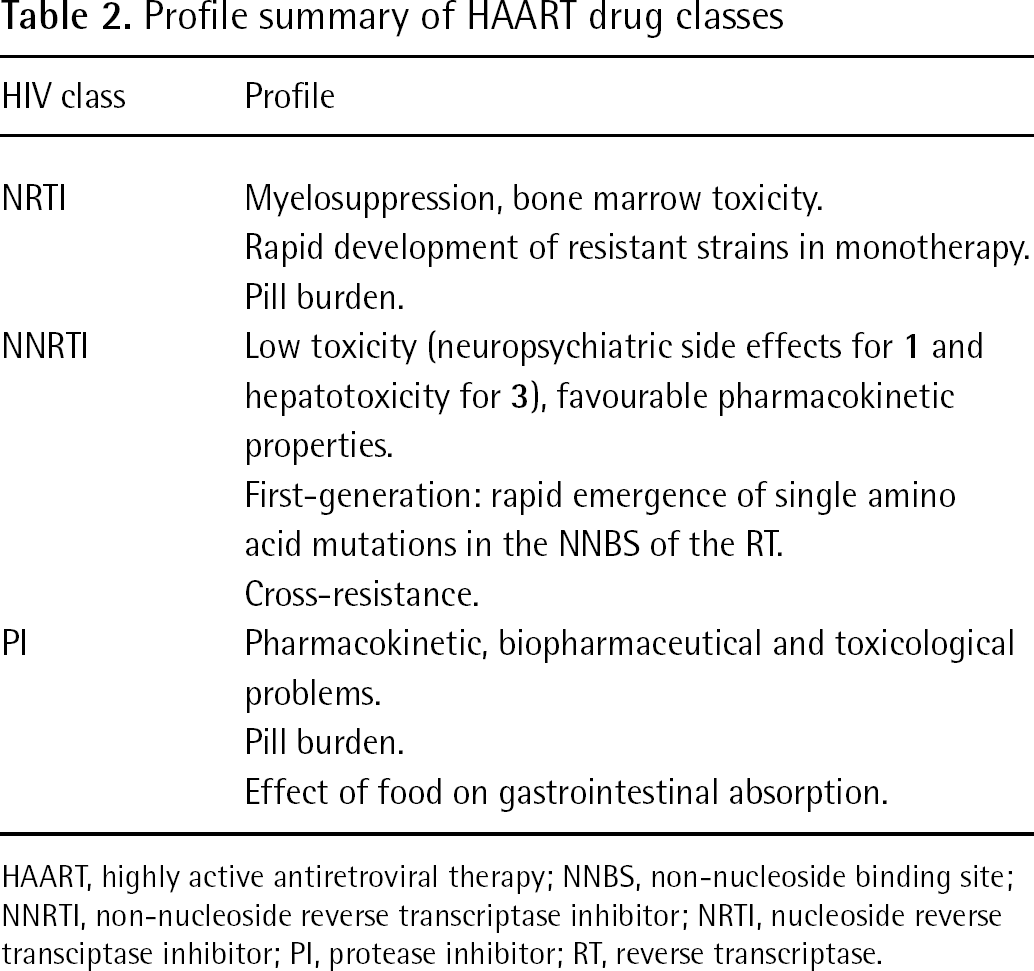
NNRTIs ameliorate the therapeutic outcome of AIDS therapy when combined with other NRTIs, NNRTIs or PIs [81]. The current HAART for the treatment of adult AIDS/HIV-infected individuals recommend three (preferred) or four drugs selected from NRTI, NNRTI and PI classes. Standard HAART is based on combinations of two NRTIs with an NNRTI or PI (drug combinations of only NNRTIs and PIs are not recommended) [82]. Both NNRTI- and PI-based HAART regimes show acceptable effectiveness in first-line AIDS/HIV treatments; however, NNRTI-based regimens are preferred because of significantly better viral suppression [83], reduced cytotoxic effects and better pharmacokinetic properties [84] (Table 2).
Profile summary of HAART drug classes
HIV class
Profile
NRTI
Myelosuppression, bone marrow toxicity.
Rapid development of resistant strains in monotherapy.
Pill burden.
NNRTI
Low toxicity (neuropsychiatric side effects for 1 and hepatotoxicity for 3), favourable pharmacokinetic properties.
First-generation: rapid emergence of single amino acid mutations in the NNBS of the RT.
Cross-resistance.
PI
Pharmacokinetic, biopharmaceutical and toxicological problems.
The major limitation of the NNRTIs in clinical use is the selection of mutations that hamper the binding of the inhibitor to the RT. A single amino acid mutation in the NNBS is sufficient to induce resistance to 1 or 3 [85,86]. Cross-resistance might rise by forming contacts with the same residues of the NNBS. The need for new therapeutic agents that are able to overcome resistance and safety problems prompted the development of new NNRTIs. ETV (4) was the first second-generation FDA-approved NNRTI. The work presented here reviews the characteristics of representative second-generation NNRTIs, 4, 15, 16 and 19–24, with particular emphasis on their binding modes into the NNBS of the HIV-1 RT (Figure 2). The development of a number of NNRTIs with promising in vitro activity has been frozen because they have proved to be ineffective or unsafe. To our knowledge, development of DCP 083, capravirine, emivirine, loviride and dapivirine (TMC-120) has been suspended [87,88]. The development of (+)-calanolide A is ongoing in the clinical phase [88].
Etravirine
Compound 4 shows potent in vitro activity against HIV-1 wild type (WT) as well as against numerous NNRTI-resistant strains [89]. It acts principally by direct binding to the RT, thus inhibiting polymerase activity [90,91]. The RT inhibition was not affected significantly by mutations in the NNBS. As proof of concept, the inhibitory concentrations against the HIV-1 K103N, Y181C, Y188L and K103N/Y181C mutant strains were >2.6x superior to that against WT [91]. In addition to the anti-RT activity, 4 might also have other inhibitory effects, for example, enhancing the intracellular processing of gag and gag–pol polyproteins, which is associated with a decrease in viral particle production [92] (Figure 4).
Three-dimensional structure of the reverse transcriptase–nucleic acid complex
Representative first- and second-generation non-nucleoside reverse transciptase inhibitors
Non-nucleoside reverse transciptase inhibitors in clinical practice
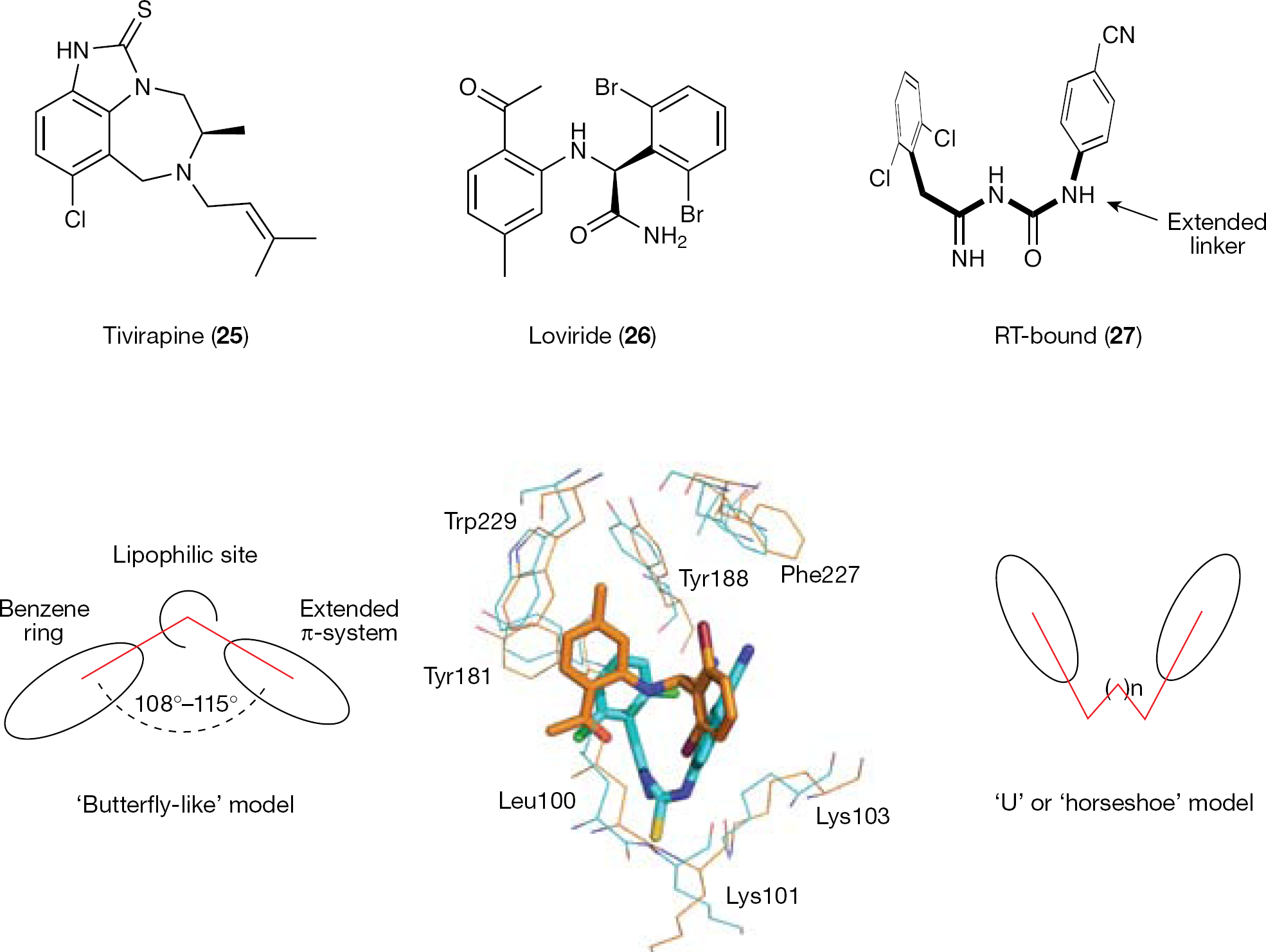
The discovery of 4 was well described by Das et al. in 2004 [93]. A lead optimization programme at Janssen Pharmaceutica led to the discovery of TIBO and α-anilinophenylacetamide (α-APA) NNRTI classes [94]. The X-ray crystal structures of TIBO tivirapine (25) and α-APA loviride (26) in complexes with the WT HIV-1 RT, and 25 with the Y181C mutant, provided structural information on the binding mode of such derivatives into the NNBS of the RT [95–97]. Both 25 and 26 were shown to adopt a similar active conformation [96], as observed for other first-generation NNRTIs (that is, 1), which are characterized by two π-systems arranged in a ‘butterfly-like’ orientation and an additional lipophilic region between the two wings (Figure 4) [98].
Schematic representations of the ‘butterly-like’ and ‘horseshoe’ models
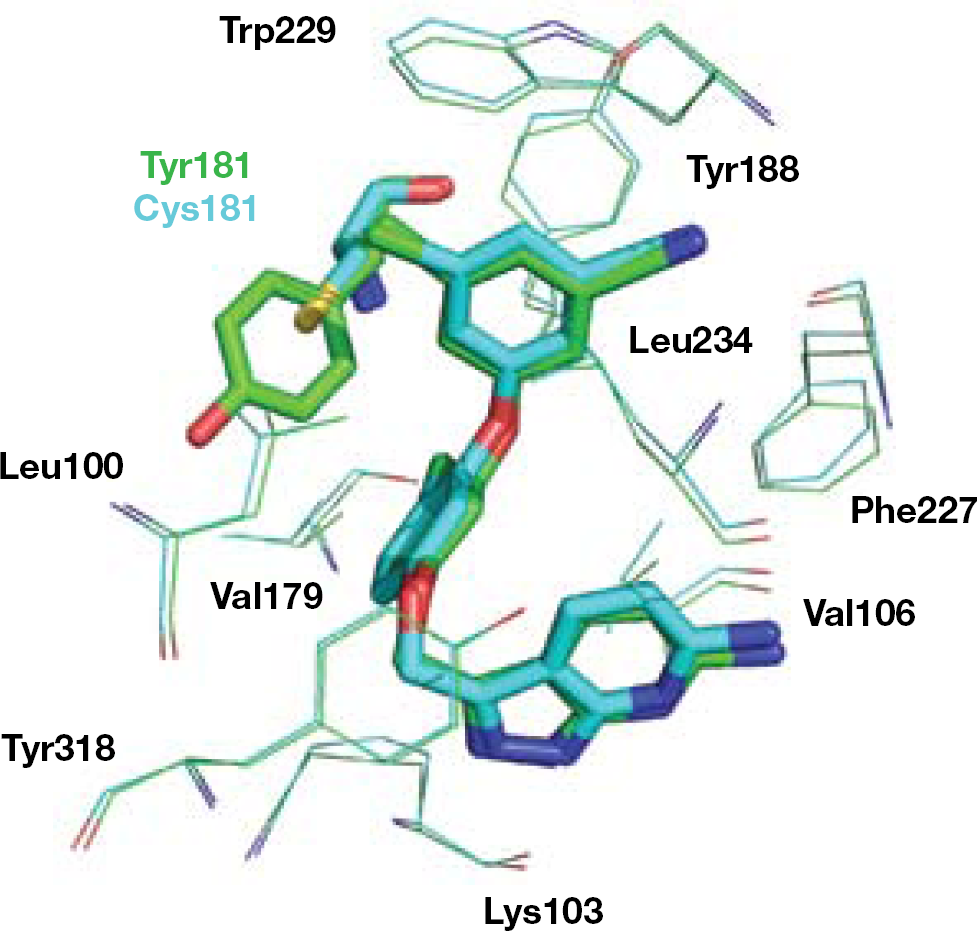
Chemical modification of the α-APA series led to imidoyl thiourea (ITU) analogues (R100943; 27) [99], where the side wings of the ‘butterfly-like’ model are connected through an extended linker. ITU derivatives are endowed with a greater flexibility than their parent α-APA compounds. In contrast to the ‘butterfly-like’ conformation displayed by TIBO, α-APA and 1, the RT-bound conformation of the ITU 27 resembles a ‘U’ or ‘horseshoe’ [93]. Superposition of 26 to the HIV-1 RT/27 complex showed some significant differences [93] (Figure 4).
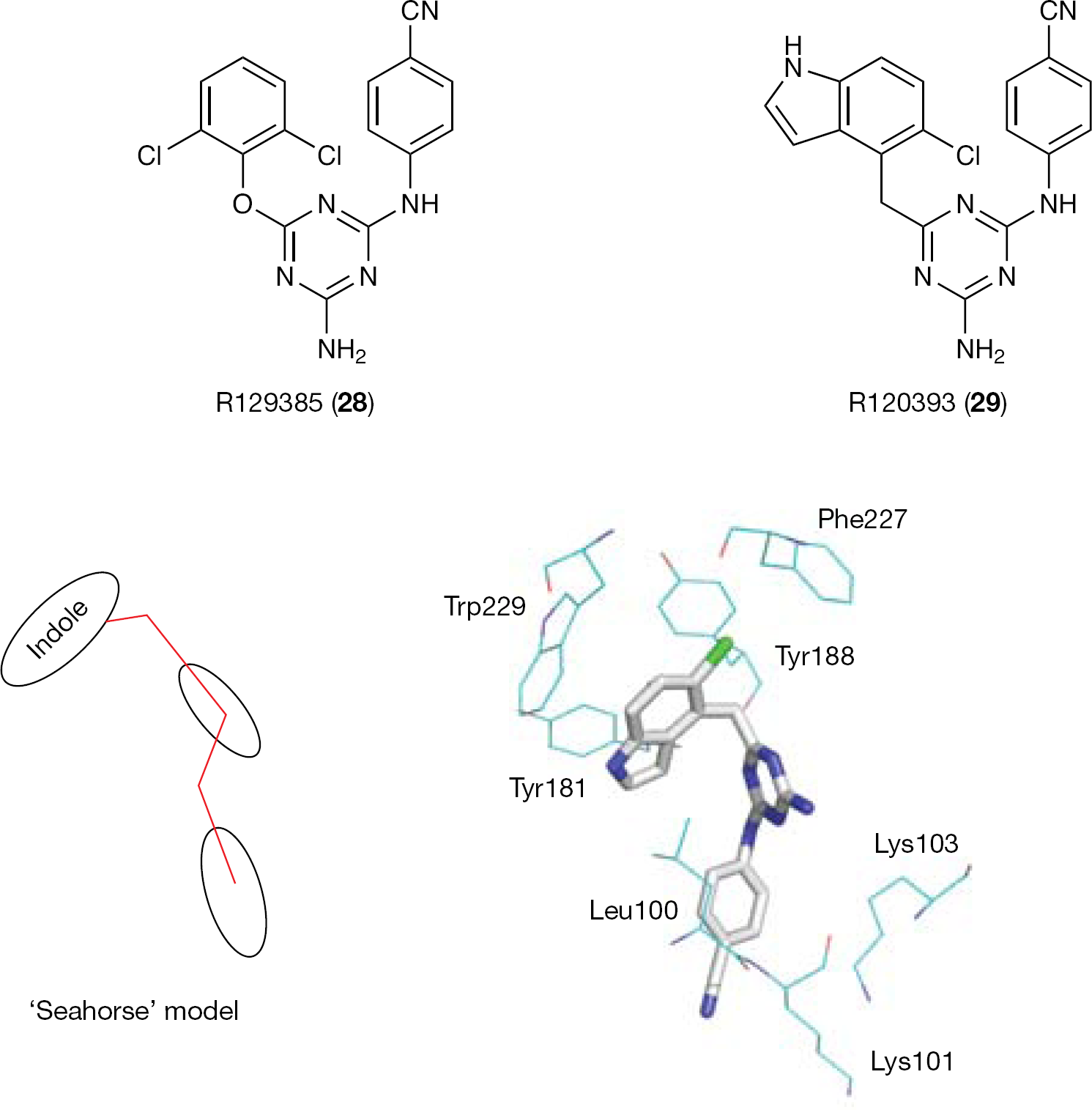
The DATA NNRTIs were obtained by intramolecular cyclization of the thiourea moiety of ITU derivatives [100]. Similarly to ITUs, DATA derivatives inhibited the HIV-1 WT at nanomolar concentrations, adopting a ‘horseshoe’ active conformation. The structure of 28 complexed with the HIV-1 RT usefully represents the structure for the RT-bound DATA complexes [93]. Replacing the 2,6-dichlorophenoxy wing of 28 with a 5-chloroindol-4-ylmethyl moiety provided R120393 (29), a compound designed to form additional interaction with the side chain of the highly conserved Trp229 (mutation of Trp229 seriously compromises the RT activity) [101,102]. The crystal structure of the HIV-1 RT/29 complex showed that this DATA analogue assumed a ‘seahorse’ active conformation [93] (Figure 5). The indole ring of 29 was positioned deeply in the NNBS of the RT (2 Å deeper than the wing I of other DATA analogues), forcing the triazine ring in a 3 Å deeper position, whereas the usual interaction with the carbonyl oxygen of Lys101 was broken. Crystallographic studies suggested that 29, similar to other DATAs, adopts multiple binding conformations. In principle, such a flexibility might provide an advantage in overcoming viral drug resistance.
The ‘seahorse’ model
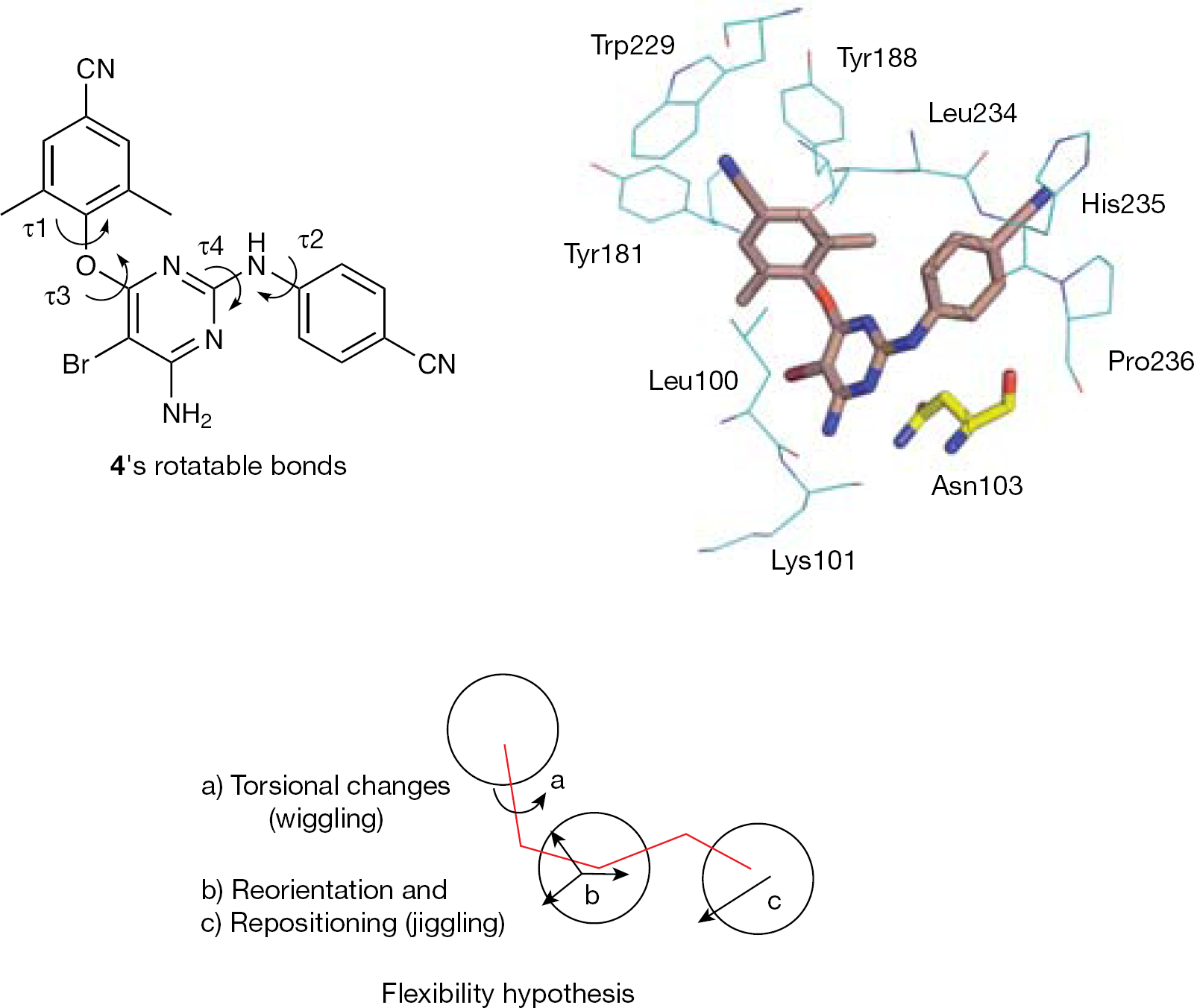
τ1–τ4 Torsional angles and binding conformation of 4 into the non-nucleoside binding site of the mutant K103N reverse transcriptase
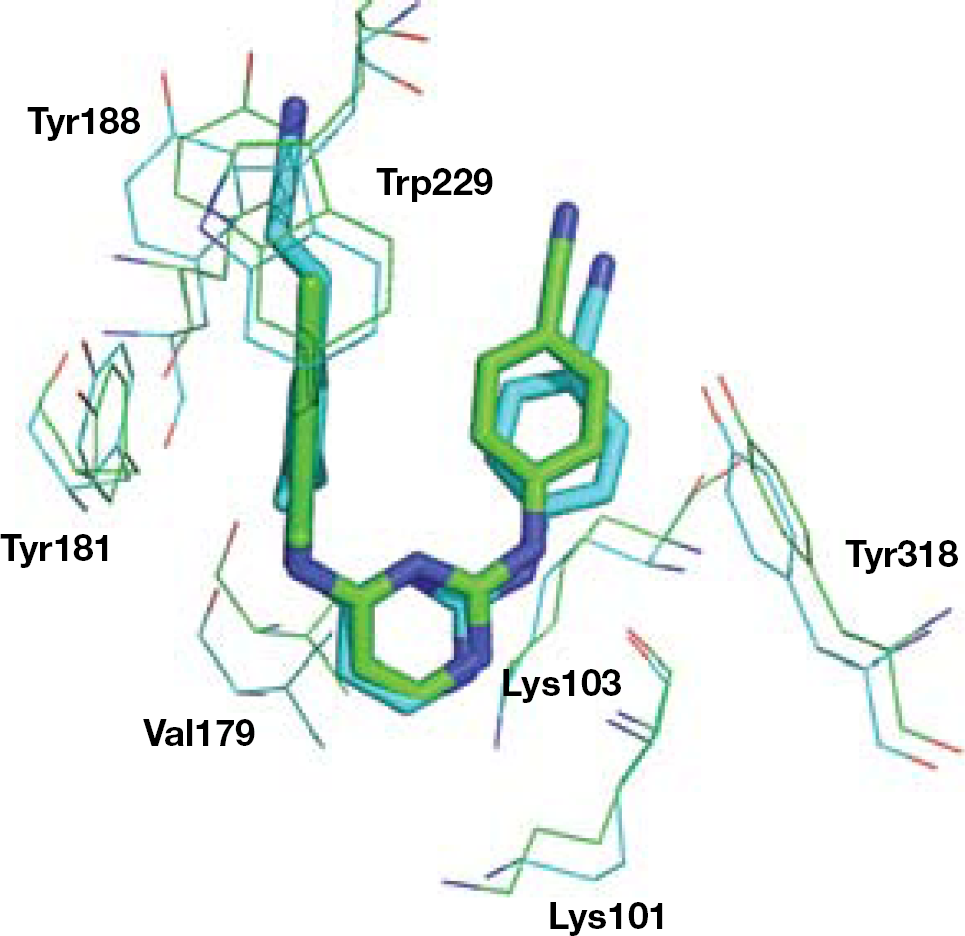
Replacement of the central triazine of DATA analogues with a pyrimidine resulted in the DAPY series. DAPYs proved to be more potent than DATAs when tested against a broad spectrum of resistant mutants. Crystallographic studies showed that 4 (as well as TMC120) in complex with the HIV-1 RT adopted the ‘horseshoe’ binding conformation [103]. Both DATA and DAPY structures allow rotational (from changes in torsion angles τ1, τ2, τ3 and τ4; Figure 6) and translational shifts inside the NNBS of the HIV-1 RT (the τ1 and τ2 angles can rotate without any significant energy variation). The dynamic adaptation of DATA and DAPY analogues into the NNBS of the HIV-1 RT might explain their ability to inhibit HIV-1 RT carrying resistance mutations.
The hypothesis of the multiple binding mode of 4 to the HIV-1 RT was consistent with the X-ray low-resolution diffraction as a result of the coexistence of >1 RT/4 complex conformations. Principal component analysis studies [103] also predicted a negligible difference of only 1.2 kcal/mol between the two lowest energy RT-bound 4 conformations [104]. In the principal pose of the K103N RT/4 cocrystal structure, the central pyrimidine ring of 4 is positioned between the side chains of Leu100 and Asn103 and, similar to 3, it forms favourable protein–ligand van der Waals interactions [105]. Simulations of molecular dynamics supported the hypothesis that the molecular flexibility allows torsional changes (wiggling), reorientation and repositioning (jiggling) of 4 into the NNBS of the RT [106]. The flexibility and easy conformational interconversion might help to explain the effectiveness of DAPY series against drug-resistant mutations (Figure 6).
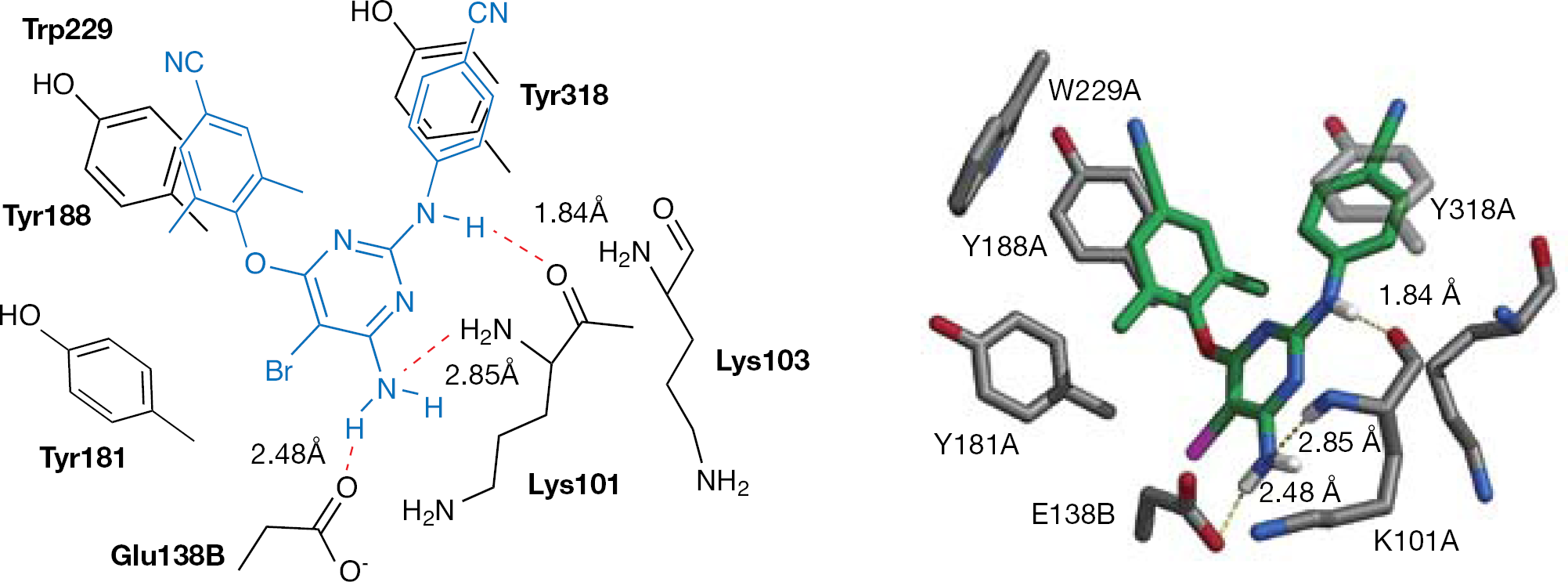
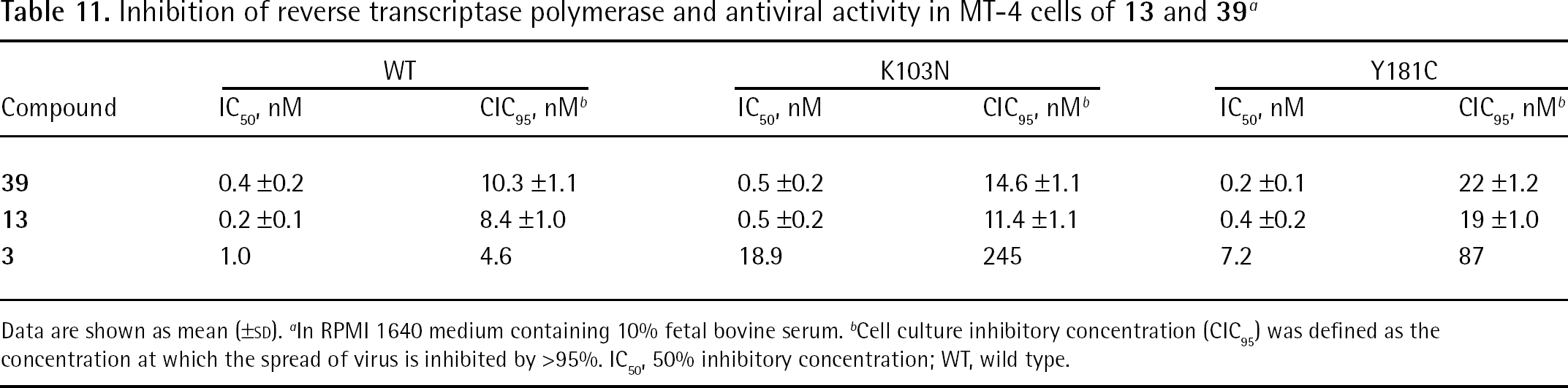
Binding mode analysis of 4 into the NNBS of the RT highlighted some interactions common to other NNRTIs and a number of additional interactions [107]. Compound 4 formed an H-bond with the Lys101 backbone carbonyl; another H-bond occurred between the N1 amino group of Lys101 and the pyrimidine C6 amino nitrogen. An additional H-bond was predicted to occur between the C6 amino group and the carboxylate of Glu138 of the p51 subunit. The ether and amino linkages of the two cyanophenyl substituents provide sufficient flexibility to allow favourable aryl–aryl interactions with Tyr181, Tyr188, Trp229 and Tyr318 [107] (Figure 7).
Compound 4 modelled into the non-nucleoside binding site of the HIV type-1 wild-type reverse transcriptase
Molecular modelling studies [107,108] showed that in the L100I mutation, 4 retained all key stabilizing interactions and the H-bonds with Lys101 and Glu138 showed similar lengths as in the WT RT. The weaker interaction with Tyr181 caused a little decrease of the antiviral potency against the Y181C mutation (the same was also observed for 3), although π-stacking interactions with Tyr188 occurred [109]. The H-bond of 4 with Lys101 was not affected by the K103N mutation within 1.76 Å of H–O bond length (in the case of 3, the K103N the mutation abolished the same contact). All the other favourable interactions were retained. The ether linkage of 4 produced molecular flexibility, which allowed stable binding interactions to the Y188L mutation.
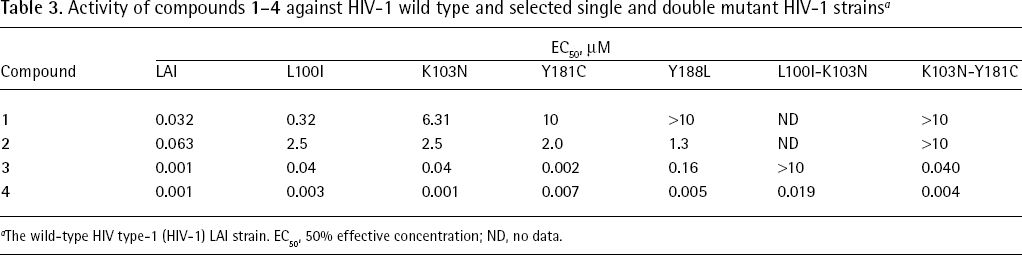
In vitro, compound 4 inhibited, at nanomolar concentrations, drug-resistant mutants carrying either single (K103N and Y181C) or double (K103N-Y181C and L100I-K103N) amino acid mutations (Table 3) [68] and showed limited cytotoxicity [90,93]. Compound 4 retained a 50% effective concentration (EC50) value of <100 nM against 97% of 1,081 clinically derived recombinant viruses that are resistant to at least one of the currently marketed NNRTIs [90]. The potential for HIV to develop resistance to 4 appears to be lower than for first-generation NNRTIs [89,110]. Compound 4 demonstrated a synergistic interaction with AZT and additive interactions with other antiretroviral agents, including NNRTIs (1–3), NRTIs and PIs [90]. In clinical studies, treatment of patients harbouring NNRTI-resistant HIV-1 variants with 4 (900 mg twice daily for 7 days) resulted in a median decrease in plasma HIV-1 RNA of −0.89 log10 copies/ml [111]. The exceptional spectrum of activity of 4 might be correlated to its capability to adopt >1 active binding conformations to the HIV-1 RT [3,4,12].
Activity of compounds 1–4 against HIV-1 wild type and selected single and double mutant HIV-1 strainsa
EC50, μM
Compound
LAI
L100I
K103N
Y181C
Y188L
L100I-K103N
K103N-Y181C
1
0.032
0.32
6.31
10
>10
ND
>10
2
0.063
2.5
2.5
2.0
1.3
ND
>10
3
0.001
0.04
0.04
0.002
0.16
>10
0.040
4
0.001
0.003
0.001
0.007
0.005
0.019
0.004
The wild-type HIV type-1 (HIV-1) LAI strain. EC50, 50% effective concentration; ND, no data.
Rilpivirine
Rilpivirine (TMC278; 16) is a cyanovinyl analogue of DAPY TMC120. The binding conformation of 16 into the NNBS was modelled from the crystallographic structure of the RT/TMC120 complex (Figure 8). The potent activity of 16 against HIV-1 WT and mutant strains might be correlated to the binding interaction between the 4-cyanovinyl arm at wing I and the indole ring of Trp229. Moreover, the superior torsional capability of 16 with respect to earlier DAPY analogues allow stronger and stable interactions of the molecule into the mutated NNBS of the RT [93,113]. As for compound 4, the flexible structure of 16 allows torsional changes, reorientation and repositioning into the NNBS of the RT (Figure 6) [106].
Binding conformation of 16 into the non-nucleoside binding site of the HIV type-1 reverse transcriptase
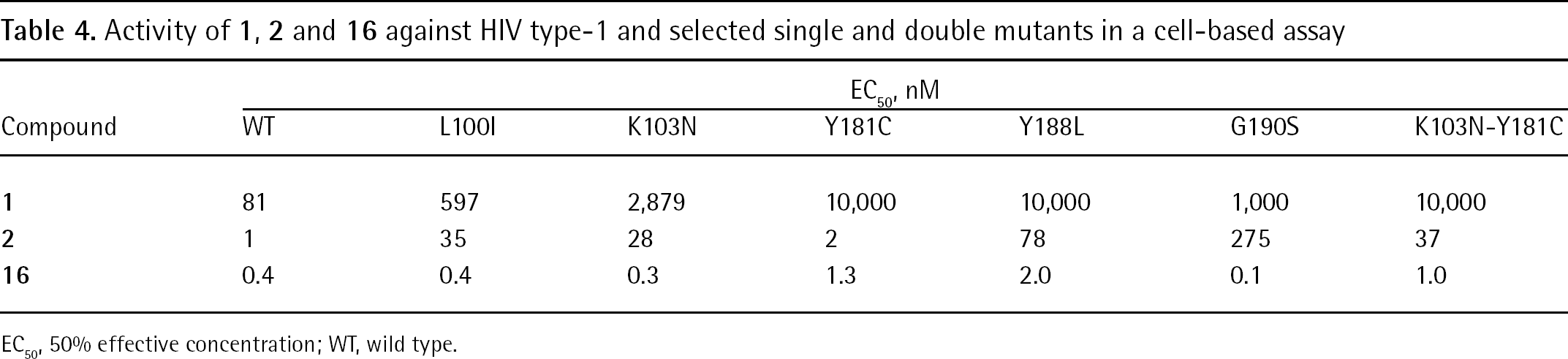
Compound 16 was more active than 1, 3 and 4 (approximately 10–20x) against HIV-1 WT and drug-resistant HIV-1 mutants carrying single and double mutations (Table 4). Compound 16 inhibited 81% of 1,200 clinical isolates at EC50<1 nM and showed low cytotoxicity [113–116]. Viral breakthrough was not observed with 16 at 1 μM during 30 days of treatment. By contrast, 3 showed viral breakthrough after 6 days of treatment. Per os administration of 16 in PEG 400 led to half-lives ranging from 2.8 h in rat and 39 h in dog, and bioavailability was 32% and 31% in rat and dog, respectively. As for other DAPY analogues, plasma protein binding was high [91]: >99% of 16 binds to human plasma proteins in a concentration-independent manner [113].
Activity of 1, 2 and 16 against HIV type-1 and selected single and double mutants in a cell-based assay
EC50, nM
Compound
WT
L100I
K103N
Y181C
Y188L
G190S
K103N-Y181C
1
81
597
2,879
10,000
10,000
1,000
10,000
2
1
35
28
2
78
275
37
16
0.4
0.4
0.3
1.3
2.0
0.1
1.0
EC50, 50% effective concentration; WT, wild type.
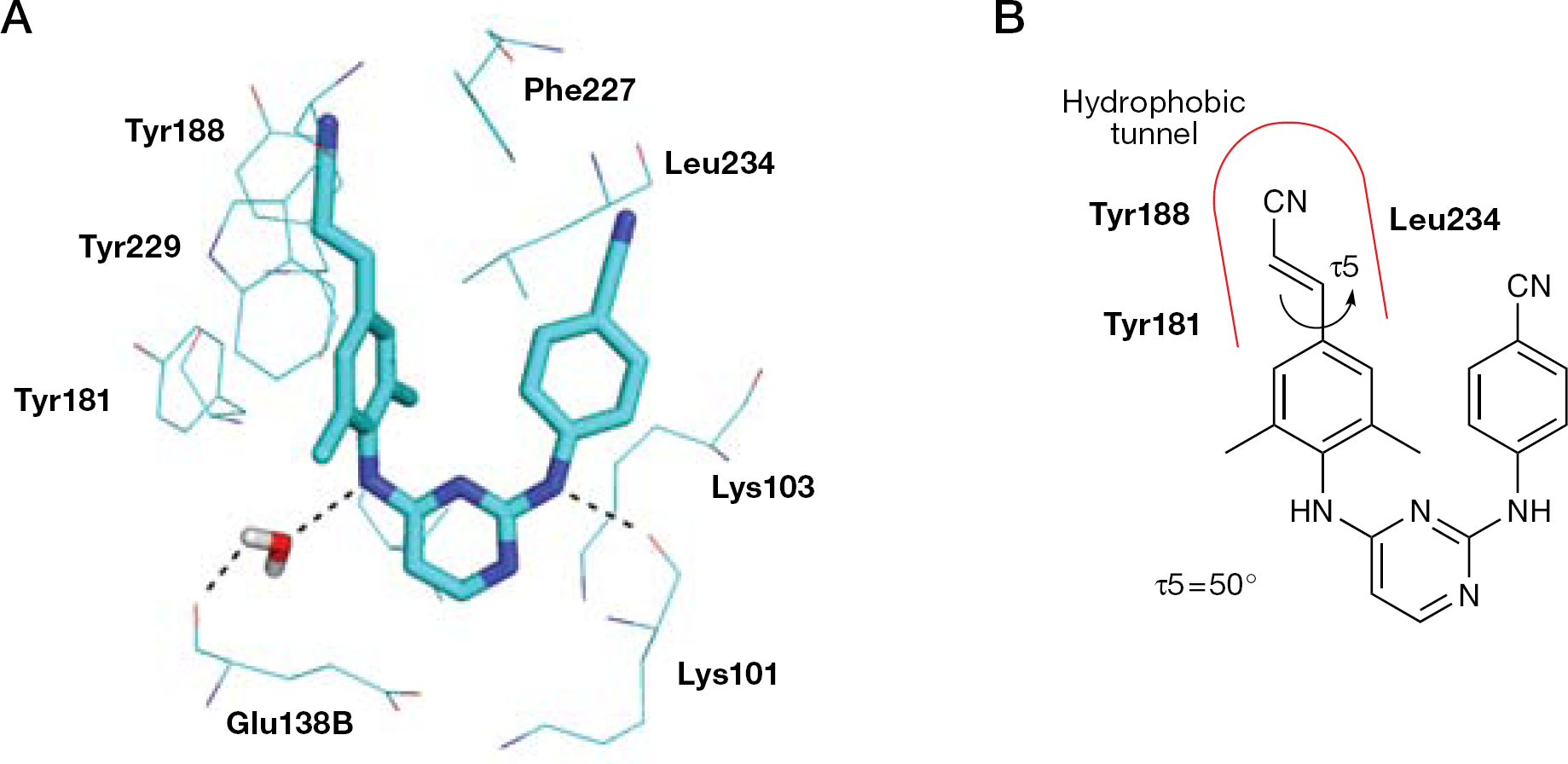
Attempts to obtain the structure of an HIV-1 RT/16 complex have failed [93,117] (the best crystals diffracted X-rays to only 6.0 Å resolution) [118] as a consequence of the high molecular flexibility. A mutant form of RT obtained through a systematic protein engineering approach yielded better diffracting crystals of the RT/16 complex. The structure resolved to 1.8 Å clearly defined position and conformation of 16 into the crystallographic complex. Compound 16 showed the typical ‘horseshoe’ binding conformation with the three aromatic rings connected through two amino bridges and the characteristic E-cyanovinyl group [93,118]. Compound 16 formed an H-bond between the N–H bridge and the carbonyl of Lys101, and another water-mediated H-bond with the main carbonyl group of Glu138 of the p51 subunit. The 4-(E-cyanovinyl)-2,6-dimethylphenyl group was positioned into a hydrophobic pocket, formed by Tyr181, Phe227, Trp229 and Leu234 amino acid side chains, in a cylindrical tunnel connecting the NNRTI-binding pocket to the nucleic-acid-binding cleft that resembles a ‘piston-and-ring’ structure (Figure 9). Although determined differently, the torsion angles of the rotatable bonds τ1–τ4 of 16 had values similar to those of the RT-bound TMC120. In the RT-bound conformation, the E-cyanovinyl group was rotated 50° with respect to the dimethylphenyl ring.
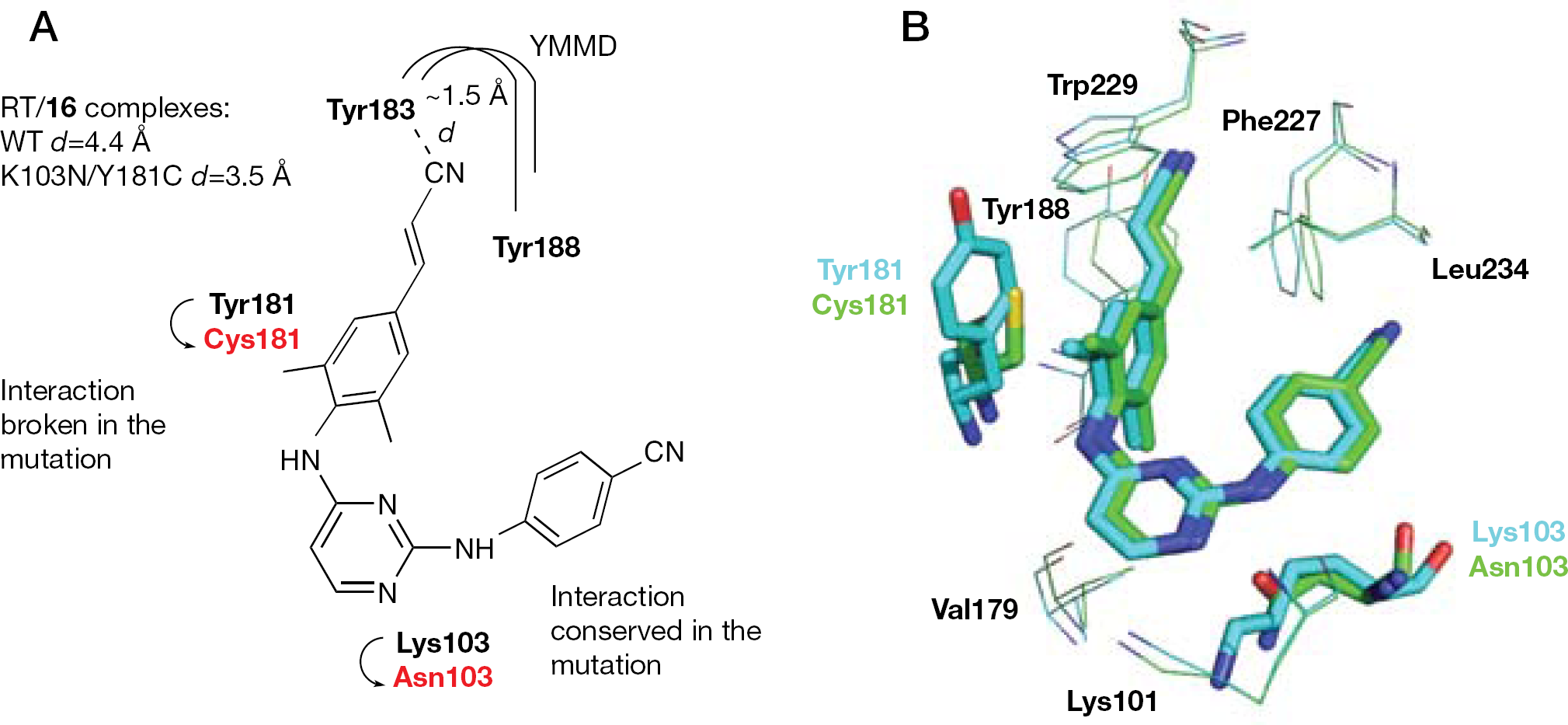
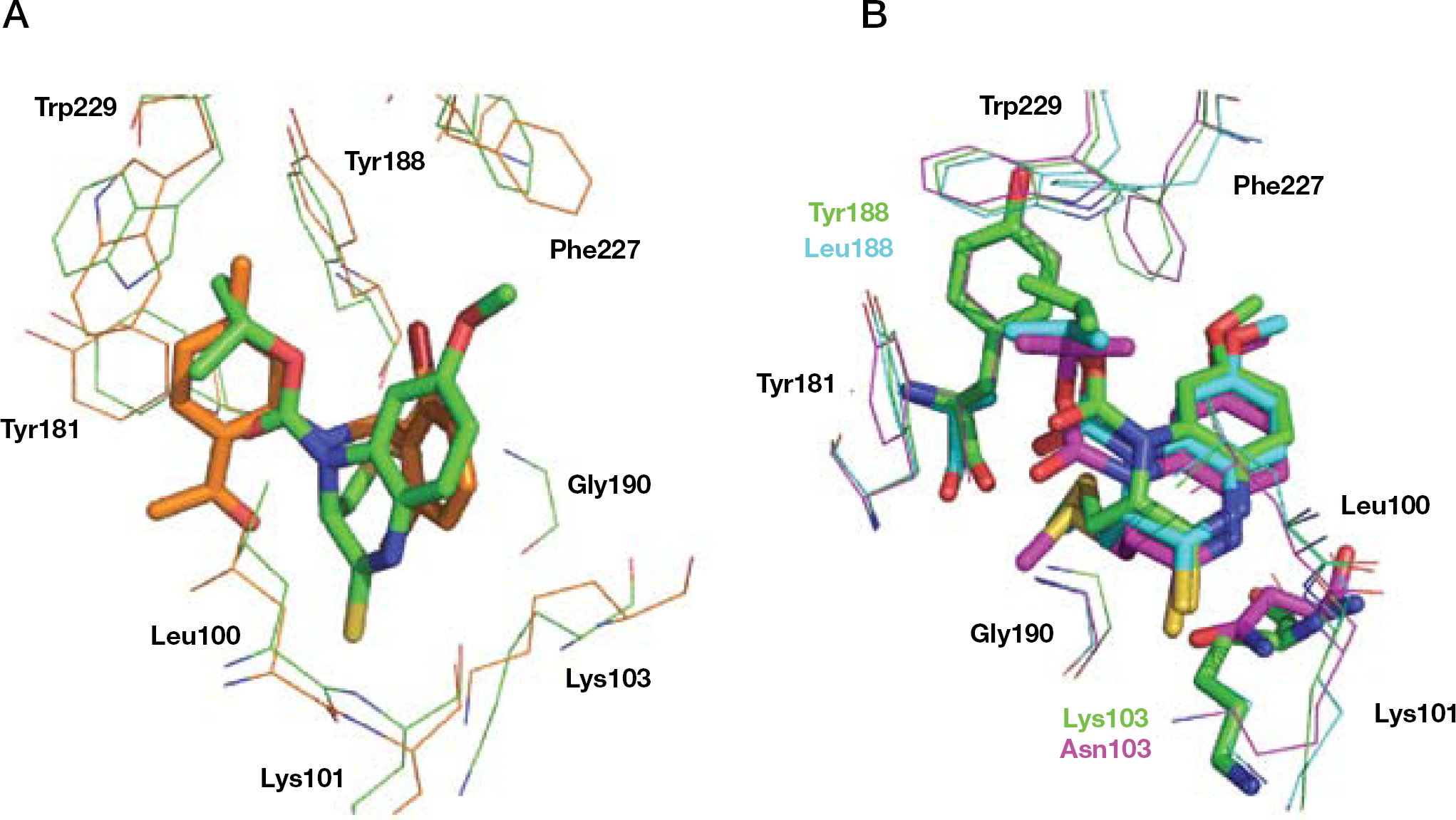
In comparing the WT RT-bound 16 conformation with the K103N/Y181C RT/16 structure, no significant conformational changes were observed. The number of distances <4.5 Å between pairs of atoms in the two complexes was almost identical. The interaction of the 2,6-dimethylphenyl ring with the aromatic side chain of Tyr181 is broken. Tyr183 was shifted by approximately 1.5 Å toward the NNBS, thus allowing 16 to form a binding interaction with the E-cyanovinyl group, which might compensate for the interactions broken by the Y181C mutation. The interaction of 16 with Asn103 was conserved in the double mutation (Figure 10A).
Binding mode of compound 16
In the L100I/K103N mutation, significant conformational and positional rearrangements of 16 with respect to the WT RT complex were not observed. Compound 16 shifted from Ile100 towards Asn103 because of the steric hindrance of the L100I mutation. The torsion angles τ1–τ5 were rotated with respect to the WT complex and, in contrast to the complexes with the WT and K103N/Y181C RTs, the E-cyanovinyl group was coplanar (Figure 10B) [118].
Two-dimensional infrared spectroscopy experiments indicated that the two arms of 16 sensed quite different environments within the hydrophobic pocket. The vibrational relaxation of the two arms were almost equal at 3 ps from model studies. The slow spectral diffusion of the cyanovinyl arm was attributed to its interaction with the backbone and side chains in the hydrophobic tunnel [119].
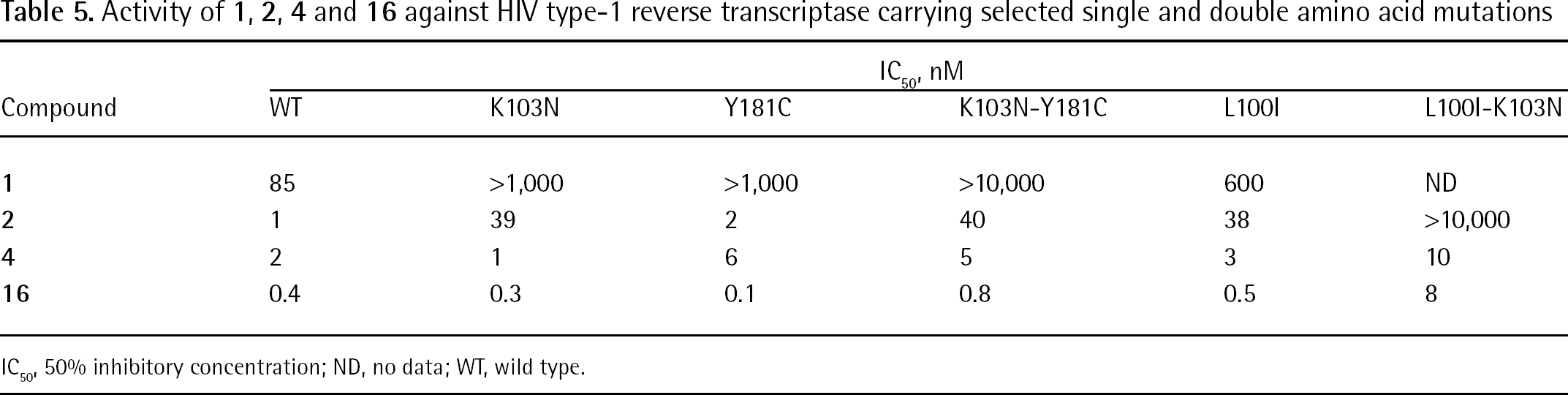
Compound 16 proved to potently inhibit single (K103N and Y181C) and double (K103N-Y181C) drug-resistant mutant HIV-1 RTs with IC50 values in the subnanomolar range of concentration, as reported in Table 5 [120].
Activity of 1, 2, 4 and 16 against HIV type-1 reverse transcriptase carrying selected single and double amino acid mutations
IC50, nM
Compound
WT
K103N
Y181C
K103N-Y181C
L100I
L100I-K103N
1
85
>1,000
>1,000
>10,000
600
ND
2
1
39
2
40
38
>10,000
4
2
1
6
5
3
10
16
0.4
0.3
0.1
0.8
0.5
8
IC50, 50% inhibitory concentration; ND, no data; WT, wild type.
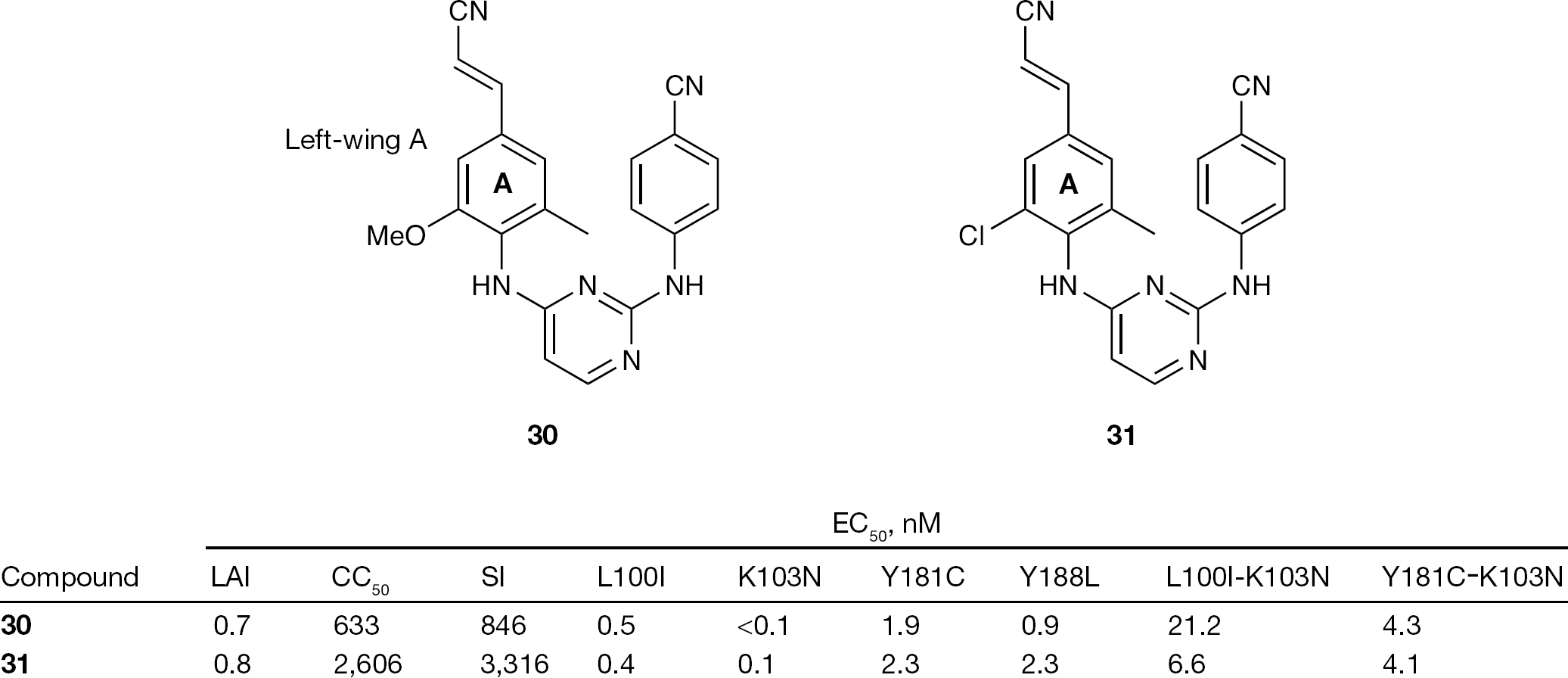
One or both methyl groups of 16 were succesfully replaced by either a chlorine atom or a methoxy group. Two new compounds, 30 and 31, demonstrated excellent antiretroviral activity against a panel of clinically relevant NNRTI drug-resistant mutant strains and presented interesting pharmacokinetic characteristics in dogs (Figure 11) [121].
RDEA-806
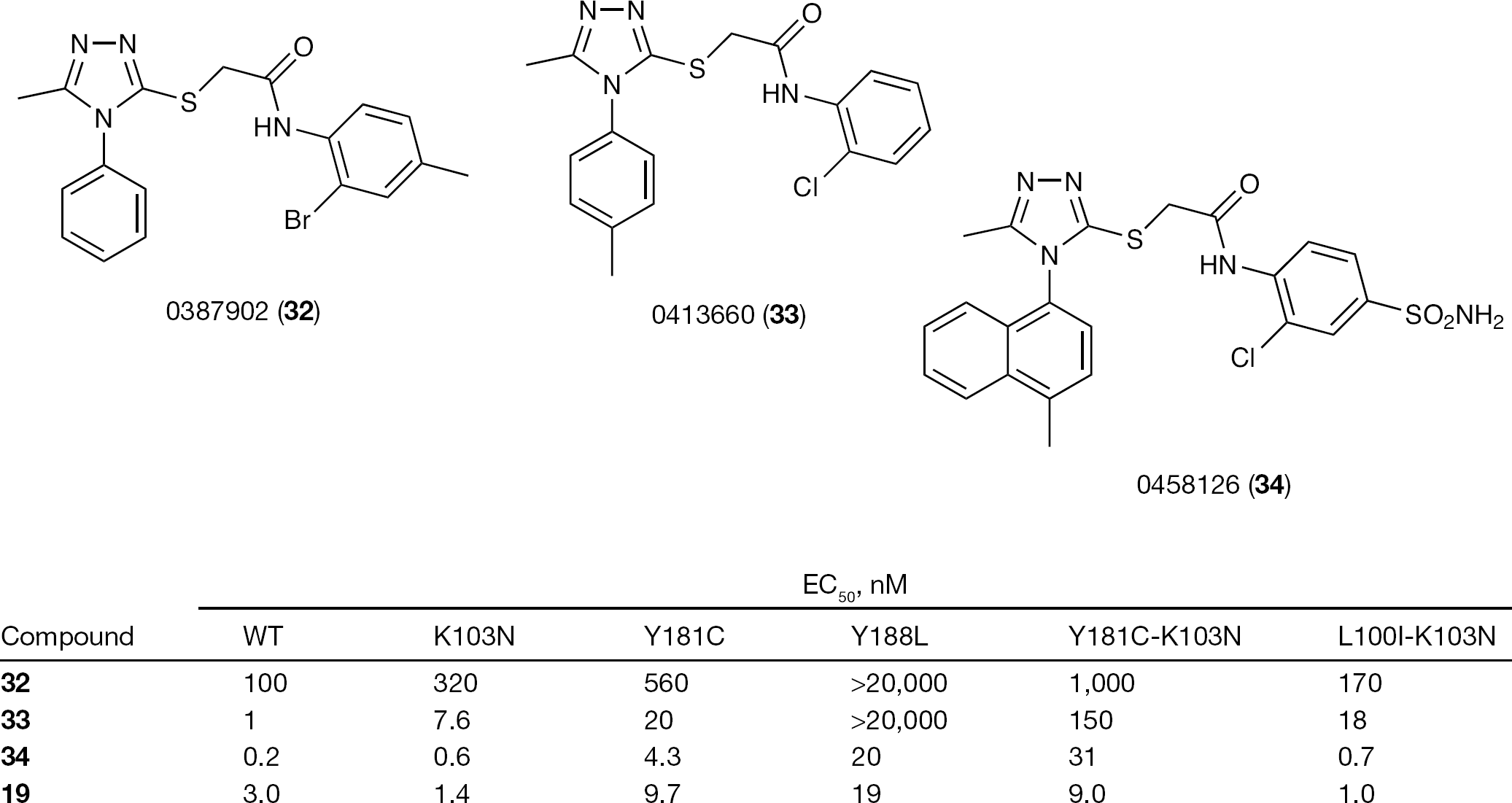
A large scale library screening of 87,000 compounds using a cell-based assay led to the discovery of 0387902 (32), a triazole compound endowed with moderate activity against HIV-1 WT and the K103N-Y181C mutant strain [122]. Ardea Biosciences, Inc., synthesized >1,000 analogues of 32 in an effort to optimize potency against HIV-1 WT and the most prevalent mutant viruses after NNRTI treatment failure (K103N, Y181C, K103NY181C and K103N-L100I) and pharmacokinetic properties.
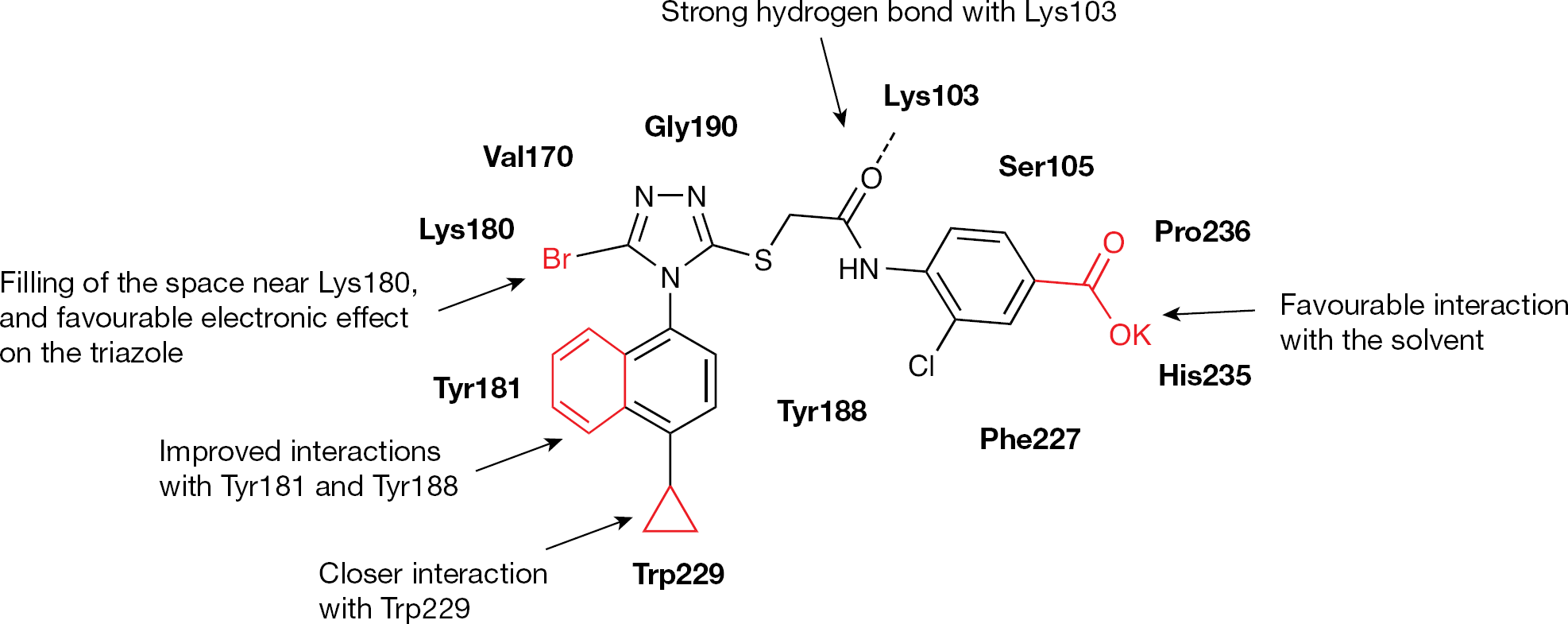
The structure of 33 cocrystallized with the HIV-1 WT RT showed the presence of interactions between the 1-(4-methylphenyl) group and Tyr181, Tyr188 and Trp229, and interactions between the extended chlorophenyl group and Pro236, Phe227 and Val106. Analysis of the binding interactions led to the design of 19 in three ways: by enlarging the substituent at position 4 of the 1-phenyl group to improve the interaction with Trp229, by replacing the 1-phenyl ring with a naphthalen-1-yl moiety to occupy the space between Tyr181 and Tyr188 and by introducing a chemical group at position 4 of the 2-chlorophenyl group, which might favour the interactions with water molecules (Figure 12) [122].
Schematic representation of relevant binding interactions and comparison of binding interactions of 16 into the HIV type-1 WT and K103N-Y181C complexes
Compound 19 is the potassium salt of the corresponding benzoic acid derivative, which can be easily synthesized in six steps from two starting materials [122]. The 50% inhibitory concentration (IC50) of 19 against the purified WT RT enzyme is 3.1 nM. In a cell-based assay, 19 inhibited WT, K103N and L100I-K103N to IC50 values of 3.0 nM, 1.4 nM and 1.0 nM, respectively. Compound 19 is a novel second-generation NNRTI, with the potential to be used in both treatment-experienced and treatment-naive patients, including those harbouring the K103N mutation. Compound 19 showed high genetic barrier to resistance and a broad spectrum of activity [123]. In preclinical through to Phase II studies, 19 was highly effective against mutant HIV-1 strains resistant to 3, exhibited reduced clinical adverse reactions and serum half-life for once-daily dosing. When used in combination with currently used anti-HIV-1 drugs, 19 showed limited drug interactions [124] (Figure 13).
UK-453061
UK-453,061 (lersiviride; 20) was selected as an HIV NNRTI drug candidate at Pfizer Laboratories [123] because of low clearance (Clunbound<8 ml/min/kg) and reduced octanol–water distribution coefficient (logD=1.8), which might lead to a reduction of drug metabolism [125].
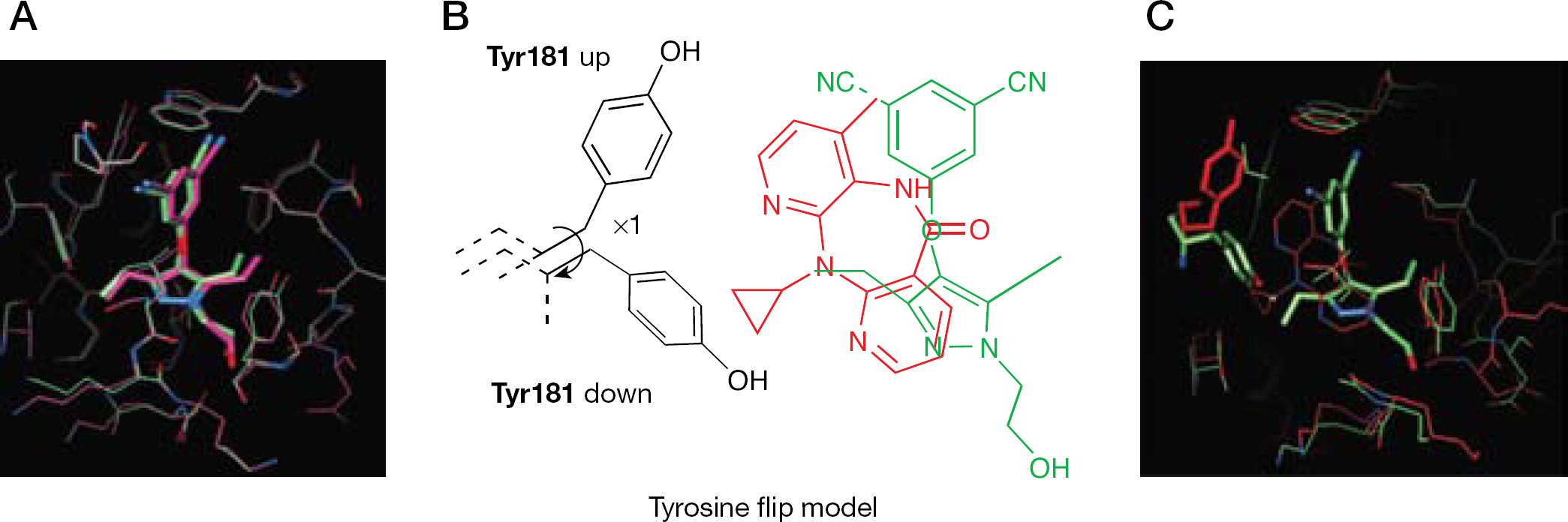
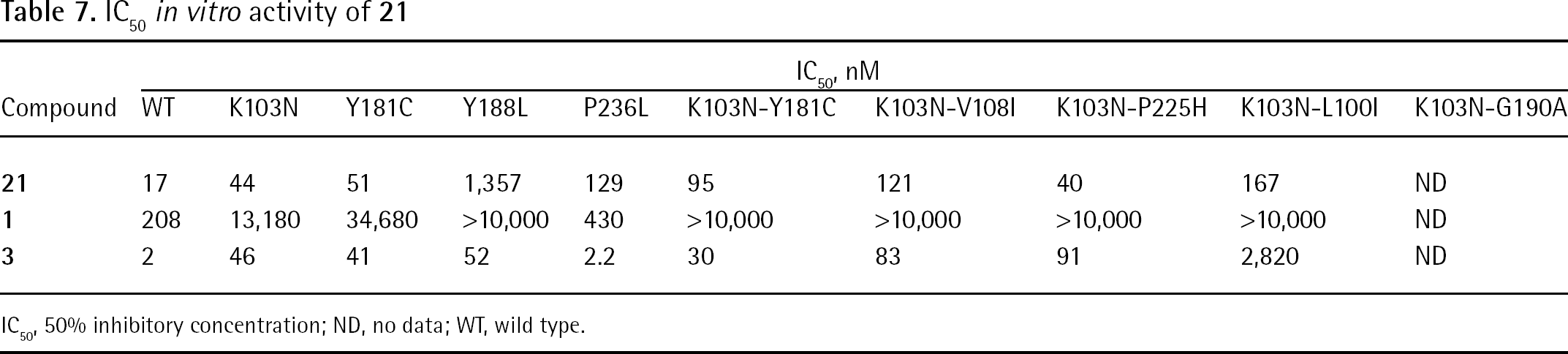
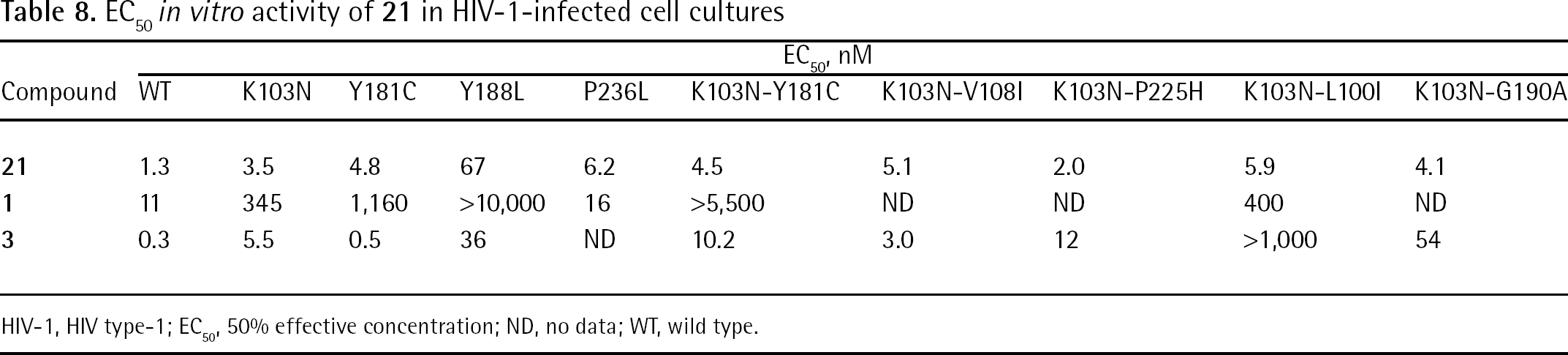
An HIV-1 RT/20 cocrystal structure revealed that inside the NNBS, the inhibitor formed interactions with residues Leu100, Val106, Tyr181, Tyr188, Phe227, Trp229, Tyr318, Leu234 and Pro236 of the p66 subunit. Compound 20 seemed to bind either HIV-1 WT RT or the RT carrying the single K103N mutation in a very similar way (Figure 14A). In comparing these two binding modes with cocrystal HIV-1 RT complexed with compound 1, the Tyr181 residue appeared to be rotated approximately 100° around χ1 (that is, flipped ‘down’). Such a conformational change of Tyr181 (‘tyrosine flip’) led to the formation of a new hydrophobic interaction with the cyclopropyl group, which was thought to be responsible for the 10-fold increase in potency (Figure 14B and 14C). The excellent antiviral profile of 20 against HIV-1 NNRTI resistance mutants was correlated to the flexibility of this tyrosine subpocket and the H-bonds and lipophilic contacts formed within it [125].
Activity of 30 and 31 against HIV type-1 wild-type LAI strain and mutant strains
Drug design of 19 from the HIV type-1 wild type reverse transcriptase/33 cocrystal structure
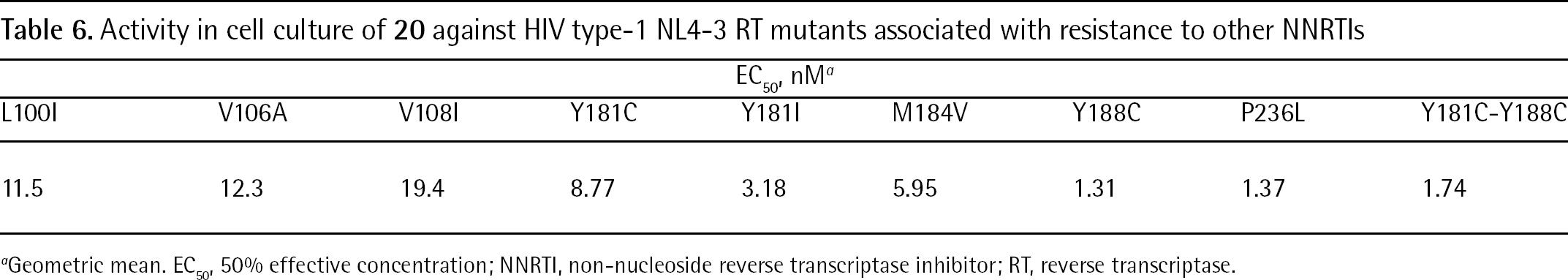
The evaluation of the enzymatic inhibiton by the RT assay shows that the K103N mutation did not markedly affect the antiviral potency of 20 (HIV-1 WT RT IC50=118 nM and HIV-1 mutant K103N RT IC50=215 nM). Against 15 of 18 single and double amino acid mutant HIV-1 RTs, 20 almost retained WT potency showing <10-fold increase in IC50 values. The biological evaluation on infected cellular lines show that compound 20 inhibited 14 of 18 HIV-1 NL4-3 strain viruses harbouring mutations associated with resistance to commercially available NNRTIs (inhibition data against some representative mutant HIV-1 strains are shown in Table 6) [125].
Activity in cell culture of 20 against HIV type-1 NL4-3 RT mutants associated with resistance to other NNRTIs
Cell-based activity of 19 and 32–34 against HIV type-1 WT and mutant strains
Binding mode of compound 20 and schematic representation of the tyrosine flip
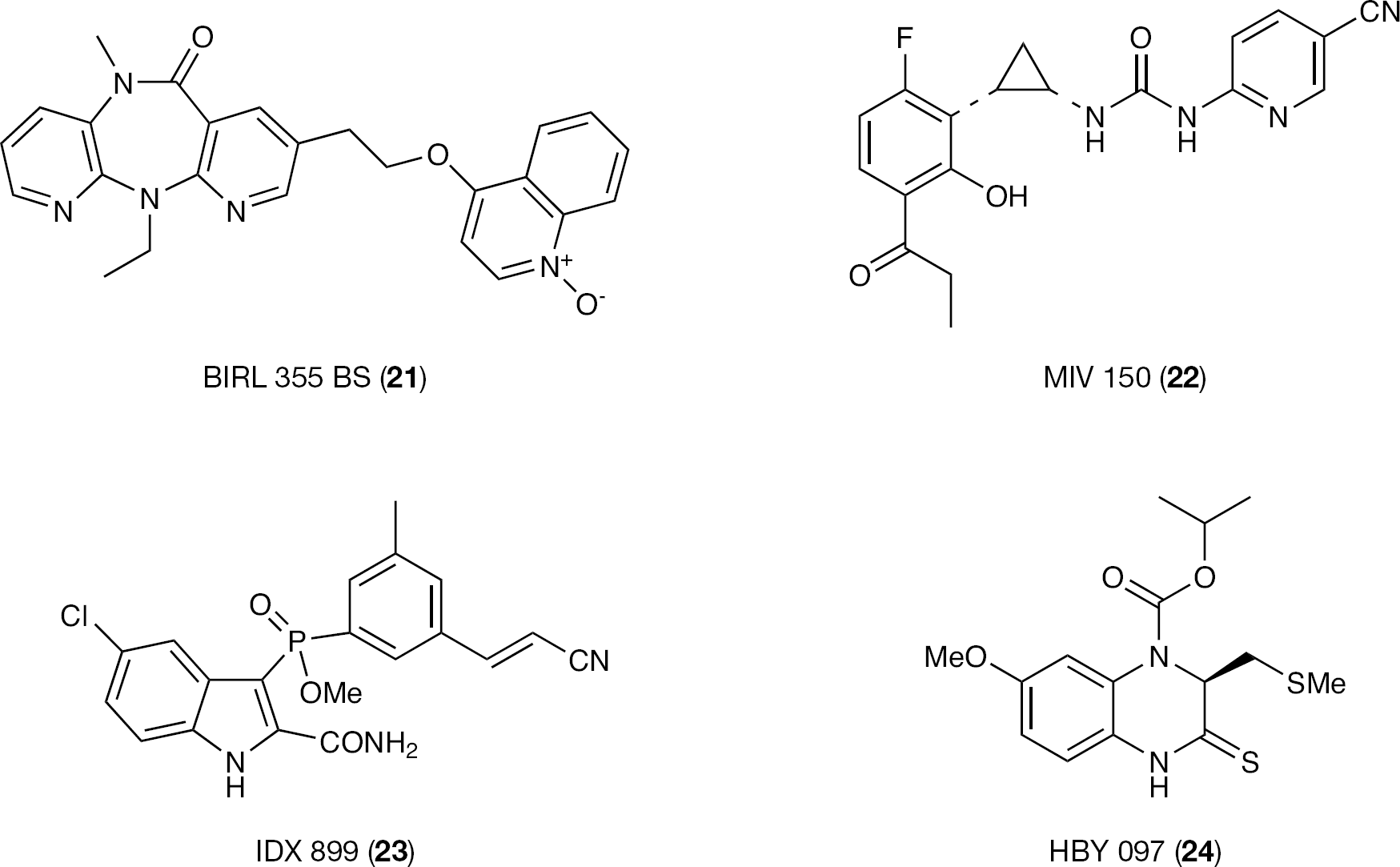
BIRL 355 BS
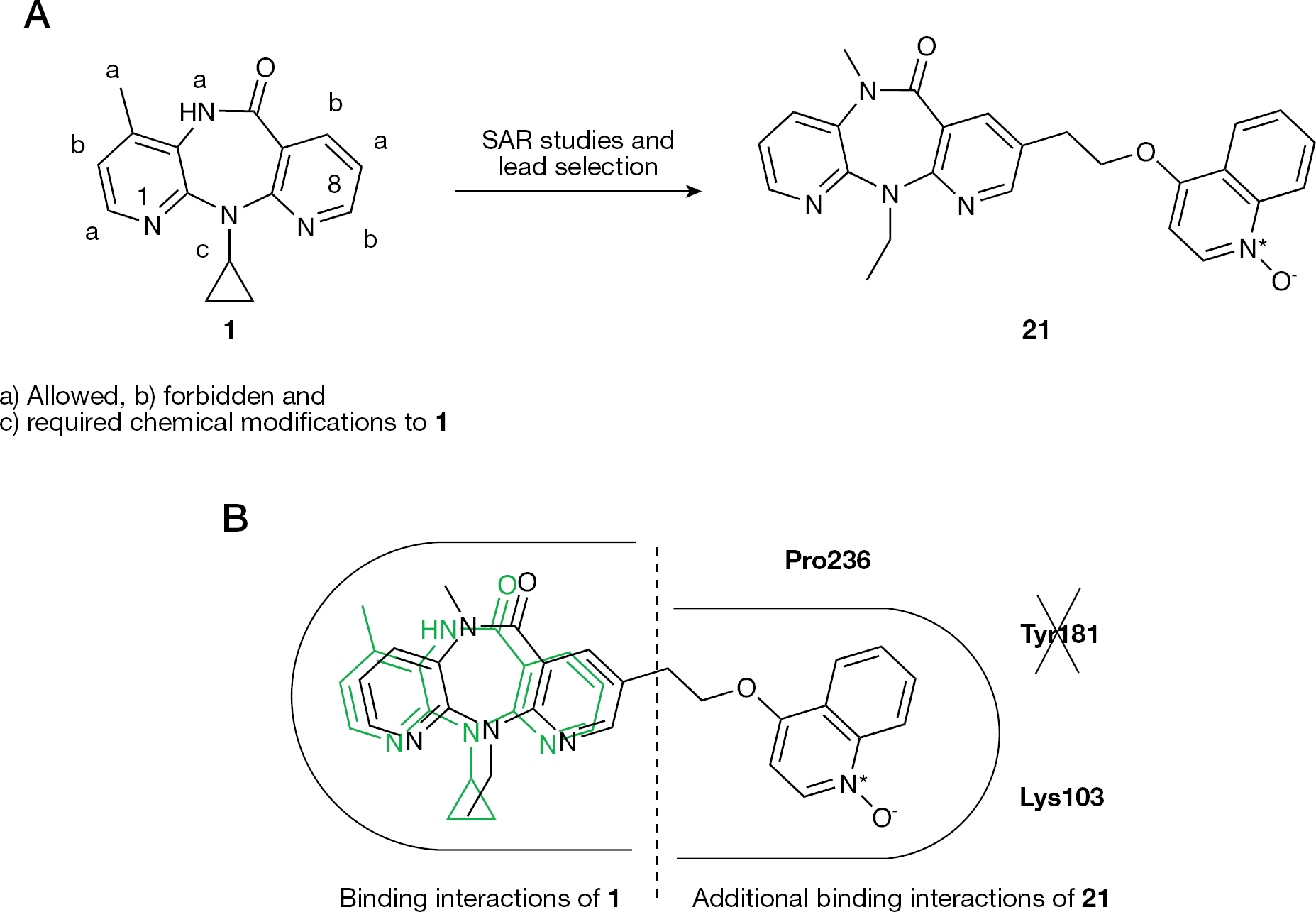
Extensive structure–activity relationship studies by Boehringer Ingelheim on the dipyridodiazepinone scaffold of 1 led to the discovey of a new HIV-1 NNRTI class endowed with potent antiviral activity against clinically relevant HIV-1 NNRTI-resistant strains and acceptable biopharmaceutical properties. Among the new derivatives, BIRL 355 BS (21) was selected for an advanced development (Figure 15A) [126–128].
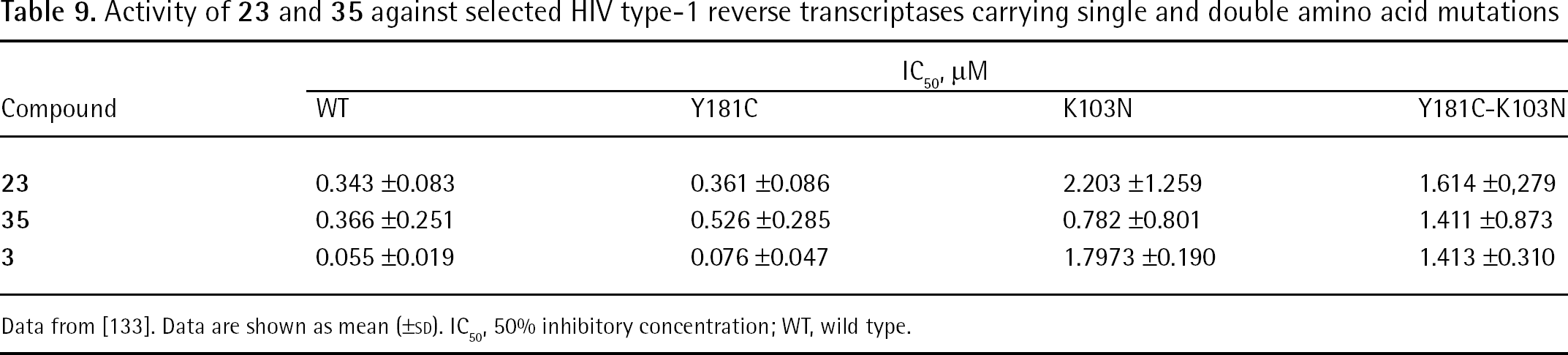
Compound 21 displayed potent anti-HIV-1 activity against HIV-1 isolates that are resistant to the currently used NNRTIs (Table 7 and Table 8) [27,29,30]. Following a single dose of oral solution, the mean half-life was 2–4 h, with peak concentrations occurring at 30–60 min post-administration. The mean apparent clearance ranged from 79.2 to 246 l/h for administered doses of 12.5–100 mg [129].
IC50in vitro activity of 21
IC50, nM
Compound
WT
K103N
Y181C
Y188L
P236L
K103N-Y181C
K103N-V108I
K103N-P225H
K103N-L100I
K103N-G190A
21
17
44
51
1,357
129
95
121
40
167
ND
1
208
13,180
34,680
>10,000
430
>10,000
>10,000
>10,000
>10,000
ND
3
2
46
41
52
2.2
30
83
91
2,820
ND
IC50, 50% inhibitory concentration; ND, no data; WT, wild type.
EC50in vitro activity of 21 in HIV-1-infected cell cultures
EC50, nM
Compound
WT
K103N
Y181C
Y188L
P236L
K103N-Y181C
K103N-V108I
K103N-P225H
K103N-L100I
K103N-G190A
21
1.3
3.5
4.8
67
6.2
4.5
5.1
2.0
5.9
4.1
1
11
345
1,160
>10,000
16
>5,500
ND
ND
400
ND
3
0.3
5.5
0.5
36
ND
10.2
3.0
12
>1,000
54
HIV-1, HIV type-1; EC50, 50% effective concentration; ND, no data; WT, wild type.
X-ray crystallography studies at 2.7–2.1 Å resolution provided detailed information of the inhibitor binding modes. Molecular modelling studies have shown that the tricyclic core of 21 was able to bind into the NNBS of the RT in similar manner to 1; however, the heterocyclic moiety at position 8 in 21 improves the ability to form favourable binding interactions with the NNBS of the mutated RTs. In particular, the new binding interactions provided by the heterocyclic moiety at position 8 (including Pro236 and Lys103 backbone) allowed 21 to retain its antiviral potency despite disruption of interactions with the aromatic ring of Tyr181. Structure models strongly supported the hypothesis that the entrance to the NNRTI NNBS is located near the Pro236 and Val106 of the p66 subunit (Figure 15B) [131].
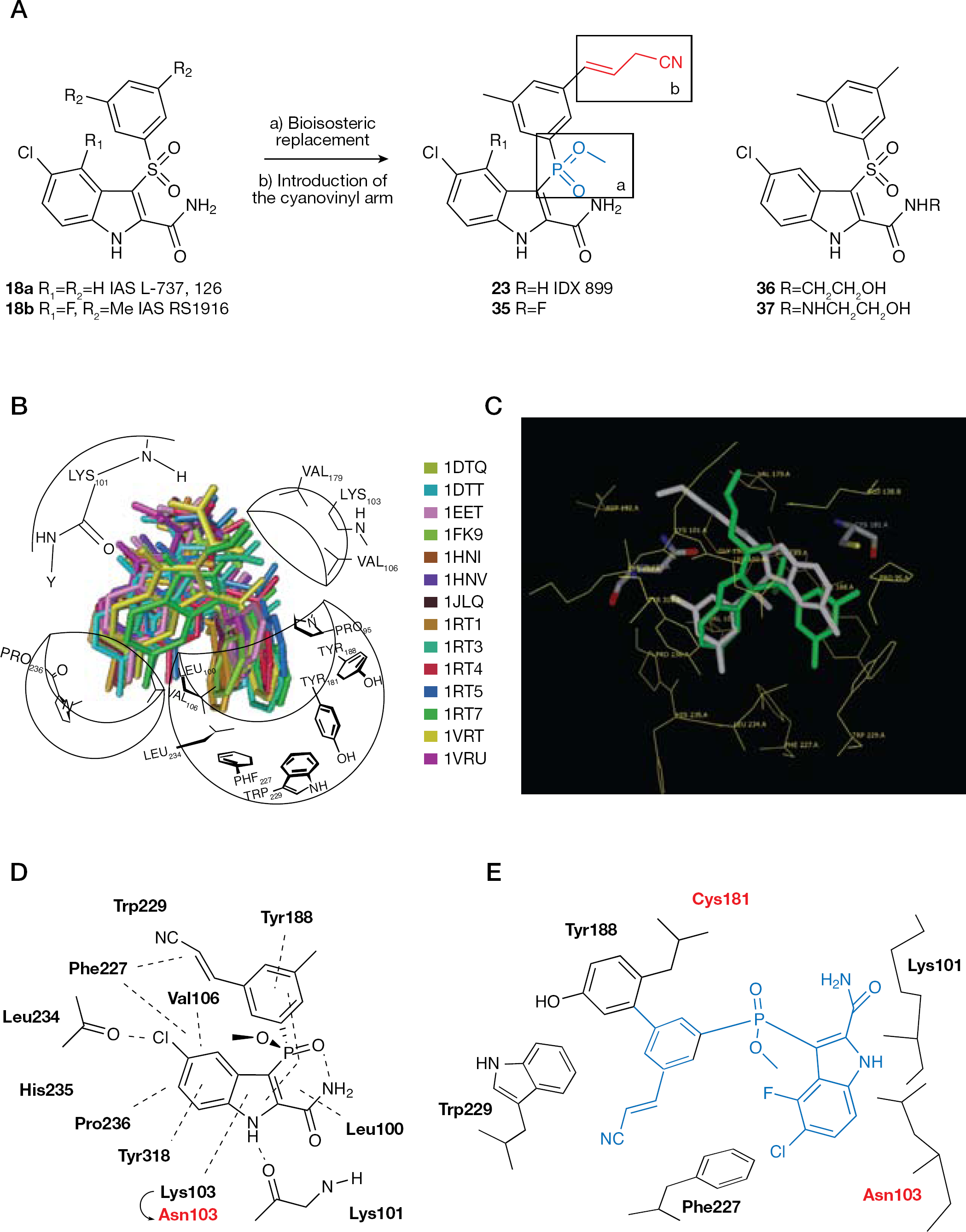
IDX 899
IDX 899 (23) is a new HIV-1 NNRTI initially developed by Idenix Pharmaceuticals Laboratories by bioisosteric replacement of the 3-sulfonyl bridging group of IAS NNRTIs (that is, 18) [73,,–80] with a phosphinic acid methyl ester group (Figure 16A). The 3′-Z-cyanovinyl moiety was introduced in order to achieve a broad spectrum of activity against HIV-1 NNRTI-resistant mutants, as it did for 16 [93,113]. In 2009, Idenix Pharmaceuticals Laboratories signed a license agreement granting GlaxoSmithKline exclusive worldwide rights to 23.
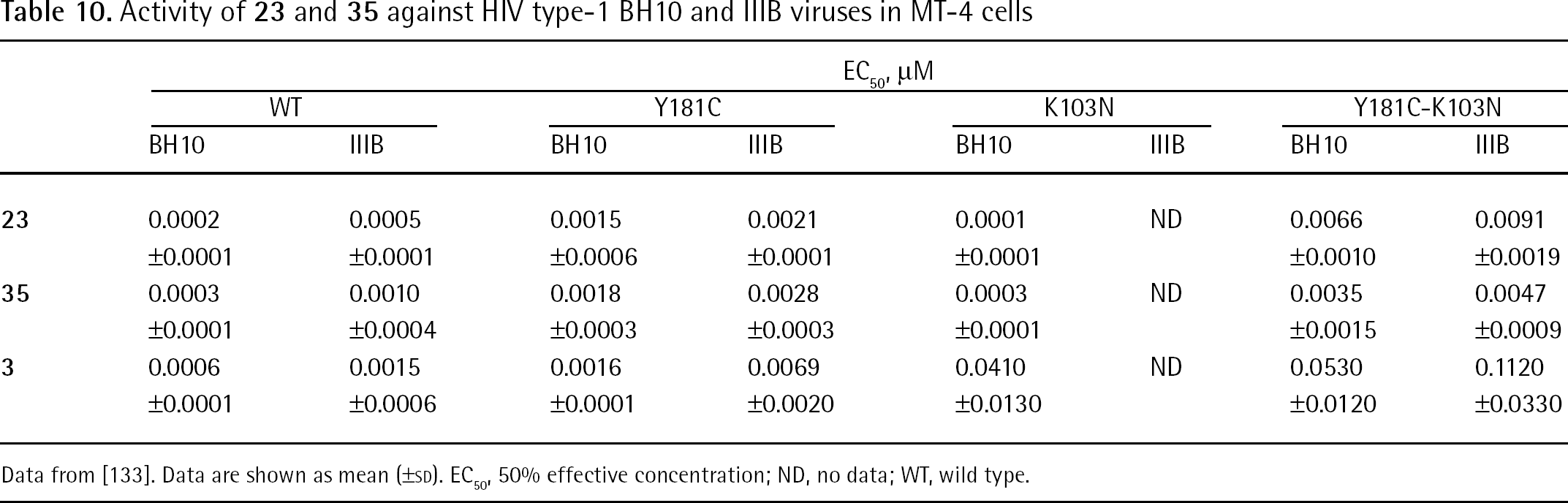
Compound 23 is a drug candidate endowed with potent in vitro antiretroviral activity and a barrier to resistance that is superior to 3 [132]. Breakthrough studies in MT-2 cells demonstrated slower development of resistance to 23 (45 days) than to 3 (17 days) and resistance mutations selected with 23 differed from those selected with 3 [132]. Table 9 shows the in vitro anti-HIV activity of enantiomerically pure (S)-3-phosphoindoles 23 and 35 against a panel of mutant HIV-1 RT enzymes. Compound 23 inhibited HIV-1 WT RT (subtype B; BH10 strain) at an IC50 value of 0.34 μM and the K103N-Y181C mutant strain at an IC50 value of 1.61 μM; compound 35 IC50 values ranged from 0.37 μM (WT RT) to 1.41 μM (K103N-Y181C RT). In MT-4 cells, compounds 23 and 35 inhibited HIV-1 (subtype B; BH10 strain) reproduction with EC50 values in the nanomolar range (Table 10). The selectivity index (SI; calculated by the 50% cytotoxic concentration/EC50 ratio) values determined in MT-4 cells for compounds 23 and 35 were >18,000 and >22,000, respectively [133].
Activity of 23 and 35 against selected HIV type-1 reverse transcriptases carrying single and double amino acid mutations
IC50, μM
Compound
WT
Y181C
K103N
Y181C-K103N
23
0.343 ±0.083
0.361 ±0.086
2.203 ±1.259
1.614 ±0,279
35
0.366 ±0.251
0.526 ±0.285
0.782 ±0.801
1.411 ±0.873
3
0.055 ±0.019
0.076 ±0.047
1.7973 ±0.190
1.413 ±0.310
Data from [133]. Data are shown as mean (±SD). IC50, 50% inhibitory concentration; WT, wild type.
Activity of 23 and 35 against HIV type-1 BH10 and IIIB viruses in MT-4 cells
EC50, μM
WT
Y181C
K103N
Y181C-K103N
BH10
IIIB
BH10
IIIB
BH10
IIIB
BH10
IIIB
23
0.0002
0.0005
0.0015
0.0021
0.0001
ND
0.0066
0.0091
±0.0001
±0.0001
±0.0006
±0.0001
±0.0001
±0.0010
±0.0019
35
0.0003
0.0010
0.0018
0.0028
0.0003
ND
0.0035
0.0047
±0.0001
±0.0004
±0.0003
±0.0003
±0.0001
±0.0015
±0.0009
3
0.0006
0.0015
0.0016
0.0069
0.0410
ND
0.0530
0.1120
±0.0001
±0.0006
±0.0001
±0.0020
±0.0130
±0.0120
±0.0330
Data from [133]. Data are shown as mean (±SD). EC50, 50% effective concentration; ND, no data; WT, wild type.
The binding mode of IAS derivatives was investigated by docking studies into the HIV-1 NNBS of 14 RTs, using compound L-737,126 (18a) as a reference compound [134]. The only uncertainty was caused by the 2-carboxyamide function: the amide carbonyl was either in a cis position with respect to the indole NH or rotated by 180°. The other chemical features of 18a shared a common binding mode, namely the indole NH made an H-bond with Lys101 carbonyl, the phenyl ring of the benzenesulfonyl moiety occupied a hydrophobic aromatic-rich pocket formed mainly by the side chains of Tyr181, Tyr188, Phe227 and Trp229, the solfonyl group fitted in a little hydrophobic pocket formed by Val106, Lys103 (only α- and β-CH2) and Val179, and the 5-chlorine atom established favourable contacts with Pro236 (Figure 16B). Docking and cross-docking studies showed that the double K103N-Y181C mutation led to two different docked conformations with respect to those observed for the single Y181C or K103N mutation. The binding mode of 36 into the K103N-Y181C RT was similar to 18a into the WT RT. By contrast, derivative 37 showed different binding interactions (Figure 16C). According to previous findings [135], IAS 36 was superior to 37 as a K103NY181C RT inhibitor.
Structure–activity relationship analysis on compound 21
In comparing the cocrystal structure of 23 with the docked conformation of 36 into the HIV-1 K103N-Y181C RT, similar binding interactions are visible and both inhibitors clearly adopt a ‘butterfly-like’ active conformation [132,136] (Figure 16D and 16E, respectively).
Development of compound 23
Development of 15 and 39 from 38
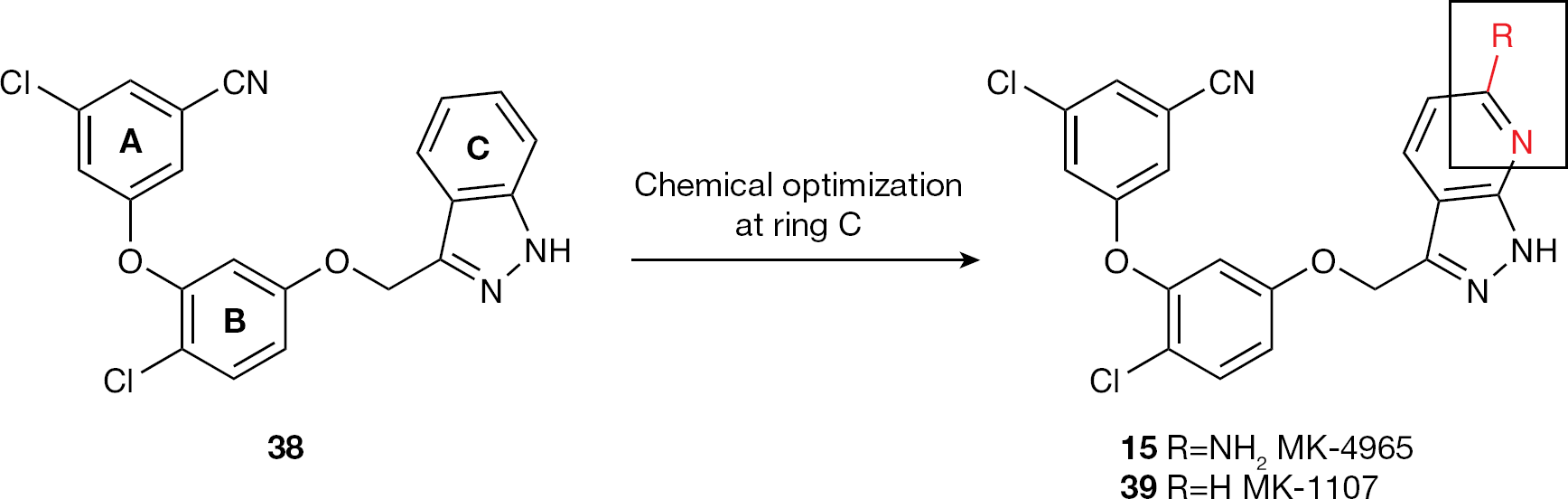
MK-4965
Merck Research Laboratories discovered indazole 38, a potent antiviral agent against HIV-1 WT and clinically relevant mutant strains, but one that is endowed with low solubility and low oral bioavailability [137]. Crystallographic studies suggested replacement of the phenyl ring of indazole with a pyridine ring and the introduction of a number of polar substituents, with the goal to improve solubility and oral bioavailability while retaining the desirable overall biological profile of the parent compound (Figure 17) [138].
The 7-aza analogue 39 was a potent inhibitor of HIV-1 WT and a panel of clinically relevant mutant viruses (Table 11), and showed promising pharmacokinetics after both intravenous and oral dosing in several species; however, further in vivo studies showed that 39 was orally bioavailable only when dosed as a solution. By adding an amino group in the 6 position of the pyridine ring, further improvement of the basicity (and then solubility) was reached while maintaining the excellent overall antiviral profile. Compound 15 showed antiviral activity similar to that of the parent compound 39 and was much more soluble than 39, especially at an acidic pH.
Inhibition of reverse transcriptase polymerase and antiviral activity in MT-4 cells of 13 and 39a
In RPMI 1640 medium containing 10% fetal bovine serum.
Cell culture inhibitory concentration (CIC95) was defined as the concentration at which the spread of virus is inhibited by >95%. IC50, 50% inhibitory concentration; WT, wild type.
The WT RT/15 cocrystal structure showed all binding interactions into the NNBS previously described in this class of NNRTIs. The binding mode into the Y181C RT was not affected by the mutation, as the inhibitor did not form any direct binding interaction with Tyr181. The cocrystal structures showed similar binding modes and only small differences in the central aryl ether conformation and slight movements of the compounds relative positions in the site were observed. The results obtained by the Merck team were in agreement with the ‘flexibility’ hypothesis previously reported for DAPY derivatives [93]; however, in this case, the differences in binding modes between the WT and mutant enzymes were more subtle [138]. Crucial features of the binding mode of 15 into the NNBS were its ability to form direct interactions with the Lys103 backbone and to avoid direct interaction with Tyr181 (Figure 18).
HBY 097
HBY 097 (24) was developed by Hoechst–Roussel in collaboration with Bayer from compound S-2720, a quinoxaline NNRTI that showed favourable property of an improved resistance profile against G1903E mutant HIV-1 RT in vitro [139,140]. Compound 24 inhibited HIV-1 WT replication with an IC50 value of 6 nM (with no activity against the HIV type-2 strain) and showed a potent anti-HIV-1 activity against a panel of 41 clinical isolates with IC50 and IC90 values in the nanomolar range of concentration. Compound 24 also showed favourable bioavailability and reduced cytotoxic concentration with an SI of 104 [141]. Assuming that any NNRTI is associated with specific HIV-1 drug-resistant mutations, 24 selected the unusual RT mutation G190E along with the mutations L74V/I and V75L/I, which are ordinarily associated with NRTI-based therapy. Interestingly, the G190E mutation causes reduction of the RT activity and the rate of HIV proliferation, whereas either Leu74-Gly190 or Val75-Gly190 double mutations partially restore the enzymatic activity. Binding mode inspection by means of the crystal structure (entry code 1BQM; resolution 3.10 Å) showed significant differences from the ‘butterfly-like’ model, which might account for the peculiar drug resistance profile [141]. In particular, it is possible to note that 24 adopts a pseudo V-shaped arrangement into the NNBS of the RT. The aromatic region of the quinoxaline nucleus lies in the butterfly wing I zone together with the methoxy group and thus form hydrophobic interactions with Leu100, Tyr318 and Phe227. Two hydrogen bonds are formed between the N2 and Lys101 backbone oxygen and the compound 24 sulfur (S1) and Lys101 backbone nitrogen. Wing II of the model is represented by the iso-propoxycarbonyl moiety, which forms hydrophobic interactions with Leu100, Tyr181, Tyr188 and Trp229 (similarly to 1, no aromatic contacts are detectable; Figure 19A). Analysis of the crystal structures of 24 complexed with either Y188L (entry code 1BQN; resolution 3.30 Å [141]) or K103N mutated RTs (entry code 1HQU; resolution 2.8 Å [142]) showed that these mutations did not affect the binding mode (Figure 19B). The 100-fold reduction activity against the Y188L (IC50=0.6 μM) mutation with respect to the WT RT might be correlated with the loss of interaction with Y188; all the other binding interactions described for the WT RT are retained. The flexibility of both isopropoxycarbonyl and methyl-thiomethylene moieties might contribute to improve the performance against the Y188L mutation.
Binding mode of compound 5 in wild-type and mutated RT
Binding mode of compounds 26 and 24
Conclusions
Second-generation NNRTIs currently under development are potent inhibitors of HIV-1 WT and the most clinically relevant HIV-1 mutant strains, and show inhibitory activities in the nano- or subnanomolar of concentration range. Although all compounds are endowed with potent and (more or less) similar antiretroviral potency, they do not share a common binding mode to the NNBS of the RT. In the past five years, NNRTIs based on the ‘butterfly-like’ model seemed obsolete with respect to the newer ‘horseshoe-like’ inhibitors; however, new drug candidates renewed the interest for the ‘butterfly-like’ active conformation. In addition, a new concept of binding interaction with the HIV-1 drug-resistant mutant strains emerged: the ‘flexibility hypothesis’. According to this idea, a powerful inhibition of the mutant variants of the HIV-1 should be correlated to the ability of the molecule to adopt variable binding conformations, which would be not affected by the interchanges of the amino acid residues into the NNBS of the mutant RT. These findings support the opinion that a newer era for the HIV-1 NNRTIs has just begun. Future NNRTIs should be capable to adopt multi-binding conformations for a powerful inhibition of HIV-1 WT and the most relevant drug-resistant mutant strains.
Footnotes
The authors declare no competing interests.
References
1.
UNAIDS. Status of the global HIV epidemic. Report on the global AIDS epidemic. Geneva: UNAIDS2008; pp. 29–62.
2.
SchererEDouekDMcMichaelA. 25 Years of HIV research on virology, virus restriction, immunopathogenesis, genes and vaccines. Clin Exp Immunol2008; 154:6–14.
3.
De ClercqE. Toward improved anti-HIV chemotherapy: Therapeutic strategies for intervention with HIV infections. J Med Chem1995; 38:2491–2517.
4.
De ClercqE. Highlights in the development of new antiviral agents. Mini Rev Med Chem2002; 2:163–175.
5.
WilliamsIG. Enfuvirtide (Fuzeon): The first fusion inhibitor. Int J Clin Pract2003; 57:890–897.
6.
OversteegenLShahMRoviniH. HIV combination products. Nat Rev Drug Discov2007; 6:951–952.
7.
MitsuyaHWeinholdKJFurmanPA. 3′-Azido-3′-deoxythymidine (BWA509U): An antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc Natl Acad Sci U S A1985; 82:7096–7100.
8.
De ClercqE. The design of drugs for HIV and HCV. Nat Rev Drug Discov2007; 6:1001–1018.
9.
BarbaroGScozzafavaAMastrolorenzoASupuranCT. Highly active antiretroviral therapy: Current state of the art, new agents and their pharmacological interactions useful for improving therapeutic outcome. Curr Pharm Des2005; 11:1805–1843.
10.
OrkinCStebbingJNelsonM. A randomized study comparing a three- and four-drug HAART regimen in first-line therapy. J Antimicrob Chemother2005; 55:246–251.
11.
LiXMargolickJBConnoverCS. Interruption and discontinuation of highly active antiretroviral therapy in the multicenter AIDS cohort study. J Acquir Immune Defic Syndr2005; 38:320–328.
ChengYCDutschmanGEBastowKFSarngadharanMGTingRY. Human immunodeficiency virus reverse transcriptase. General properties and its interactions with nucleoside triphosphate analogs. J Biol Chem1987; 262:2187–2189.
14.
Jacobo-MolinaAClarkADJr.WilliamsRL. Crystals of a ternary complex of human immunodeficiency virus type 1 reverse transcriptase with a monoclonal antibody Fab fragment and double-stranded DNA diffract X-rays to 3.5-Å resolution. Proc Natl Acad Sci U S A1991; 88:10895–10899.
15.
KohlstaedtLAWangJFriedmanJMRicePASteitzTA. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science1992; 256:1783–1790.
16.
Jacobo-MolinaADingJNanniRG. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 Å resolution shows bent DNA. Proc Natl Acad Sci U S A1993; 90:6320–6324.
17.
RodgersDWGamblinSJHarrisBA. The structure of unliganded reverse transcriptase from the human immunodeficiency virus type 1. Proc Natl Acad Sci U S A1995; 92:1222–1226.
18.
CohenKAHopkinsJIngrahamRH. Characterization of the binding site for nevirapine (BI-RG-587), a nonnucleoside inhibitor of human immunodeficiency virus type-1 reverse transcriptase. J Biol Chem1991; 266:14670–14674.
19.
TantilloCDingJJacobo-MolinaA. Locations of anti-AIDS drug binding sites and resistant mutations in the three dimensional structure of HIV-1 reverse trancriptase. J Mol Biol1994; 243:369–387.
20.
BabaMTanakaHDe ClercqE. Highly specific inhibition of human immunodeficiency virus type 1 by a novel 6-substituted acyclouridine derivative. Biochem Biophys Res Commun1989; 165:1375–1381.
21.
MiyasakaTTanakaHBabaM. A novel lead for specific anti-HIV-1 agents: 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine. J Med Chem1989; 32:2507–2509.
22.
PauwelsRAndriesKDesmyterJ. Potent and selective inhibition of HIV-1 replication in vitro by a novel series of TIBO derivatives. Nature1990; 343:470–474.
23.
DebyserZPauwelsRAndriesK. An antiviral target on reverse transcriptase of human immunodeficiency virus type 1 revealed by tetrahydroimidazo-[4,5,1-jk] [1,4] benzodiazepin-2(1H)-one and -thione derivatives. Proc Natl Acad Sci U S A1991; 88:1451–1455.
24.
KuklaMJBreslinHJPauwelsR. Synthesis and anti-HIV-1 activity of 4,5,6,7-tetrahydro-5-methylimidazo[4,5,1-jk] [1,4]benzodiazepin-2(1H)one (TIBO) derivatives. J Med Chem1991; 34:746–751.
25.
KuklaMJBreslinHJDiamondCJ. Synthesis and anti-HIV-1 activity of 4,5,6,7-tetrahydro-5-methylimidazo[4,5,1-jk] [1,4]benzodiazepin-2(1H)one (TIBO) derivatives. 2. J Med Chem1991; 34:3187–3197.
26.
BreslinHJKuklaMJLudoviciDW. Synthesis and anti-HIV-1 activity of 4,5,6,7-tetrahydro-5-methylimidazo[4,5,1-jk] [1,4]benzodiazepin-2(1H)one (TIBO) derivatives. 3. J Med Chem1995; 38:771–793.
27.
MerluzziVJHargraveKDLabadiaM. Inhibition of HIV-1 replication by a nonnucleoside reverse transcriptase inhibitor. Science1990; 250:1411–1413.
28.
HargraveKDProudfootJRGrozingerKG. Novel non-nucleoside inhibitors of HIV-1 reverse transcriptase. 1. Triciclyc pyridobenzo- and dipiridodiazepinones. J Med Chem1991; 34:2231–2241.
29.
KlunderJMHargraveKDWestM. Novel nonnucleoside inhibitors of HIV-1 reverse transcriptase. 2. Tricyclic pyridoxazepinones and dibenzoxazepinones. J Med Chem1992; 35:1887–1897.
30.
ProudfootJRPatelURKapadiaSRHargraveKD. Novel non-nucleoside inhibitor of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase. 3. Dipyrido[2,3-b:2′,3′-c]diazepinones. J Med Chem1995; 38:1406–1410.
31.
RomeroDLMorgeRAGeninMJ. Bis(heteroaryl) piperazine (BHAP) reverse transcriptase inhibitors: Structure–activity relationships of novel substituted indole analogs and the identification of 1-[(5-methanesulfonamido-1H-indol-2-yl)carbonyl]-4-[3-[(1-methylethyl)amino] pyridinyl]piperazinemonomethanesulfonate (U-90152S), a second-generation clinical candidate. J Med Chem1993; 36:1505–1508.
32.
RomeroDLMorgeRABilesC. Discovery, synthesis and bioactivity of bis(heteroaryl)piperazines. 1. A novel class of non-nucleoside reverse transcriptase inhibitors. J Med Chem1994; 37:999–1014.
33.
RomeroDLOlmstedRAPoelTJ. Targeting delarvidine/atervidine resistant HIV-1: Identification of (alkylamino)piperidine-containing bis(heteroaryl) piperazines as broad spectrum HIV-1 reverse transcriptase inhibitors. J Med Chem1996; 39:3769–3789.
34.
GeninMJPoelTJYagiY. Syntehsis and bioactivity of novel bis(heteroaryl)piperazine (BHAP) reverse transcriptase inhibitors: Structure–activity relationship and increased metabolic stability of novel substituted pyridine analogs. J Med Chem1996; 39:5267–5275.
35.
FreimuthWW. Delavirdine mesylate, a potent non-nucleoside HIV-1 reverse transcriptase inhibitor. Adv Exp Med Biol1996; 394:279–289.
36.
GoldmanMENunbergJHO'BrienJA. Pyridinone derivatives: Specific human immunodeficiency virus type 1 reverse transcriptase inhibitors with antiviral activity. Proc Natl Acad Sci U S A1991; 88:6863–6867.
37.
CorbettJWKresgeKJPanS. Trifluoromethyl-containing 3-alkoxymethyl and 3-aryloxymethyl-2-pyridones are potent inhibitors of HIV-1 non-nnucleoside reverse transcriptase. Bioorg Med Chem Lett2001; 11:309–312.
38.
ArticoM. Non-nucleoside anti-HIV-1 reverse transcriptase inhibitors (NNRTIs): A chemical survey from lead compounds to selected drugs fro clinical trials. Farmaco1996; 51:305–331.
De ClercqE. Antivirals: Current state of art. Future Virol2008; 3:393–405.
44.
De ClercqE. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int J Antimicr Ag2009; 33:307–320.
45.
FujiwaraTSatoAel-FarrashM. S-1153 inhibits replication of known drug-resistant strains of human immunodeficiency virus type 1. Antimicrob Agents Chemother1998; 42:1340–1345.
46.
MaoCSudbeckEAVenkatachalamTKUckunFM. Rational design of N-[2-(2,5-dimethoxyphenylethyl)]-N'-[2-(5-bromopyridyl)]-thiourea (HI-236) as a potent non-nucleoside inhibitor of drug-resistant human immunodeficiency virus. Bioorg Med Chem Lett1999; 9:1593–1598.
47.
TaylorDLAhmedPSChambersP. Pyrido [1,2a] indole derivatives identified as novel non-nucleoside reverse transcriptase inhibitors of human immunodeficiency virus type 1. Antivir Chem Chemother1999; 10:79–86.
48.
TuckerTJLyleTAWiscountCM. Synthesis of a series of 4-(arylethynyl)-6-chloro-4-cyclopropyl-3,4-dihydroquinazolin-2(1H)-ones as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. J Med Chem1994; 37:2437–2444.
49.
YoungSDBrichterSFTranLO. L-743,726 (DMP-266): A novel nonnucleoside inhibitor of human immunodeficiency virus type 1 reverse transcriptase. Antimicrob Agents Chemother1995; 39:2602–2605.
CorbettJWKoSSRodgersJD. Inhibition of clinically relevant mutant variants of HIV-1 by quinazolinone nonnucleoside reverse transcriptase inhibitors. J Med Chem2000; 43:2019–2030.
52.
CocuzzaAJChidesterDRCordovaBC. Synthesis and evaluation of efavirenz (Sustiva™) analogues as HIV-1 reverse transcriptase inhibitors: Replacement of the cyclopropylacetylene side chain. Bioorg Med Chem Lett2001; 11:1177–1179.
53.
CorbettJWKoSSRodgersJD. Expanded-spectrum nonnucleoside reverse transcriptase inhibitors inhibit clinically relevant mutant variants of human immunodeficiency virus type 1. Antimicrob Agents Chemother1999; 43:2893–2897.
54.
CorbettJWRodgersJD. Discovery of second generation quinazoline non-nucleoside reverse transcriptase inhibitors. Prog Med Chem2002; 40:63–105.
55.
BellFWCantrellASHogbergM. Phenethylthiazolethiourea (PETT) compounds, a new class of HIV-1 reverse transcriptase inhibitors. 1. Synthesis and basic structure–activity relationship studies of PETT analogs. J Med Chem1995; 38:4929–4936.
56.
CantrellASEngelhardtPHogbergM. Phenethylthiazolylthiourea (PETT) compounds as a new class of HIV-1 reverse transcriptase inhibitors. 2. Synthesis and further structure-activity relationship studies of PETT analogs. J Med Chem1996; 39:4261–4274.
57.
HogbergMSahlbergCEngelhardtP. Urea-PETT compounds as a new class of HIV-1 reverse transcriptase inhibitors. 3. Synthesis and further structure-activity relationship studies of PETT analogues. J Med Chem1999; 42:4150–4160.
58.
BalzariniJBrouwerWGDaoDCOsikaEMDe ClercqE. Identification of novel thiocarboxanilide derivatives that suppress a variety of drug-resistant mutant human immunodeficiency virus type 1 strains at a potency similar to that for wild-type virus. Antimicrob Agents Chemother1996; 40:1454–1466.
59.
BalzariniJPelemansHAquaroS. Highly favorable antiviral activity and resistance profile of the novel thiocarboxanilide pentenyloxy ether derivatives UC-781 and UC-82 as inhibitors of human immunodeficiency virus type 1 replication. Mol Pharmacol1996; 50:394–401.
60.
BottaMArticoMMassaS. Synthesis, antimicrobial and antiviral activities of isotrimethoprim and some related derivatives. Eur J Med Chem1992; 27:251–257.
61.
DanelKNielsenCPedersenEB. Anti-HIV active naphthyl analogues of HEPT and DABO. Acta Chem Scand1997; 51Suppl 3:426–430.
62.
SudbeckEAMaoCVigRVenkatachalamTKTuel-AhlgrenLUckunFM. Structure-based design of novel dihydroalkoxybenzyloxopyrimidine derivatives as potent nonnucleoside inhibitors of the human immunodeficiency virus reverse transcriptase. Antimicrob Agents Chemother1998; 42:3225–3233.
63.
MaiAArticoMSbardellaG. 5-Alkyl-2-(alkylthio)-6-(2,6-dihalophenylmethyl)-3, 4-dihydropyrimidin-4(3H)-ones: novel potent and selective dihydro-alkoxy-benzyl-oxopyrimidine derivatives. J Med Chem1999; 42:619–627.
64.
MaiAArticoMSbardellaG. Dihydro(alkylthio) (naphthylmethyl)oxopyrimidines: Novel non-nucleoside reverse transcriptase inhibitors of the S-DABO series. J Med Chem1997; 40:1447–1454.
65.
VigRMaoCVenkatachalamTKTuel-AhlgrenLSudbeckEAUckunFM. 5-Alkyl-2-[(methylthiomethyl) thio]-6-(benzyl)-pyrimidin-4-(1H)-ones as potent non-nucleoside reverse transcriptase inhibitors of S-DABO series. Bioorg Med Chem Lett1998; 8:1461–1466.
66.
RadiMMagaGAlongiM. Discovery of chiral cyclopropyl dihydro-alkylthio-benzyl-oxopyrimidine (S-DABO) derivatives as potent HIV-1 reverse transcriptase inhibitors with high activity against clinically relevant mutants. J Med Chem2009; 52:840–851.
67.
TuckerTJSiskoJTTyneborRM. Discovery of 3-{5-[(6- amino-1H-pyrazolo[3,4-b]pyridine-3-yl)methoxy]-2-chlorophenoxy}-5-chlorobenzonitrile (MK-4965): A potent, orally bioavailable HIV-1 non-nucleoside reverse transcriptase inhibitor with improved potency against key mutant viruses. J Med Chem2008; 51:6503–6511.
68.
De CorteBL. From 4,5,6,7-tetrahydro-5-methylimidazo[4,5,1-jk] [1,4]benzodiazepin-2(1H)-one (TIBO) to etravirine (TMC125): fifteen years of research on non-nucleoside inhibitors of HIV-1 reverse transcriptase. J Med Chem2005; 48:1689–1696.
69.
JanssenPAJLewiPJArnoldE. In search of a novel anti-HIV drug: Multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (R278474, Rilpivirine). J Med Chem2005; 48:1901–1909.
70.
ArticoMSilvestriRStefancichG. Synthesis of pyrryl aryl sulfones targeted at the HIV-1 reverse transcriptase. Arch Pharm (Weinheim)1995; 328:223–229.
71.
ArticoMSilvestriRMassaS. 2-Sulfonyl-4-chloroanilino moiety: A potent pharmacophore for the anti-human immunodeficiency virus type 1 activity of pyrrolyl aryl sulfones. J Med Chem1996; 39:522–530.
72.
ArticoMSilvestriRPagnozziE. Structure-based design, synthesis and biological evaluation of novel pyrrolyl aryl sulfones (PASs), HIV-1 non-nucleoside reverse transcriptase inhibitors active at nanomolar concentrations. J Med Chem2000; 43:1886–1891.
73.
SilvestriRDe MartinoGLa ReginaG. Novel indolyl aryl sulfones active against HIV-1 carrying NNRTI resistance mutations: Synthesis and SAR studies. J Med Chem2003; 46:2482–2493.
74.
SilvestriRArticoMDe MartinoG. Simple, short peptide derivatives of a sulfonylindolecarboxamide (L-737,126) active in vitro against HIV-1 wild-type and variants carrying non-nucleoside reverse transcriptase inhibitor resistance mutations. J Med Chem2004; 47:3892–3896.
75.
RagnoRArticoMDe MartinoG. Docking and 3-D QSAR studies on indolyl aryl sulfones (IASs). Binding mode exploration at the HIV-1 reverse transcriptase non-nucleoside binding site and design of highly active N-(2-hydroxyethyl) carboxyamide and N-(2-hydroxyethyl)carboxyhydrazide derivatives. J Med Chem2005; 48:213–223.
76.
SilvestriRArticoM. Indolyl aryl sulfones (IASs): Development of highly potent NNRTIs active against wt-HIV-1 and clinically relevant drug resistant mutants. Curr Pharm Des2005; 11:3779–3806.
77.
De MartinoGLa ReginaGRagnoR. Indolyl aryl sulfones as HIV-1 non-nucleoside reverse transcriptase inhibitors: Synthesis, biological evaluation and binding mode studies of new derivatives at indole-2-carboxamide. Antivir Chem Chemother2006; 57:59–77.
78.
RagnoRColucciaALa ReginaG. Design, molecular modeling, synthesis and anti-HIV-1 activity of new indolyl aryl sulfones. Novel derivatives of the indole-2-carboxamide. J Med Chem2006; 49:3172–3184.
79.
La ReginaGColucciaAPiscitelliF. Indolyl aryl sulfones as HIV-1 non-nucleoside reverse transcriptase inhibitors: Role of two halogen atoms at the indole ring in developing new analogues with improved antiviral activity. J Med Chem2007; 50:5034–5038.
80.
PiscitelliFColucciaABrancaleA. Indolylarylsulfones bearing natural and unnatural aminoacids. Discovery of potent inhibitors of both HIV-1 non-nucleoside wild type and resistant mutant strains reverse transcriptase, and Coxsackie B4 virus. J Med Chem2009; 52:1922–1934.
81.
KingRWKlabeRMReidCDErickson-ViitanenSK. Potency of non nucleoside reverse transcriptase inhibitors (NNRTIs) used in combination with other human immunodeficiency virus NNRTIs, NRTIs, or protease inhibitors. Antimicrob Agents Chemother2002; 46:1640–1646.
82.
Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the clinical management of AIDS/HIV infection in adult people. US Department of Health and Human services. 1 December 2009. (Updated 01 December 2009. Accessed 07 February 2010.) Available from http://aidsinfo.nih.gov/contentfiles/AdultandAdolescentGL.pdf.
83.
StaszewskiSMorales-RamirezJTashimaKT. Efavirenz plus zidivudine and lamivudine, efavirenz plus indinavir, and indinavir plus zidivudine and lamivudine in the treatment of HIV-1 infection in adults. N Engl J Med1999; 341:1865–1873.
84.
HuiDY. Effects of HIV protease inhibitor therapy on lipid metabolim. Prog Lipid Res2003; 42:81–92.
85.
BachelerLJeffreySHannaG. Genotypic correlates of phenotypic resistance to efavirenz in virus isolated from patients failing non-nucleoside reverse transcriptase inhibitor therapy. J Virol2001; 75:4999–5008.
86.
DelaugerreCRohbanRSimonA. Resistance profile and cross-resistance of HIV-1 among patients failing a non-nucleoside reverse transcriptase inhibitor-containing regimen. J Med Virol2001; 65:445–448.
87.
Abdel-MalakMGallatiCMousaSA. The rise of second-generation non-nucleoside reverse transcriptase inhibitor: Etravirine, rilpivirine, UK-453061 and RDEA-806. Drug Fut2008; 33:691–699.
88.
AIDS InfoNet. Non-nucleoside reverse transcriptase inhibitors in development. Fact sheet number 430. (Updated 23 November 2009. Accessed 07 February 2010.) Available from http://www.aidsinfonet.org/categories/view/430.
AndriesKAzijnHThielemansT. TMC125, a novel next generation nonnucleoside reverse transcriptase inhibitor active against nonnucleoside reverse transcriptase inhibitor-resistant human immunodeficiency virus type 1. Antimicrob Agents Chemother2004; 48:4680–4686.
91.
HangJQLiYYangY. Substrate-dependent inhibition or stimulation of HIV RNase H activity by non-nucleoside reverse transcriptase inhibitors (NNRTIs). Biochem Biophys Res Commun2007; 352:341–350.
DasKClarkADJr.LewiPJ. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J Med Chem2004; 47:2550–2560.
94.
PauwelsRAndriesKDebyserZ. New tetrahydroimidazo[4,5,1-jk] [1,4]benzodiazepin-2(1H)-one and -thione derivatives are potent inhibitors of human immunodeficiency virus type 1 replication and are synergistic with 2′,3′-dideoxynucleoside analogs. Antimicrob Agents Chemother1994; 38:2863–2870.
95.
DingJDasKTantilloC. Structure of HIV-1 reverse transcriptase in a complex with the non-nucleoside inhibitor R-APA R 95845 at 2.8 Å resolution. Structure1995; 3:365–379.
96.
DingJDasKMoereelsH. Structure of HIV-1 RT/TIBO R 86183 complex reveals similarity in the binding of diverse nonnucleoside inhibitors. Nat Struct Biol1995; 2:407–415.
97.
DasKDingJHsiouY. Crystal structures of 8-Cl and 9-Cl TIBO complexed with wild-type HIV-1 RT and 8-Cl TIBO complexed with the Tyr181Cys HIV-1 RT drug-resistant mutant. J Mol Biol1996; 264:1085–1100.
98.
SchäferWFriebeW-GLeinertH. Non-nucleoside inhibitors of HIV-1 reverse transcriptase: Molecular modeling and X-ray structure investigations. J Med Chem1993; 36:726–732.
99.
LudoviciDWKuklaMJGrousPG. Evolution of anti-HIV drug candidates. Part 1: From α-anilinophenylacetamide (α-APA) to imidoyl thiourea (ITU). Bioorg Med Chem Lett2001; 11:2225–2228.
100.
LudoviciDWKavashRWKuklaMJ. Evolution of anti-HIV drug candidates part 2: Diaryltriazine (DATA) analogues. Bioorg Med Chem Lett2001; 11:2229–2234.
101.
PelemansHEsnoufRMJonckheereHDe ClercqEBalzariniJ. Mutational analysis of Tyr-318 within the non-nucleoside reverse transcriptase inhibitor binding pocket of human immunodeficiency virus type I reverse transcriptase. J Biol Chem1998; 273:34234–34239.
102.
PelemansHEsnoufRDe ClercqEBalzariniJ. Mutational analysis of Trp-229 of human immunodeficiency virus type 1 reverse transcriptase (RT) identifies this amino acid residue as a prime target for the rational design of new non-nucleoside RT inhibitors. Mol Pharmacol2000; 57:954–960.
103.
LewisPJde JongeMDaeyaertF. On the detection of multiple-binding modes of ligands to proteins, from biological, structural, and modeling. J Comput Aided Mol Des2003; 17:129–134.
104.
Udier-BlagovicMTirado-RivesJJorgensenWL. Validation of a model for the complex of HIV-1 reverse transcriptase with nonnucleoside inhibitor TMC125. J Am Chem Soc2003; 125:6016–6017.
105.
Udier-BlagovicMWatkinsEKTirado-RivesJJorgensenWL. Activity predictions for efavirenz analogues with the K103N mutant of HIV reverse transcriptase. Bioorg Med Chem Lett2003; 13:3337–3340.
106.
BrungerATAdamsPDRiceLM. Recent developments for the efficient crystallographic refinement of macromolecular structures. Curr Opin Struct Biol1998; 8:606–611.
107.
Udier-BlagovicMTirado-RivesJJorgensenWL. Validation of a model for the complex of HIV-1 reverse transcriptase with nonnucleoside inhibitor TMC125. J Am Chem Soc2003; 125:6016–6017.
108.
RenJNicholsCBirdLE. Binding of the second generation non-nucleoside inhibitor S-1153 to HIV-1 reverse transcriptase involves extensive main chain hydrogen bonding. J Biol Chem2000; 275:14316–14320.
109.
RizzoRCWangD-PTirado-RivesJJorgensenWL. Validation of a model for the complex of HIV-1 reverse transcriptase with sustiva through computation of resistance profiles. J Am Chem Soc2000; 122:12898–12900.
110.
VingerhoetsJAzijnHFransenE. TMC125 displays a high genetic barrier to the development of resistance: Evidence from in vitro selection experiments. J Virol2005; 79:12773–12782.
111.
GazzardBGPozniakALRosenbaumW. An open-label assessment of TMC 125, a new, next-generation NNRTI, for 7 days in HIV-1 infected individuals with NNRTI resistance. AIDS2003; 17:F49–F54.
112.
DasKLewiPJHughesSHArnoldE. Crystallography and the design of anti-AIDS drugs: Conformational flexibility and positional adaptability are important in the design of nonnucleoside HIV-1 reverse transcriptase inhibitors. Prog Biophys Mol Biol2005; 88:209–231.
113.
JanssenPALewiPJArnoldE. In search of a novel anti-HIV drug: Multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (R278474, rilpivirine). J Med Chem2005; 48:1901–1909.
114.
CliveDCasparyWKirbyP. Guide for performing the mouse lymphoma assay for mammalian cell mutagenicity. Mutat Res1987; 189:143–156.
115.
SmetFD'AubioulJvan GervenWXhonneuxRRenemanRS. A chronically implantable catheter-tip micromanometer (JSI 0400) that can be calibrated after implantation. Cardiov Res1979; 13:601–605.
116.
WolfordSTSchroerRAGohsFX. Reference range database for serum chemistry and hematology values in laboratory animals. J Toxicol Environ Health1986; 18:161–188.
117.
PauwelsR. Aspects of successful drug discovery and development. Antiviral Res2006; 71:77–89.
118.
DasKBaumanJDClarkADJr., High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: Strategic flexibility explains potency against resistance mutations. Proc Natl Acad Sci U S A2008; 105:1466–1471.
119.
FangCBarmanJDDasKRumorinoAArnoldEHochstrasserRM. Two-dimensional infrared spectra reveal relaxation of the nonnucleoside inhibitor TMC278 complexed with HIV-1 reverse transcriptase. Proc Natl Acad Sci U S A2008; 105:1472–1477.
120.
Van HerrewegeYVanhamGMichielsJ. A series of diaryltriazines and diarylpyrimidines are highly potent nonnucleoside reverse transcriptase inhibitors with possible applications as microbicides. J Antimicrob Chemother2004; 48:3684–3689.
121.
MordantCSchmittBPasquierE. Synthesis of novel diarylpyrimidine analogues of TMC278 and their antiviral activity against HIV-1 wild-type and mutant strains. Eur J Med Chem2007; 42:567–579.
122.
GirardetJ-LKohY-HLa RosaM. The discovery of RDEA806, a potent new HIV NNRTI in phase 1 clinical trials. 47th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy. 17–20 September 2007, Chicago, IL, USA. Abstract 3285.
123.
HamtakeRZhangZXuWBellowsDRaneyAGiradetJL. RDEA806, a potent, new HIV NNRTI with a high genetic barrier to resistance and a broad spectrum of activity. 47th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy. 17–20 September 2007, Chicago, IL, USA. Poster 300.
124.
MoyleGBoffitoMShenZ. RDEA806, a novel HIV non-nucleoside reverse transcriptase inhbitor, shows positive outcome in treatment of naive HIV patients. 46th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy. 25–28 October 2008, Washington, DC, USA. (Accessed 5 October 2009.) Available from http://www.ardeabio/com/Docs/icaac_rdea201_presentations_FINAL.pdf.
125.
CorbauRAllanGBurtC. UK-453,061: a non-nucleoside reverse transcriptase inhibitor for the treatment of drug-resistant HIV infections. 47th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy. 17–20 September 2007, Chicago, IL, USA. Abstract 2751.
126.
BonneauPDoyonLDuanJ. Characterization of a novel series of NNRTIs with broad antiviral potency against NNRTI-resistant HIV. 11th Conference on Retroviruses and Opportunistic Infections. 8–11 February 2004, San Francisco, CA, USA. Poster 530.
127.
BonneauPRobinsonPADuanJ. Antiviral characterizazion and human experience with BIRL 355 BS, a novel next-generation non-nucleoside reverse transcriptase inhibitor (NNRTI) with a broad anti-HIV-1 profile. 12th Conference on Retroviruses and Opportunistic Infections. 22–25 February 2005, Boston, MA, USA. Poster 558.
128.
BusaccaCACerretaMDongY. Development of a pilot-plant process for a nevirapine analogue HIV NNRT inhibitor. Org Process Res Dev2008; 12:603–613.
129.
HuangFKoenen-BergmannMMacGregorTRRingAHattoxSRobinsonP. Pharmacokinetic and safety evaluation of BILR 355, a second-generation nonnucleoside reverse transcriptase inhibitor, in healthy volunteers. Antimicrob Agents Chemother2008; 52:4300–4307.
130.
HuangFDrdaKMacGregorTR. Pharmacokinetics of BILR 355 after multiple oral doses coadministered with a low dose of ritonavir. Antimicrob Agents Chemother2009; 53:95–103.
131.
CoulombeRFinkDLandryS. Crystallographic study with BILR 355 BS, a novel nonnucleoside reverse transcriptase inhibitor (NNRTI) with a broad anti HIV-1 profile. 3rd International AIDS Society Conference on HIV Pathogenesis and Treatment. 24–27 July 2005, Rio de Janeiro, Brazil. Abstract WePp0105.
132.
JakubikJSeiferMGrayL. IDX12899 anti-HIV-1 activity and resistance profile is superior to efavirenz. Antivir Ther2007; 12Suppl 1:S32.
133.
StorerRAlexandreF-RDoussonC, inventors; Idenix Pharmaceuticals, Inc. assignee. Enatiomerically pure phosphoindoles as HIV inhibitors. United States patent WO2008/042240.10 April 2008.
134.
RagnoRColucciaALa ReginaGSilvestriR. Indolyl aryl sulphones as HIV-1 reverse transcriptase inhibitors: Docking and 3D QSAR studies. Exp Opin Drug Disc2007; 2:87–114.
135.
RagnoRMaiASbardellaG. Computer-aided design, synthesis, and anti-HIV-1 activity in vitro of 2-alkylamino-6-[1-(2,6-difluorophenyl)alkyl]-3,4-dihydro-5-alkylpyrimidin-4(3H)-ones as novel potent non-nucleoside reverse transcriptase inhibitors, also active against the Y181C variant. J Med Chem2004; 47:928–934.
136.
MayersDMurphyRZalaC. IDX899: A novel once-a-day second generation NNRTI for the treatment of HIV/AIDS. HIV DART. 9–12 December 2008, Rio Grande, Puerto Rico. Abstract 43.
137.
TuckerTJSaggarSSiskoJT. The design and synthesis of diaryl ether second generation NNRTIs with enhanced potency versus key clinical mutations. Bioorg Med Chem Lett2008; 18:2959–2966.
138.
TuckerTJSiskoJTTyneborRM. Discovery of 3-{5-[(6-amino-1H-pyrazolo[3,4-b]pyridine-3-yl)methoxy]-2-chlorophenoxy}-5-chlorobenzonitrile (MK-4965): a potent, orally bioavailable HIV-1 non-nucleoside reverse transcriptase inhibitor with improved potency against key mutant viruses. J Med Chem2008; 51:6503–6511.
139.
PauwelsRAndriesKDebyserZ. Potent and highly selective human immunodeficiency virus type 1 (HIV-1) inhibition by a series of alpha-anilinophenylacetamide derivatives targeted at HIV-1 reverse transcriptase. Proc Natl Acad Sci U S A1993; 90:1711–1715.
140.
KleimJ-PBenderRKirschR. Preclinical evaluation of HBY 097, a new nonnucleoside reverse transcriptase inhibitor of human immunodeficiency virus type 1 replication. J Antimicrob Chemother1995; 39:2253–2257.
141.
HsiouYDasKDingJ. Structures of Tyr188Leu mutant and wild-type HIV-1 reverse transcriptase complexed with the non-nucleoside inhibitor HBY 097: Inhibitor flexibility is a useful design feature for reducing drug resistance. J Mol Biol1998; 284:313–323.
142.
HsiouYDingJDasK. The Lys103Asn mutation of HIV-1 RT: A novel mechanism of drug resistance. J Mol Biol2001; 309:437–445.