
Abstract
The chemokine coreceptor 5 (CCR5) antagonists are antiretroviral agents with an extracellular, host-targeted mechanism of action against HIV. Maraviroc, the first-in-class CCR5 antagonist, received regulatory approval in 2007, becoming the first oral antiretroviral from a new class in more than 10 years. Other compounds in this class are in various stages of clinical development. In 2005, we reviewed the limited clinical data then available on CCR5 antagonists. In this follow-up review, we revisit the field and assess the clinical and virological data that have emerged in the 4 years since, with particular reference to maraviroc for which the most comprehensive data currently exist.
Introduction
Chemokine coreceptor 5 (CCR5) antagonists are a new class of antiretroviral agents that inhibit HIV replication by binding to CCR5 on the surface of host target cells and preventing entry of viral strains reliant on this coreceptor for infection. The first and only currently approved agent within this class, maraviroc, received marketing authorisation in the USA, Europe and other regions of the world in 2007, with an indication for treatment-experienced adult patients with only CCR5-tropic (R5) HIV-1 detectable. Other compounds in this class are currently at various stages of clinical development. In 2005, we reviewed the role of the CCR5 receptor in HIV-1 entry and the potential of CCR5 receptor antagonists as therapeutic anti-HIV-1 agents [1]. We reviewed the various categories of agents and compared the three small-molecule CCR5 antagonists that had progressed to clinical development: maraviroc, vicriviroc and aplaviroc. At that time, several key questions needed to be addressed by large-scale clinical trials, concerning not only the clinical efficacy and long-term safety of these agents, but also the pathways to CCR5 antagonist resistance in vivo and the effect of therapeutic administration of CCR5 antagonists on CXCR4-using virus populations. The purpose of the current review was to provide an update on developments in the field of CCR5 antagonist therapy since 2005, including the outcomes of the various Phase II/III trials conducted over the past 4–5 years, the progress that has been made in our understanding of resistance to CCR5 antagonists and the evolving body of evidence supporting the current and future role of small-molecule CCR5 antagonists in the treatment of HIV infection.
Status of CCR5 antagonists in clinical development in 2005
Of the three small-molecule CCR5 antagonists in clinical development in 2005, only maraviroc (Figure 1A) is currently approved for clinical use. Development of aplaviroc was discontinued in 2006, following reports of severe idiosyncratic hepatotoxicity in Phase IIb treatment-naive and Phase III treatment-experienced studies [2–4]. These reports of hepatotoxicity included cases of simultaneous increases in serum alanine aminotransferase and bilirubin breaching the ‘Hy's Law’ threshold, considered by the US Food and Drug Administration (FDA) to identify drug-induced hepatotoxicity of particular clinical concern [5], and a case of increased alanine aminotransferase that demonstrated a clear dechallenge-rechallenge reaction [2].
Development of vicriviroc [6] (Figure 1B) has been slowed by poor early virological results in dose-ranging studies and by safety concerns over a cluster of unexplained malignancies in a Phase II trial of treatment-experienced patients. Development of vicriviroc for treatment-naive patients was suspended for several years following interim results from a Phase II dose-ranging study in which treatment-naive patients with CCR5-tropic (R5) virus received either 600 mg of efavirenz once daily or 25, 50 or 75 mg vicriviroc once daily, each in combination with a then standard-of-care Combivir (zidovudine/lamivudine) backbone [7]. Patients in both the 25 mg and 50 mg vicriviroc arms had significantly higher rates of virological failure than did patients in the efavirenz comparator arm and the trial was terminated in late 2005. An interim review of a Phase II study in treatment-experienced patients (ACTG 5211), which compared the effects of 5, 10 or 15 mg vicriviroc once daily plus optimized background therapy (OBT) with the effects of OBT plus placebo, recommended the termination of the 5 mg treatment arm because of suboptimal responses. The study was subsequently unblinded early following reports of six cases of malignancy in the vicriviroc arms and two cases in the placebo arm (one of whom received placebo for 7 months followed by vicriviroc for 3 months prior to developing a malignancy whilst off treatment 1 month after virological failure on vicriviroc) [8]. Vicriviroc Phase II and III clinical development in treatment-experienced patients, includes a rollover study for ACTG 5211 (study P04100AM4), at a revised dose of 30 mg once daily in combination with background therapies containing a ritonavir-boosted protease inhibitor (PI) at a dose schedule modelled to provide a vicriviroc exposure equivalent to 200 mg once daily in the absence of ritonavir boosting [9]. No systemic malignancies were reported during 2 years of follow-up of patients in the ACTG 5211 rollover study [10]. At the time this review was written, a new Phase II/III study in treatment-naive patients, in which vicriviroc (30 mg once daily) plus atazanavir/ritonavir was being compared with atazanavir/ritonavir plus tenofovir/emtricitabine (Truvada), had entered its second stage following an interim safety analysis at 24 weeks (clinicaltrials.gov identifier: NCT00551018).
Maraviroc was approved for clinical use by the US FDA in August 2007 and by the European Agency for the Evaluation of Medicinal Products the following month. Maraviroc is licensed for administration twice daily with other antiretrovirals in treatment-experienced patients with R5 HIV-1. Approval was based on favourable 48-week results from two large Phase III studies (Maraviroc Plus Optimized Therapy in Viremic Antiretroviral Treatment Experienced Patients [MOTIVATE] 1 and 2) of maraviroc administered once or twice daily plus OBT compared with placebo plus OBT in treatment-experienced patients with R5 virus determined at screening with phenotypic testing [11,12]. Maraviroc has also demonstrated efficacy in treatment-naive patients in the Phase III MERIT (Maraviroc versus Efavirenz Regimens as Initial Therapy) study, although non-inferiority to efavirenz was not demonstrated in the primary analysis at week 48 [13]. However, in a post-hoc analysis, favourable results for the MERIT end points at weeks 48 [13] and 96 [14] were seen in a subset of MERIT patients eligible for study inclusion based on a more sensitive screening tropism assay. Maraviroc was granted a treatment-naive indication by the US FDA in November 2009.
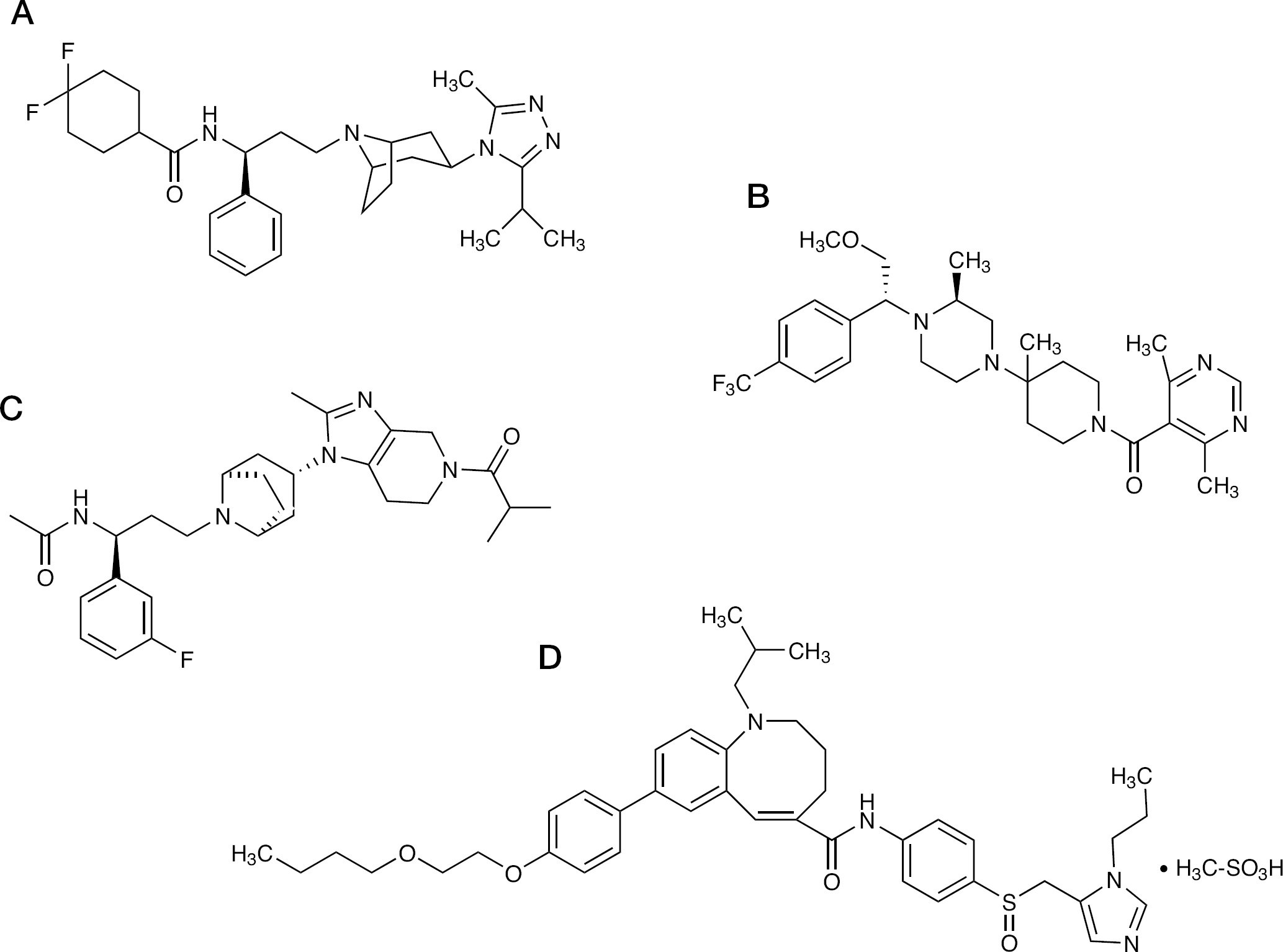
Chemical structures of CCR5 antagonists
Four other agents targeting the CCR5 coreceptor are known to be in Phase II development. PF-232798 (Figure 1C) is a second-generation small-molecule oral CCR5 antagonist with potency similar to maraviroc and the potential for once daily dosing. PF-232798 has shown activity against a laboratory-generated maraviroc-resistant R5 virus [15]. Another small-molecule oral CCR5 antagonist, INCB9471 (Incyte, Wilmington, DE, USA), has shown promising results in a Phase IIa trial in treatment-naive and treatment-experienced patients with once daily dosing over 14 days [16]. However, Incyte announced last year that they would not be initiating Phase IIb trials and were instead looking for a licensing partner to take the drug's development forward. PRO 140 is a humanized monoclonal antibody that binds to CCR5 and potently inhibits R5 HIV-1 replication in vitro. A proof-of-concept study demonstrated potent antiviral efficacy after a single intravenous dose [17], and promising results from a short-term evaluation of weekly and biweekly subcutaneous administration were recently presented [18]. PRO 140 has also shown evidence of activity against escape mutants that demonstrate cross-class resistance to the small-molecule CCR5 antagonists [19]; however, as a complex biological molecule requiring parenteral administration PRO 140 faces a different set of development challenges to the orally available small-molecule compounds. Finally, TBR-652 (Figure 1D; previously designated as TAK-652) is a small-molecule CCR5 antagonist licensed by the Takeda Pharmaceutical Company to Tobira Pharmaceuticals. TBR-652 has pharmacokinetic data supportive of once daily dosing and a good tolerability profile in single-dose studies in healthy volunteers [20].
Virological and immunological efficacy
An overview of published virological efficacy data for vicriviroc, maraviroc and aplaviroc is shown in Table 1. Data from studies of aplaviroc are somewhat limited because of its early withdrawal from development. Early (12 week) descriptive data from the ASCENT [4] and EPIC [3] studies in treatment-naive patients indicate lower virological response rates in patients receiving aplaviroc once or twice daily at doses between 200 and 800 mg in combination with nucleoside analogue reverse transcriptase inhibitors (NRTIs) or lopinavir/ritonavir than in patients receiving an efavirenz or lopinavir/ritonavir active control. Efficacy data for vicriviroc in its final dosing modality of 30 mg once daily in combination with a boosted PI are also limited, with 48-week blinded therapy data [21] and 96-week open-label extension data [22] from the 116 patient Phase II VICTOR-E1 trial the most complete available at time of writing. This study compared the safety and efficacy of vicriviroc (20 or 30 mg once daily) with placebo, each with an OBT containing a boosted PI in treatment-experienced patients with R5 virus. Mean changes from baseline in HIV-1 RNA and the proportions of patients with <50 copies/ml at week 48 were significantly greater in both vicriviroc arms than in the placebo arm, and virological and immunological responses were preserved through week 96 in those who were rolled over to open-label vicriviroc (30 mg) at week 48. Better vicriviroc pharmacokinetic exposure and higher response rates among patients with high baseline viraemia and fewer active OBT component drugs were seen at week 48 in those receiving the 30 mg dose that was carried forward into Phase III testing.
Summary of HIV-1 virological responses in published clinical studies of CCR5 antagonists
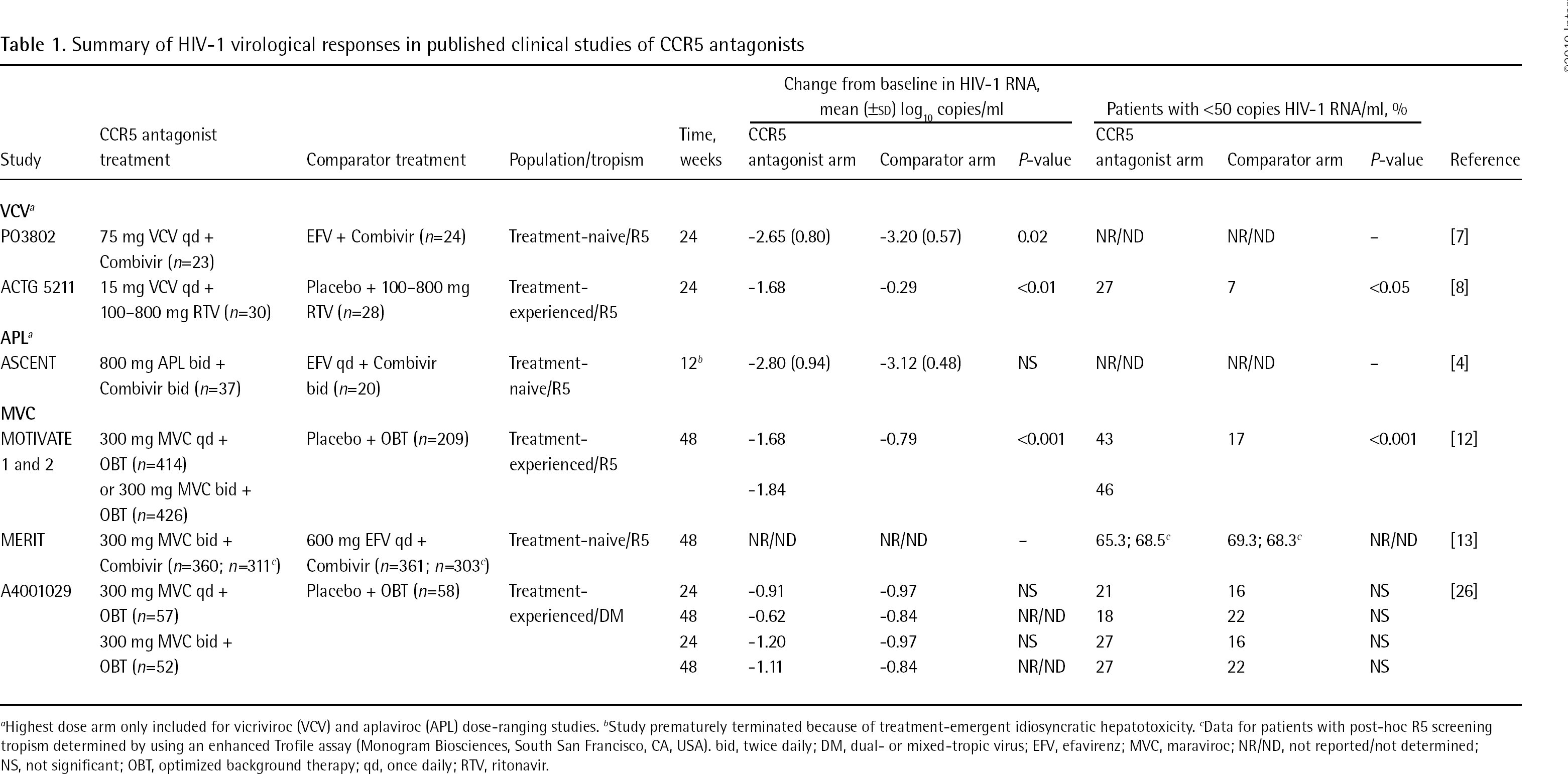
Highest dose arm only included for vicriviroc (VCV) and aplaviroc (APL) dose-ranging studies.
Study prematurely terminated because of treatment-emergent idiosyncratic hepatotoxicity.
Data for patients with post-hoc R5 screening tropism determined by using an enhanced Trofile assay (Monogram Biosciences, South San Francisco, CA, USA). bid, twice daily; DM, dual- or mixed-tropic virus; EFV, efavirenz; MVC, maraviroc; NR/ND, not reported/not determined; NS, not significant; OBT, optimized background therapy; qd, once daily; RTV, ritonavir.
Currently, the largest clinical data set for evaluating CCR5 antagonist efficacy is from the maraviroc clinical development programme, which to date has involved more than 2,000 individuals who have received maraviroc. Randomized comparative data on maraviroc treatment over at least 48 weeks are available for more than 1,900 treatment-experienced and treatment-naive patients in three Phase III and one Phase IIb studies (MOTIVATE 1 and 2, A4001029 and MERIT). Individual and pooled data from the pivotal Phase III MOTIVATE studies showed a more than twofold greater decrease in viral load at week 48 in treatment-experienced patients with R5 virus who received OBT with maraviroc (once or twice daily) than in patients who received OBT with a placebo [12]; this benefit was maintained in all studied subgroups, including those with low CD4+ T-cell counts, a high level of viraemia at baseline and weak or inactive OBT regimens [11]. Additional MOTIVATE subanalyses that assessed the association between OBT activity and maraviroc outcomes have identified a 70% virological response rate (<50 copies HIV-1 RNA/ml) in those receiving maraviroc with at least two fully active agents other than NRTIs, increasing to 80% in those initiating maraviroc with two active agents and a baseline CD4+ T-cell count >50 cells/mm3 [23]. These results are similar to those reported for other new agents, such as darunavir [24] and raltegravir [25] in treatment-experienced patients, and extend the range of options now available to this patient population to enable the goal of maximal virus suppression to be achieved.
In contrast with the results in treatment-experienced patients with R5 virus, no virological benefit of treatment with maraviroc over that with placebo was observed when both were given with OBT to patients with non-R5 virus (dual- or mixed-tropic [DM] or CXCR4-tropic [X4] virus) in the Phase IIb study A4001029 [26]. However, a significantly greater increase in CD4+ T-cell count was seen with maraviroc than with placebo in the primary analysis at week 24. This difference was also noted at week 48, but it was not statistically significant [26].
Efficacy in treatment-naive patients with R5 virus was demonstrated in the MERIT Phase III study [13], in which patients were randomly assigned to receive maraviroc once or twice daily or efavirenz once daily each with a fixed background of Combivir. Once daily maraviroc was discontinued following an interim analysis in the first 205 patients at week 16, which concluded that non-inferiority to efavirenz had not been established in this arm. Randomization to the efavirenz and twice daily maraviroc arms continued unchanged. In the primary analysis at week 48, the maraviroc arm was found to be non-inferior to efavirenz at the coprimary study end point of <400 HIV-1 RNA copies/ml, but not non-inferior at the <50 copies/ml coprimary end point. Subsequent rescreening of the MERIT patient set with a more sensitive tropism assay than that used at initial enrolment identified a 15% incidence of non-R5 virus in screening samples that had been present below the limit of detection of the original assay. Exclusion of these patients from a post-hoc reanalysis of the main study end points resulted in greater response rates in the maraviroc arm, which fell within the criteria defining non-inferiority [13]. Overall, the number of discontinuations in each treatment arm at both time points were similar, with more virological discontinuations on maraviroc balanced by a higher number of adverse event discontinuations on efavirenz. With study centres that included sites in both South Africa and South America, this study enrolled a significant proportion of patients with non-clade B infections (including more than 400 patients [∼30%] infected with clade C HIV-1). Thus, the study provided clinical confirmation of the in vitro data showing maraviroc to be active against viruses with envelopes of both B and non-B origin [27].
Consistently greater increases in CD4+ T-cells have been noted in patients who received maraviroc than in those who received either a placebo or efavirenz comparator across all patient groups [12,26,28–30] (Table 2). This effect of CCR5 antagonism, which was also apparent in a meta-analysis of recent Phase II and III trials evaluating newer agents in treatment-experienced individuals [31], appears to be independent of virological suppression [28,31] and is not linked to the presence of PIs or other potent background agents [29]. Factors that potentially contribute to this immune benefit of CCR5 antagonists include alterations in cell trafficking [32], reductions in HIV-1-mediated syncytia [33], apoptosis associated with CCR5 surface density [34] and suppression of chronic cytokine-associated T-cell activation [35,36], although no definite mechanism has been established.
Summary of CD4+ T-cell count increases in published clinical studies of CCR5 antagonists
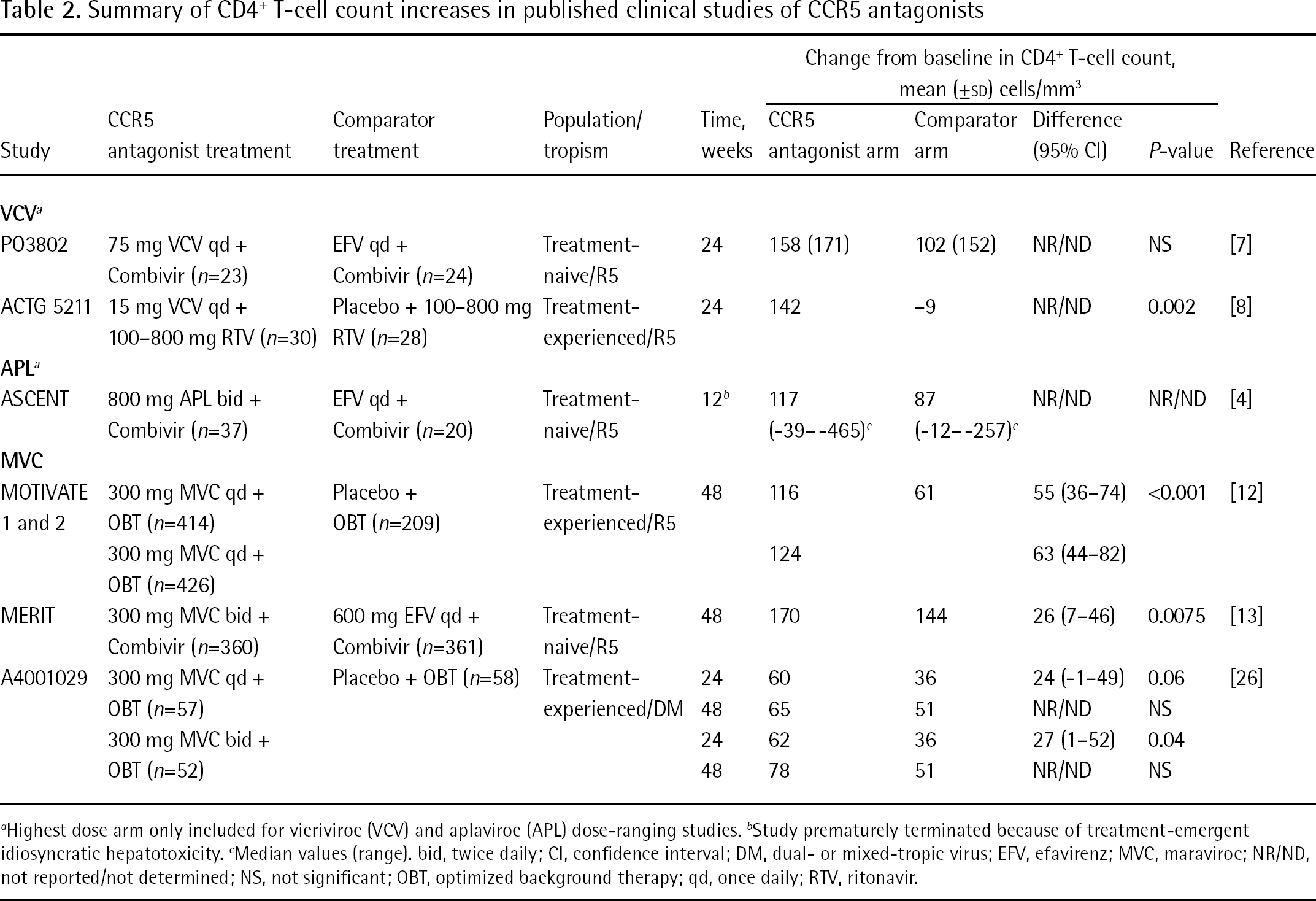
Highest dose arm only included for vicriviroc (VCV) and aplaviroc (APL) dose-ranging studies.
Study prematurely terminated because of treatment-emergent idiosyncratic hepatotoxicity.
Median values (range). bid, twice daily; CI, confidence interval; DM, dual- or mixed-tropic virus; EFV, efavirenz; MVC, maraviroc; NR/ND, not reported/not determined; NS, not significant; OBT, optimized background therapy; qd, once daily; RTV, ritonavir.
Although large-scale data are currently limited to maraviroc, the results indicate that the CCR5 antagonist class confers significant virological and immunological efficacy in treatment-experienced patients with R5 virus, similar to that observed recently with other agents. Although all members of the class appear to share this efficacy, meaningful comparisons between them are not yet possible. Comprehensive Phase III clinical trial data for vicriviroc will be needed for comparison with maraviroc and, similarly, Phase IIb/III data on PRO 140 in combination regimens will be needed before comparisons with either of these small-molecule CCR5 antagonists can be made.
CCR5 antagonist safety and tolerability
CCR5 antagonists represent the first class of antiretroviral agents to target a host cellular receptor rather than the virus and concerns have been raised about the safety of long-term CCR5 blockade. CCR5 is implicated in the immunological response to a variety of pathogens [37] and evidence indicates that, despite having an apparently normal phenotype, individuals homozygous for the CCR5-Δ32 deletion who lack functional CCR5 might have an increased susceptibility to disease following West Nile virus infection [38] or tick-borne encephalitis [39]. In addition, conflicting reports of associations between CCR5-Δ32 and various malignancies [40–45] combined with a cluster of lymphomas observed in the Phase IIb ACTG 5211 study of vicriviroc have also caused concern regarding the potential for an increase in malignancies. Data demonstrating that CCR5-Δ32 mice have an increased susceptibility to concanavalin A-induced hepatitis [46,47], when considered together with the severe idiosyncratic hepatotoxicity observed during aplaviroc development, have also raised concerns that hepatotoxicity could be a class effect.
Despite these concerns, data from Phase IIb/III studies of maraviroc following more than 2,000 patients over periods in excess of 96 weeks have shown it to be well tolerated with an adverse event profile similar to placebo [12,26,48] and superior to efavirenz [13]. Although postural hypotension (at unit doses of 600 mg or more) was identified as a signature dose-limiting adverse event for maraviroc in Phase I clinical trials, there was no evidence of an excess of adverse events related to postural hypotension in the Phase IIb/III clinical development programme at FDA-approved dosing [48].
The hepatotoxicity demonstrated by aplaviroc, although still of unknown cause, appears to be a compound-related rather than a mechanism-related event [2]. Two confirmed cases of severe hepatotoxicity – one in a patient receiving 600 mg maraviroc once daily in a Phase I study and one in a patient from the discontinued once daily maraviroc arm of MERIT – were confounded by concomitant infection and the use of other potentially hepatotoxic agents, respectively [49]. Unconfirmed hepatic failure in one patient with a history of chronic hepatitis C virus (HCV) infection and hepatic angioma, who discontinued treatment on the twice daily maraviroc arm of MOTIVATE 2 on day 337, was considered to be possibly drug-related. Exposure-adjusted data from the MOTIVATE studies showed similar or slightly lower incidences of transaminase and bilirubin increases in patients receiving maraviroc than in those receiving placebo [50]. Similarly, no systematic signs of hepatotoxicity with maraviroc were observed in treatment-naive patients in the MERIT study or in studies of vicriviroc to date.
An association between vicriviroc and malignancy remains unproven and additional clusters have not been reported during its further development involving up to 4 years of patient follow-up to date [51]. There is no evidence that treatment with vicriviroc increases replication of Epstein–Barr virus, which has been linked to lymphomas in immunocompromised patients [52]. Ninety-six week exposure-adjusted data from the MOTIVATE studies showed a lower rate of malignancies and a significantly lower risk of overall malignancies, AIDS-defining malignancies and coinfection-related malignancies in patients who received maraviroc than in those who received placebo, despite a 10-fold greater exposure to maraviroc than to placebo. Multivariate analysis demonstrated that the greater increase in CD4+ T-cells with maraviroc treatment was protective against the development of new malignancies [53]. Similarly, a low incidence of malignancies was reported in treatment-naive MERIT patients who received maraviroc with a Combivir backbone [14]. Finally, there was no evidence of treatment-related malignancies in the terminated aplaviroc clinical development programme [3,4].
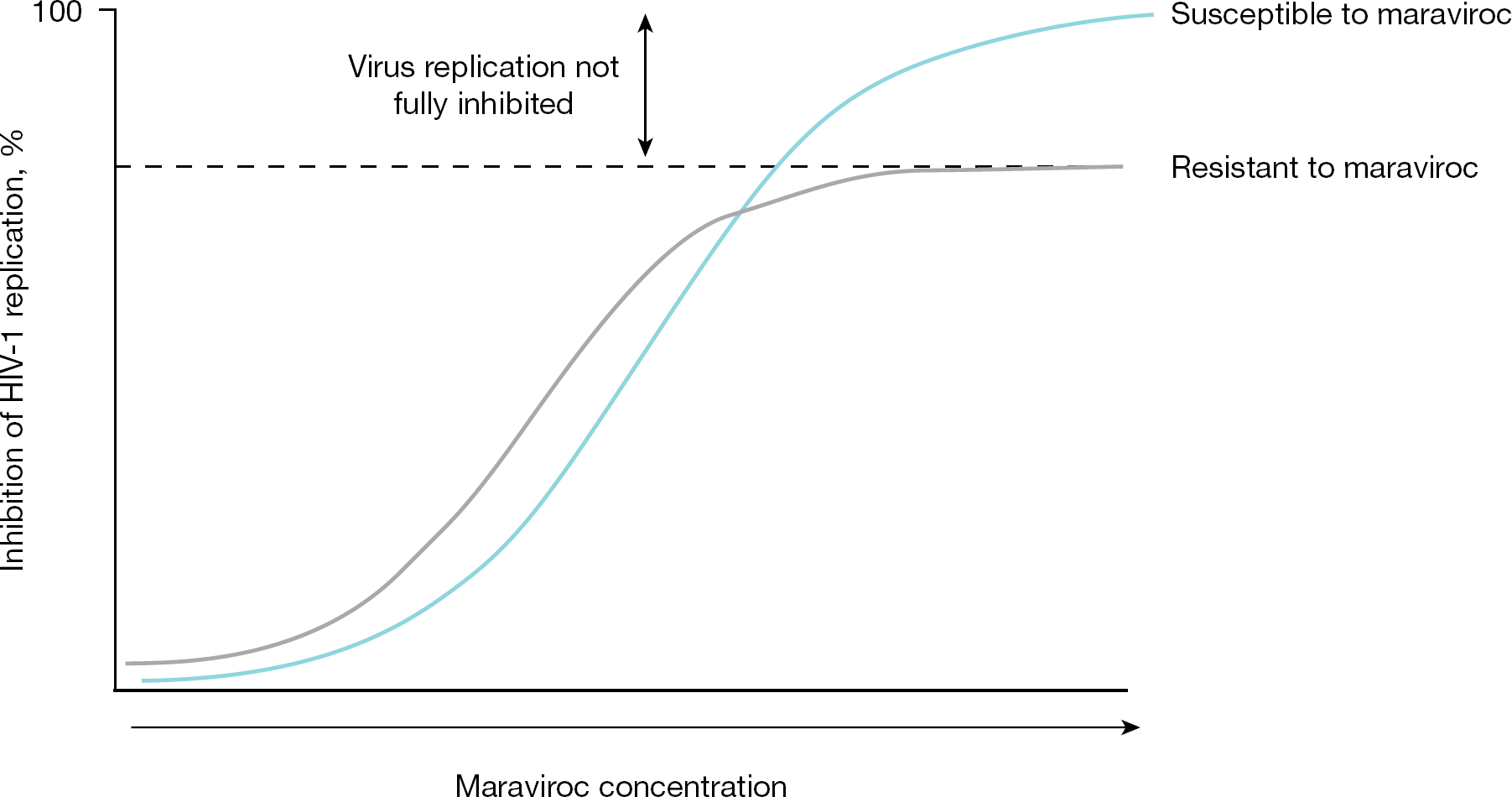
Maraviroc resistance is characterized by dose–response curves with plateaus in maximal percentage inhibition
Taken together, these data are reassuring and suggest that the antagonism of CCR5 does not give rise to class-wide signature toxicities over the durations of treatment studied thus far.
Development of resistance to CCR5 antagonists
Given the innate variability of the HIV envelope gene [54] and the complexity of interactions between the virus and host cell during HIV entry, resistance to CCR5 antagonists is complex [55]. As would be anticipated, mechanisms of viral escape from CCR5 antagonists, which target an immutable host protein, are fundamentally different from the resistance seen with antiretroviral drugs that bind viral targets. Two pathways of virological escape from CCR5 antagonists have been identified: selection of R5 virus that can use drug-bound CCR5 and ‘un-masking’ of pre-existing minority populations of dual-tropic or X4 virus (collectively called ‘CXCR4-using’ viruses). These pathways are summarized below and are reviewed in greater detail elsewhere [56,57].
CCR5 antagonist treatment failure with R5 virus
For CCR5 antagonists, shifts in the 50% HIV inhibitory concentration (IC50), characteristic of resistance to other antiretrovirals, have only rarely been observed at virological failure [58–60]. Such shifts probably represent variants with an increased affinity for CCR5 that are able to enter cells even when the density of antagonist-free receptors is low [61]. These mutants would be expected to have a lower susceptibility to CCR5 antagonists than would variants with lower affinity for free receptors, regardless of the CCR5 antagonist used, and indeed this has been observed in laboratory analyses of panels of viruses [56].
Resistance to CCR5 antagonists more typically manifests itself phenotypically as a reduction in the maximum level of viral inhibition achievable at any concentration of drug (Figure 2) [62]. Such reductions in the maximum percentage inhibition (MPI) are characteristic of viruses that have acquired the ability to use antagonist-bound receptors for cellular entry and, therefore, cannot be fully inhibited even when no unbound receptors are present [63]. The level of MPI in drug susceptibility assays reflects the difference in the affinity of the virus for antagonist-free as opposed to antagonist-bound forms of CCR5, with the MPI becoming progressively lower as the efficiency of the virus at using antagonist-bound receptor increases [55,56,63].
Reductions in MPI, which have been observed with maraviroc [62], vicriviroc [7,64] and aplaviroc [58], are consistent with these drugs acting as allosteric, noncompetitive entry inhibitors [65]. Small-molecule CCR5 antagonists are thought to interact with coreceptors in a way that stabilizes or induces a CCR5 receptor conformation not recognised efficiently by the HIV envelope [62,66–68]. Although very different in structure and shape, the CCR5 antagonists investigated to date appear to share a common binding site that is located in the second extracellular loop of CCR5 [65,67,69]. The specific interactions between each compound and amino acid lining the binding pocket differ between the agents, as might be anticipated from their different electrostatic shapes and polarities [69].
Although demonstrated both in vitro and in vivo, the results of clinical studies suggest that R5 virological failure with reduced susceptibility to CCR5 antagonists is not the most common mechanism of failure. In the MOTIVATE studies in treatment-experienced patients, a reduced MPI was observed in only ∼40% of R5 virological failures with maraviroc over the first 24 weeks of treatment; poor or intermittent adherence was the most probable reason for failure in the remaining 60% of R5 failures without maraviroc resistance [59]. Similarly, low numbers of R5 failure isolates with a reduced MPI were observed in the MERIT study of treatment-naive patients [70] and in studies of aplaviroc [58,71] and vicriviroc [7].
The observation of a low clinical incidence of R5 virus with reduced susceptibility to CCR5 antagonists is consistent with the complexity of the genetic changes involved in its development. Unlike resistance to virally targeted antiretrovirals, CCR5 antagonist resistance in R5 virus is conferred by complex and generally unpredictable changes across a number of regions in the HIV envelope [57,62,63,72] and no predictive signature mutations or combinations have been identified to date. Interpatient sequence diversity of resistant R5 virus has been reported for clinical isolates from both maraviroc [60,73] (Figure 3) and aplaviroc [58] virological failures, which suggests the existence of multiple genetic pathways to resistance and implies that the mutations observed might be virus-specific [55]. Sequencing has also revealed different patterns of adaptive amino acid changes in the HIV envelope under selection pressure from different CCR5 antagonists, including changes outside the V3 loop [57,58,72,74,75]. These sequence changes might, however, come at a cost to viral fitness [76]. Viral affinity for CCR5 bound by one antagonist will not necessarily confer affinity for CCR5 bound by another, given the different amino acid interactions involved. Although there is evidence of cross-resistance between different CCR5 antagonists [19,77], this does not appear to be a universal phenomenon [1].
Failure with CXCR4-using virus
Although theoretical concerns about whether prolonged therapy with a CCR5 antagonist will accelerate viral evolution toward CXCR4 use still exist, this has not been observed in clinical studies to date. Emergence of CXCR4-using virus in the absence of CCR5 antagonist pressure tends to dominate later in the course of HIV disease [78–83]. Such emergence is coincident with more rapid CD4+ T-cell depletion and acceleration of disease progression, but it remains unclear whether this indicates a cause-and-effect relationship [84]. Longitudinal analyses of envelope sequences in clinical isolates that evolved to use CXCR4 in the absence of antiretroviral drug pressure indicate that R5 to X4 tropism ‘switching’ requires an ordered accumulation of multiple mutations, which differ both in composition and in the order of appearance between patients and that may or may not include changes in the V3 loop [85]. Interestingly, ‘ultra-deep’ clonal sequencing has identified CXCR4-using minority populations in most or all clinical R5 samples studied [86]. This finding, therefore, raises the question of whether a simple threshold level is required for X4 outgrowth under CCR5-antagonist selection pressure or whether more complex interactions are involved. It also raises the possibility that the emergence of CXCR4-using virus later in the course of infection could be a consequence of disease-associated changes in the host environment that favour a shift toward CXCR4-mediated replication.
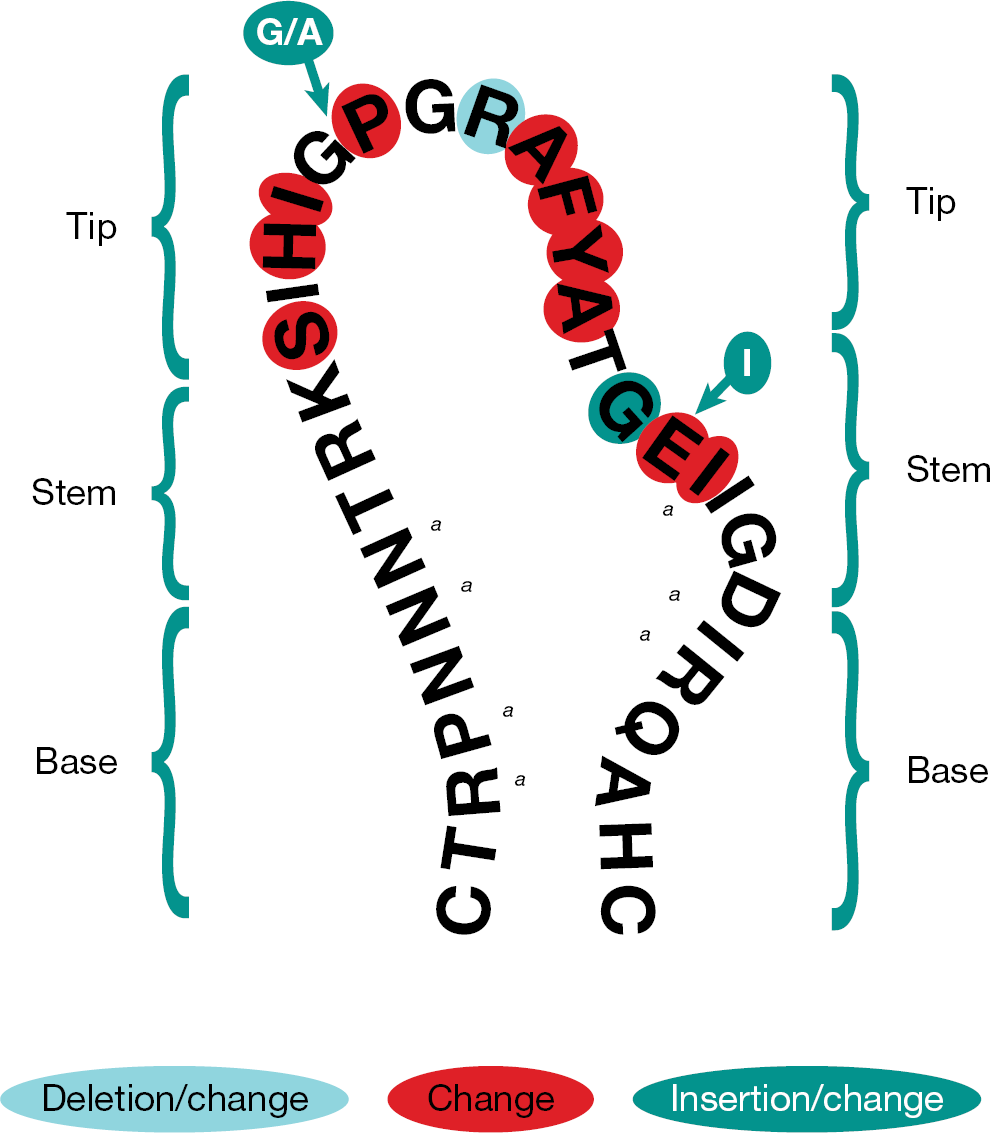
HIV envelope sequence changes associated with maraviroc-resistant R5 virus
Evaluation of tropism data over 48 weeks in the MOTIVATE studies indicated that more than 50% of treatment-experienced patients experiencing virological failure on maraviroc did so with CXCR4-using virus, which was derived from the unmasking and outgrowth of pre-existing minority populations present at baseline [11,87]. This outgrowth was not associated with any short-term adverse immunological outcomes; greater gains in CD4+ T-cell counts were seen in patients failing maraviroc with CXCR4-using virus relative to patients failing in the placebo arm with any viral tropism [11]. Similarly, in MERIT, greater CD4+ T-cell increases were seen in treatment-naive patients with CXCR4-using virological failure on maraviroc plus zidovudine/lamivudine, relative to those failing on efavirenz with the same nucleosides and any viral tropism [70]. Furthermore, in the MOTIVATE studies, no association was observed between CXCR4-using virus at failure and the development of Category C (AIDS-defining) events, and most patients with more than 1 month of off-drug follow-up reverted back to R5 virus infection following withdrawal of maraviroc [88].
Clinical data from the A4001029 study (maraviroc plus OBT compared with OBT with placebo in patients with non-R5 infection) clearly showed that, at a population level, patients with DM virus at screening did not derive a virological benefit from maraviroc [26]. However, interestingly, evidence suggests that DM virus populations are not homogeneous in their natural resistance to CCR5 antagonists. It has been shown that dual-tropic viruses from clinical samples exhibit a broad range of affinities for CCR5- and CXCR4-mediated cell entry: the majority of dual-tropic strains show a notably greater preference for CCR5 (dual-R5 variants) and the minority show an increased affinity for CXCR4 (dual-X4) [58,89–92]. Furthermore, an analysis of the proportion of CCR5-using and CXCR4-using viruses in DM populations from antiretroviral-naive patients showed that the majority of clones used CCR5 only [92]. Together, these observations indicate that some DM populations might be responsive to CCR5 antagonists in vivo to a greater or lesser extent [90]. Clinical data also appear to support this inference. For example, a retrospective analysis of A4001029 identified a greater short-term virological response (approximately twofold at week 12) among maraviroc patients with <10% CXCR4-using virus at screening, by ultra-deep sequencing, than those with >10% [93]. Furthermore, in the EPIC study of aplaviroc [3], week 12 viral load reductions among 11 evaluable patients with DM virus at screening visit ranged from −1.89 to −3.49 log10 copies/ml, suggesting that some patients experienced rapid virological declines despite detectable CXCR4-using virus [3]. Although these reports suggest that some patients with DM virus might respond to treatment with a CCR5 antagonist, the clinical relevance of these observations remains unclear given the small sample sizes, the short duration of follow-up involved and the lack of a clinically validated algorithm to distinguish ‘susceptible’ from ‘non-susceptible’ DM samples.
Future directions
The introduction of several new, potent antiretrovirals in the past few years – darunavir, tipranavir, etravirine, raltegravir and maraviroc – has significantly improved HIV treatment and made full virological suppression an achievable objective for almost all patients, including those with extensive prior treatment experience. In all cases, clinical studies have consistently demonstrated the importance of using these new drugs in combination with at least two other fully active agents to achieve durable suppression [94,–98]. The increasing availability of more potent later options, coupled with the generally favourable safety profiles of these newer agents, presents the opportunity to combine them directly and avoid the practice of recycling nucleoside analogues between regimens. Many planned and ongoing studies, including study ACTG A5241 (OPTIONS; clinicaltrials.gov identifier: NCT00537394) and the POEM observational cohort study (study A4001067 [NCT00665561]), will explore combinations of these newer agents and provide valuable information on optimal drugs to partner with maraviroc under various conditions.
Other strategies for maraviroc use are being explored, including NRTI-sparing once daily dosing in treatment-naive patients (study A4001078 [NCT00827112]) and regimen intensification in acute primary infection (MARAVIBOOST study [NCT00808002]) or in established infection in patients with virological suppression on their current regimen (study MMA-0612 [NCT00703586]). These studies will determine whether the addition of maraviroc to conventional triple therapy or its inclusion in a four-class initial regimen can accelerate HIV-1 reservoir decay in latently infected cells, reduce residual replication in gut-associated lymphoid tissue (GALT) and/or improve immune reconstitution in peripheral blood and GALT. Another study plans to examine the effects of therapeutic intensification with both maraviroc and raltegravir together with immune modulators on the proviral and GALT-associated viral reservoirs (study EraMune02 [NCT00976404]). The potential immune benefits of CCR5 blockade are being further explored in studies involving patients with low CD4+ T-cell counts despite long-term virological suppression (studies ANRS 145 MARIMUNO [NCT00944541] and CCTG 590 [NCT00925756]), as well as in an intensive GALT sampling study to examine the effect of maraviroc (with or without raltegravir) on GALT immune cell recovery (study CH-2009.01 [NCT00935480]).
Complications with current antiretroviral treatment regimens – including drug resistance, poor adherence and formulation issues – have left many children and adolescents with limited therapeutic options. Paediatric studies are therefore planned (vicriviroc study IMPAACT P1071 [NCT00766597]) or underway (maraviroc study A4001031 [NCT00791700]) to evaluate CCR5 antagonists in this important patient group. Another group of patients of special interest is those with hepatitis B virus (HBV) or HCV coinfection. These patients have an increased risk of drug-induced hepatotoxicity and, although no increases in hepatotoxic effects among coinfected patients have been observed in studies of maraviroc [49] or vicriviroc [99] to date, these analyses were based on relatively small numbers of trial participants. More detailed information on maraviroc safety in hepatitis coinfection will be gathered from an ongoing Phase IV study (A4001080 [NCT00782301]) comparing the effects of maraviroc with those of etravirine, each in combination with darunavir/ritonavir and raltegravir, in approximately 120 treatment-experienced patients with HBV or HCV coinfection.
Larger and more specific trials of CCR5 antagonists in coinfection are also warranted on the basis of recent evidence implicating CCR5 function and its modulation in the etiopathogenesis of viral hepatitis. For example, spontaneous clearance of HBV occurs more frequently among individuals without functional CCR5 and with genetically elevated levels of the CCR5 ligand RANTES [100,101]. In addition, CCR5 expression is detectable in hepatic stellate cells and in areas of liver inflammation and fibrogenesis in HIV–HCV coinfection. Moreover, stellate cell chemotaxis and secretion of the pro-inflammatory cytokine MCP-1 increase on exposure to CCR5-tropic gp120 [102]. These effects are prevented by pre-incubation with a CCR5 antagonist [102]. Functional CCR5 expression has also been observed to promote hepatic fibrosis in a mouse model [103]. Thus, there is evidence to indicate that the faster progression to liver fibrosis and cirrhosis in HCV-infected patients who are coinfected with HIV [104,105] might at least in part be attributable to CCR5–HIV interactions that are preventable with the use of CCR5 antagonists.
The potential use of CCR5 antagonists for pre- and post-exposure prophylaxis is an active area of research given that R5 viruses are by far the most common types present in sexual transmission [79–81] and early infection [78,106,107]. Proof-of-concept for CCR5 antagonist prophylaxis has been demonstrated in a non-human primate model using a novel oral CCR5 antagonist (CMPD167) [108,109]. There is also one reported case study of successful post-exposure prophylaxis with a maraviroc-containing regimen following occupational needle stick injury [110]. Pharmacokinetic support for examining prophylactic use of maraviroc in women was provided by healthy volunteer data showing that maraviroc not only reaches similar concentrations in plasma and cervicovaginal fluid to many other antiretroviral agents, but it also reaches higher levels in the female genital tract [111]. An analogous study (study IRB 08-0418 [NCT00775294]) is underway in men to determine the extent of maraviroc exposure in blood, saliva, seminal fluid and rectal mucosal tissue.
Maraviroc's extracellular mode of action also makes it a promising candidate for inclusion in a vaginal microbicide to prevent HIV transmission. In early 2008, Pfizer granted a non-exclusive, royalty-free license to develop maraviroc as a microbicide to the International Partnership for Microbicides (IPM), a non-profit product development partnership. Although no clinical trials have yet been performed, pre-clinical studies by the IPM in human genital and colorectal tissue explants have shown that maraviroc effectively blocks ex vivo infection in these tissues and that potency is increased by sustained delivery [112].
Finally, the need for a pre-therapeutic tropism assessment has been a barrier to CCR5 antagonist use in some patients. Currently, most HIV-1 tropism screening involves centrally provided recombinant phenotype assays and this remains the only form of tropism testing to have been validated in large-scale prospective clinical studies to date. However, there is also considerable interest in developing improved genotypic methods (reviewed by Low et al. [113]), which have been shown to predict outcomes after CCR5 antagonist administration [114]. Recent data [115,116] have raised hopes that ongoing improvements in genotypic methodologies and algorithms might soon render them a viable alternative to phenotypic assays. Such genotypic platforms would be analogous to genotypic resistance testing methods, in that they are typically less expensive, more portable, locally implementable, less reliant on either individual operator skill or highly specialized equipment and typically have faster turnaround times than their phenotypic counterparts. Quality-controlled local sequencing might also assist the development and validation of clinical algorithms to differentiate those DM virus subpopulations that retain significant sensitivity to CCR5 antagonists.
In conclusion, the attractiveness of viral entry as a target for HIV drug discovery has now yielded two new classes of extracellular antiretroviral agents with the introduction of fusion inhibitors in 2003 and CCR5 antagonists in 2007. The CCR5 antagonists have established the clinical viability of host-targeted interventions and have provided some interesting hints that such interventions could provide additional benefits in modifying the course of HIV disease beyond the suppression of viral replication alone. More deliberate disease-modifying host interventions are likely to undergo investigation in clinical trials over time, opening up an entirely new direction in HIV medicine. Whereas small-molecule CCR5 antagonists have always had certain theoretical advantages over intracellular and virus-targeted agents, the clinical data accumulated in the past 4 years has established this new drug class as an important therapeutic option in the long-term treatment of HIV disease.
Note added in proof
Please note that since the submission of this review, Merck has announced (17 February 2010) that it will not submit a New Drug Application for vicriviroc in treatment-experienced patients at this time. Their decision follows poor initial efficacy results in the VICTOR-E3 and E4 phase III studies reported as a late-breaking presentation at the 17th Conference on Retroviruses and Opportunistic Infections in San Francisco 2010 [117].
Footnotes
Acknowledgements
The study was sponsored by Pfizer Global Research and Development and was funded by Pfizer, Inc. Editorial assistance was provided by Nick Fitch and Caroline Minshull of Health Interactions, with funding by Pfizer, Inc.
MW and EvdR are full-time employees of Pfizer.