
Abstract
Background:
Hepatitis C virus (HCV) infection is a major cause of chronic liver disease. Here, we report a new and effective strategy for inhibiting HCV replication using an inhibitor of heat-shock protein 90, 17-AAG (17-allylamino-17demethoxygeldanamycin), and a proteasome inhibitor, MG132.
Methods:
To explore the virological basis of combination therapy, we analysed the effects of 17-AAG and MG132, singly and in combination on HCV replication in an HCV replicon cell system.
Results:
In HCV replicon cells, HCV RNA replication was suppressed by 17-AAG in a dose-dependent manner. As shown in the present study, the 50% inhibitory concentration values were 0.82 nM for 17-AAG and 0.21 nM for MG132. Low concentrations of MG132 had strong synergistic inhibitory effects with low toxicity on HCV replicon cells.
Conclusions:
The results of this study suggest that the different effects and synergistic actions of 17-AAG and MG132 could provide a new therapeutic approach to HCV infection.
Introduction
Infection by the hepatitis C virus (HCV) is a major public health problem, with 170 million chronically infected people worldwide [1,2]. Combined treatment with interferon-α and ribavirin induces sustained antiviral response in 80% of patients with HCV genotype 2 or 3 [3,4]. Chronic infection with HCV results in liver cirrhosis and can lead to hepatocellular carcinoma [5,6]. Although interferon-α plus ribavirin therapy is effective for approximately 50–80% of patients with HCV, the development of improved therapies and preventative vaccines is urgently needed [7].
Heat-shock protein 90 (Hsp90) exerts chaperone activity together with a number of cochaperones, playing an important role in the folding of at least 200 specific proteins of various signalling pathways and in the refolding of proteins that have been denatured by stress [8–11]. Hsp90 is considered to be of prime importance to the survival of cancer cells. The constitutive expression of Hsp90 is 2- to 10-fold higher in tumour cells compared with their normal counterparts, suggesting that Hsp90 is critically important for tumour cell growth and/or survival. A small molecule inhibitor of Hsp90, the benzoquinone ansamycin 17-AAG (17-allylamino-17-desmethoxy-geldanamycin; Figure 1A), exhibits antitumour activity in several human xenograft models, including colon, breast and prostate cancer [12–14]. The drug is currently completing multi-institution Phase I clinical trials, and Phase II trials are being planned.
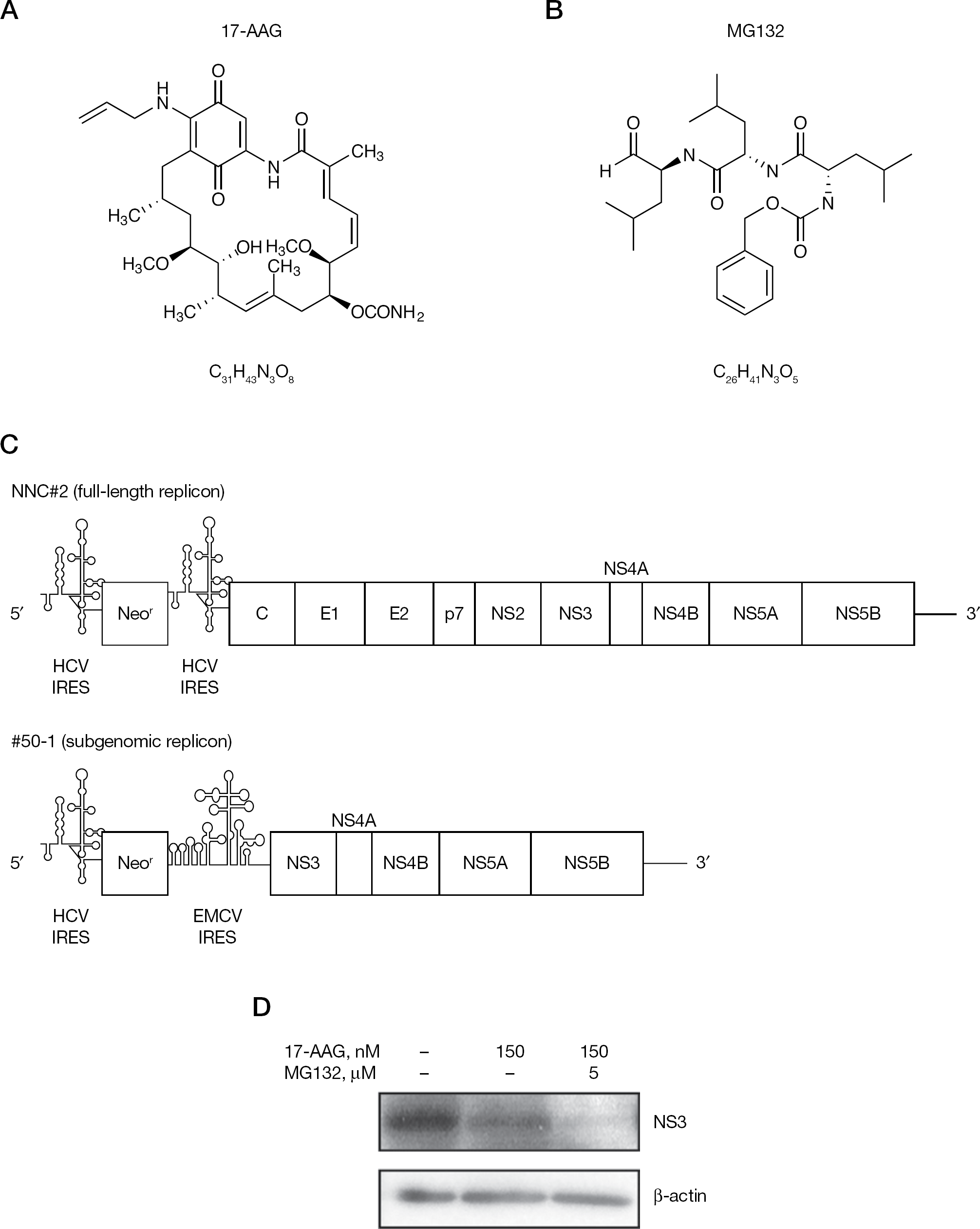
Schematic representation of the HCV replicon and the structures of 17-AAG and MG132
The HCV non-structural protein 3 (NS3) forms a complex with Hsp90 [8,10,11] that is critical for HCV replication [15]. Treatment of the #50–1 HCV replicon cells with the Hsp90 inhibitor 17-AAG [8,16,17] suppresses HCV RNA replication and NS3 is the only HCV protein degraded in these cells [15]. This finding led us to suggest a crucial role for Hsp90–NS3 complexes in the HCV life cycle. By contrast, the proteasome is a large protein complex that also participates in protein degradation [18]. Among the 28 subunits of the 20S proteasome, the α-subunit 7 (PSMA7) is one of the α-ring subunits at the barrel centre of the proteasome complex. The 20S proteasome is activated upon association with its regulatory protein complex, classified as PA700 (19S regulator) and PA28 (11S regulator) [18]. Ribozymes and small interference RNAs that specifically target the putative HCV cofactor PSMA7 inhibit HCV expression [19,20].
In the present study, we describe our findings of HCV combination therapy with the Hsp90 inhibitor 17-AAG and the proteasome inhibitor MG132 (Figure 1B). Combining these different inhibitors produced significant synergistic inhibitory effects on HCV replicons, indicating that the inhibitors might be useful as an efficient dual strategy of molecular HCV therapeutics.
Methods
Cell culture and reagents
The HCV replicon cell line #50–1 (NN/1b/SG) [21], which carries a subgenomic replicon, and NNC#2 (NN/1b/FL) [22], which carries a full genome replicon, were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, non-essential amino acids, L-glutamine, penicillin–streptomycin, and 300–1,000 μg/ml G418 (Invitrogen, Carlsbad, CA, USA) at 37°C in 5% CO2. Huh-7 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 mg/ml streptomycin. 17-AAG and MG132 were purchased from Sigma–Aldrich Chemical Co. (St Louis, MO, USA).
Real-time reverse transcriptase PCR analysis
HCV replicon cells were seeded at 1.5×105 cells in 24-well plates and cultured for 72 h. Total RNA was then isolated using Trizol (Invitrogen) according to the manufacturer's instructions. HCV RNA was quantified by real-time reverse transcriptase (RT)-PCR using an ABI 7700 sequence detector (Perkin-Elmer Applied Biosystems, Foster City, CA, USA), and the following primers and TaqMan probes located in the 5′-untranslated region: forward primer (nucleotides [nt] 130–146), 5′-CGGGAGAGCCATAGTGG-3′; reverse primer (nt 272–290), 5′-AGTACCACAAGGCCTTTCG-3′; and TaqMan probe (nt 148–168), 5′-CTGCGGAACCGGTGAGTACAC-3′ (all purchased from Applied Biosystems). The probe sequence was labelled with the reporter dye 6-carboxyflurorescein at the 5′-end and with the quencher dye TAMRA at the 3′-end [23].
Western blotting
NNC#2 cells were seeded at 1.5×105 cells in 24-well plates, treated with 17-AAG alone, MG132 alone, or combined 17-AAG and MG132, and cultured for 48 h. Western blotting analysis was performed using a previously described method [15]. The primary antibodies were monoclonal or polyclonal antibodies against PSMA7 (Abcam, Cambridge, MA, USA) and β-actine (SIGMA, Sigma–Aldrich). The anticore antibody was a kind gift from M Kohara (Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan). Anti-NS3 antibody was a kind gift from Y Matsuura (Osaka University, Osaka, Japan).
Transfection and reporter assay
Huh-7 cells were seeded at 1.5×105 cells 24 h before transfection. Huh-7 cells were treated with MG132 (100 nM) following transfection with the plasmid DNA pHCV internal ribosome entry site (IRES) luciferase (luc) or pEMCV IRES luc using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Luc activity was measured in the cell lysates using a luminometer (Berthold, Bad Wildbad, Germany).
MTS assay
HCV replicon cells were seeded in 96-well plates at 3×104 cells per well in a final culture volume of 100 μl for 72 h before adding increasing concentrations of 17-AAG and MG132. After incubation for 3 days, viable cell numbers were determined using the CellTitre 96® Aqueous non-radioactive cell proliferation assay (Promega Corp., Madison, WI, USA). The value of the background absorbance at 490 nm (A490) of wells without cells was subtracted from the A490 value of wells with cells. The percentages of viable cells were then calculated using the following formula: (A490 17-AAG-, MG132- or 17-AAG+MG132-treated cells/A490 untreated cells)×100.
Drug synergism analysis
The effect of treatment of HCV replicon cells with 17-AAG and MG132, alone and in combination, was analysed by measuring HCV RNA with real-time PCR. The combination index (CI) for each combination of 17-AAG and MG132 treatment was calculated by the following formula using the 50% inhibitory concentration (IC50): CI= IC50(17-AAG combined)/IC50(17-AAG alone) +IC50(MG132 combined)/IC50(MG132 alone). For such plots, the combined effects of the two drugs can be assessed as either additive (CI=1), synergistic (CI<1) or antagonistic (CI>1) [24].
Results
In a previous study, we examined the inhibitory effects of 17-AAG on HCV replication in an HCV replicon cell culture system. In HCV replicon cells treated with 17-AAG, HCV RNA replication was suppressed in a dose-dependent manner, and the only HCV protein degraded in these cells was NS3 [15]. To determine whether 17-AAG promoted the degradation of NS3, we evaluated the effect of 17-AAG on #50–1 cells in which proteasomal degradation was also inhibited with the proteasome inhibitor MG132. Although 17-AAG treatment reduced NS3 levels in #50–1 cells, this NS3 degradation was completely blocked in the presence of MG132 [15]. In the present study, we used HCV full-length replicon NNC#2 cells (Figure 1C) [22] instead of the HCV subgenomic replicon #50–1 cells used in previous studies [15,21]. Treatment with 17-AAG decreased NS3 levels, but the presence of MG132 did not block the reduction in HCV NS3 (Figure 1D). Indeed, MG132 in combination with 17-AAG induced the complete disappearance of the NS3 (Figure 1D), suggesting that the reduction of HCV NS3 induced by treatment with the proteasome inhibitor MG132 was dependent on the HCV IRES. NNC#2 and #50–1 cells have different virus IRESs, known as the HCV IRES [25–27] and the encephalomyocarditis virus (EMCV) IRES [28], and HCV IRES-mediated translation is induced by PSMA7 [29]. The PSMA7 activity is blocked by MG132 [30]. Similar effects were observed when PSMA7-directed ribozyme or small interfering RNAs inhibited HCV replication [19,20].
We examined the effects of combination therapy with 17-AAG and MG132 on HCV. To evaluate the MG132-mediated effect on HCV IRES activity, Huh-7 cells were treated with MG132 (100, 200 and 250 nM) and then transfected with pHCV IRES luc or pEMCV IRES luc (Figure 2A). MG132 inhibited luc activity of HCV IRES in dose-dependent manner, but did not inhibit translation derived from EMCV IRES (Figure 2B), indicating a preferential effect of MG132 on HCV IRES activity [19]. NNC#2 cells were then treated with 17-AAG or MG132 for 3 days. HCV RNA was quantified by real-time RT-PCR using an ABI 7500 Fast (Perkin-Elmer, Applied Biosystems). Dose-dependent cytotoxicity was not observed upon application of MG132 and/or 17-AAG (99% at concentrations ranging between 5 and 25 nM; Figure 3A), but was observed at MG132 concentrations of 10 μM (data not shown). Cells treated with increasing doses of 17-AAG or MG132 showed reduced levels of HCV RNA (Figure 3B). Low concentrations (1 nM) of MG132 had inhibitory effects similar to those of 15 nM 17-AAG (Figure 3B). In NNC#2 cells treated with 5 nM MG132, HCV RNA replication was suppressed by 85%, and this effect was dose-dependent (Figure 3B). The IC50 values were 0.82 nM for 17-AAG and 0.21 nM for MG132 (Table 1 and Figure 3B).
Combination index and dose reduction in inhibition of hepatitis C virus RNA replication by combining 17-AAG with MG132
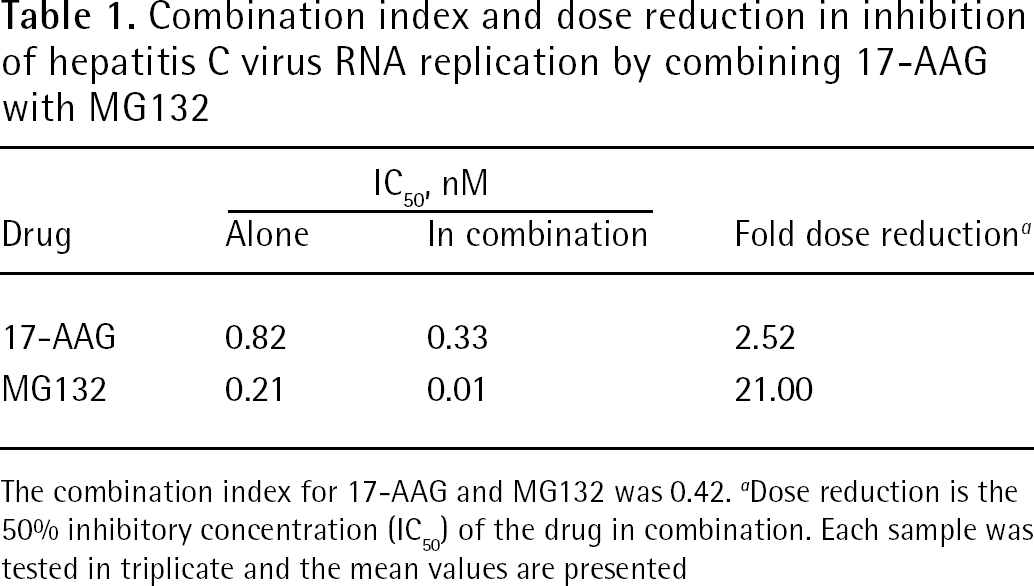
The combination index for 17-AAG and MG132 was 0.42.
Dose reduction is the 50% inhibitory concentration (IC50) of the drug in combination. Each sample was tested in triplicate and the mean values are presented
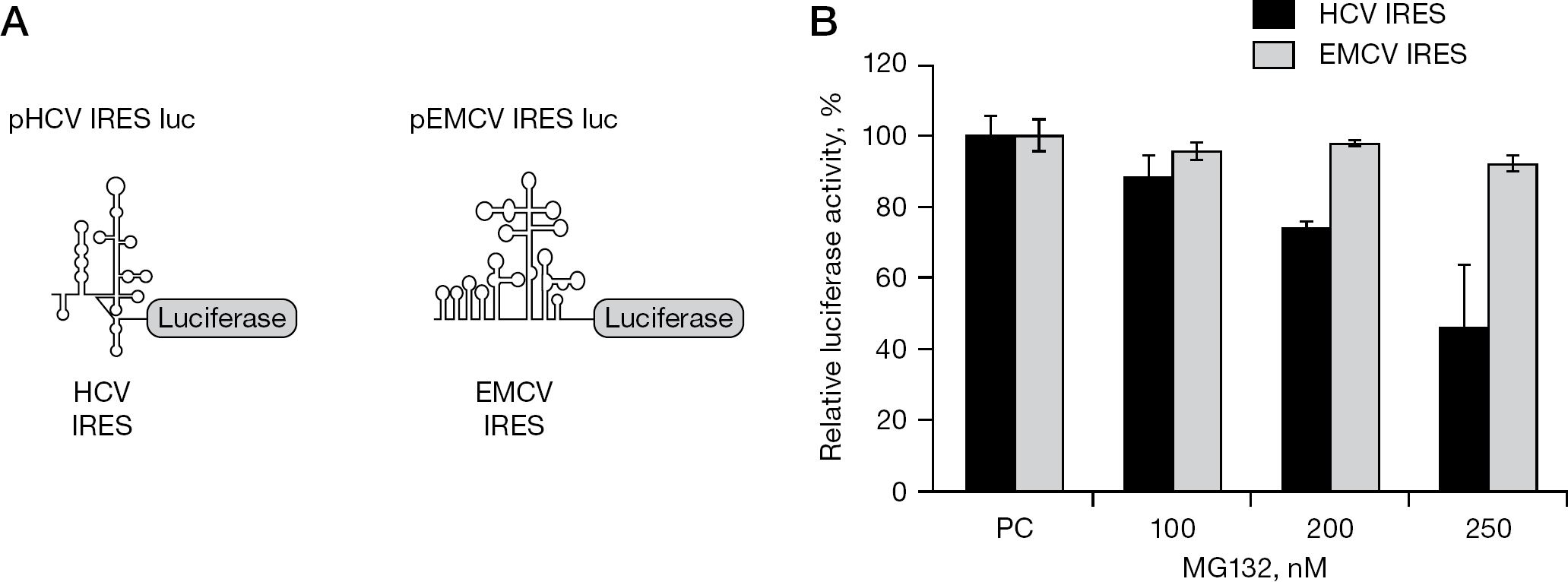
Effect of proteasome inhibitor MG132 on HCV IRES activity
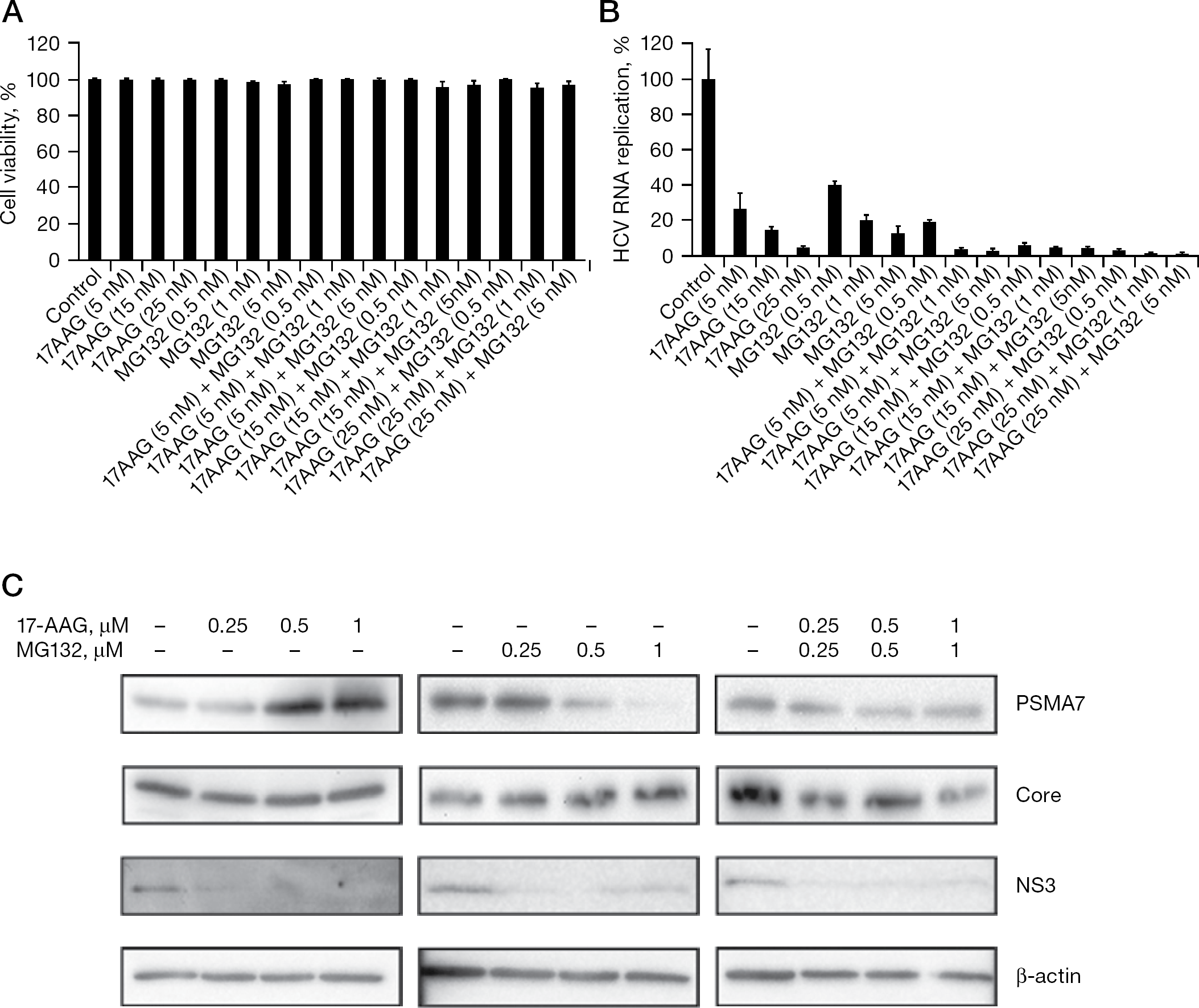
MG132 and 17-AAG inhibition of HCV RNA replication in HCV replicon cells
We also investigated combination therapy with 17-AAG and MG132 on HCV in NNC#2 cells. In combination with 5 nM, 15 nM or 25 nM of 17-AAG, 0.5 nM, 1 nM or 5 nM of MG132 was applied to NNC#2 cells, respectively. The combination of 5 nM 17-AAG and 1 nM MG132 suppressed HCV RNA replication by 90% in the NNC#2 cells (Figure 3B). The CI was 0.42, demonstrating that 17-AAG and MG132 had synergistic inhibitory effects on HCV replicon cells (Table 1). The combination of 17-AAG with MG132 inhibited HCV RNA replication with an approximately 2.5-fold reduction in the dose (Table 1). To determine the effects of 17-AAG and MG132 on the expression of core and PSMA7 proteins, NNC#2 cells were treated with various concentrations of 17-AAG or MG132 alone, and 17-AAG plus MG132 at day 2. Treatment with 17-AAG alone increased the expression of PSMA7, but did not reduce core protein expression. By contrast, treatment with MG132 alone reduced the expression of PSMA7, but not the core protein (Figure 3C), whereas expression of NS3 was reduced by treatment with 17-AAG or MG132 alone, and 17-AAG plus MG132. This result was consistent with our previous findings that NS3 levels were reduced in NNC#2 cells treated with 17-AAG for 3 days, but that core protein was detected for up to 6 days [15]. Other researchers, however, have reported that MG132 blocks the degradation of HCV core protein [31]. Interestingly, combined treatment with MG132 and 17-AAG reduced the expression of both the core and PSMA7 proteins in NNC#2 cells (Figure 3C). These results suggest that MG132 and 17-AAG are potent anti-HCV agents, and are more effective in combination therapy than as single monotherapeutic agents.
Discussion
HCV is a major cause of chronic liver disease. The results of the present study indicate that the individual effects of the Hsp90 inhibitor 17-AAG on HCV replicon cells and the synergistic action of the proteasome inhibitor MG132 might account for the improved clinical response to combination therapy. We previously reported a new and effective strategy for inhibiting HCV replication using 17-AAG to inhibit Hsp90 [15]. The mechanism by which 17-AAG so effectively suppresses HCV replication is the destabilization of NS3, which disrupts the Hsp90 chaperone complex. A previous study demonstrated that HCV NS3 degradation is greatly increased by treatment of HCV subgenomic replicon #50–1 cells with 17-AAG, but this degradation is completely blocked in the presence of the proteasome inhibitor MG132 [15]. By contrast, in the present study we used HCV full-length replicon NNC#2 cells in place of HCV subgenomic replicon #50–1 cells used in previous studies [15]. The resulting 17-AAG treatment reduced NS3 levels, but MG132 treatment of NNC#2 cells did not block the degradation of HCV NS3 (Figure 1D). This blocking efficiency significantly influenced the different activities of the virus IRES (that is, HCV IRES and EMCV IRES) [25–28]. In our assays, when Huh-7 cells exposed to MG132 were transfected with pHCV IRES luc or pEMCV IRES luc, HCV IRES activity was inhibited, but EMCV IRES was not (Figure 2B). Apcher et al. [29] reported that HCV IRES-mediated translation can be induced by PSMA7. The proteasome inhibitor MG132 inhibits PSMA7 activity. These findings demonstrate that HCV IRES activity is also reduced by MG132. By contrast, degradation of NS3 by 17-AAG is dependent on the proteasome system [15].
The present study demonstrated that NNC#2 cells containing a full HCV genome replicon treated with 17-AAG or MG132 for 3 days did not show dose-dependent cytotoxicity (Figure 3A), but 10 μM MG132 was cytotoxic (data not shown). As shown in the present study, the IC50 values were 0.82 nM for 17-AAG and 0.21 nM for MG132 (Table 1 and Figure 3B). These data provide evidence that a dual treatment strategy with 17-AAG and MG132 inhibits HCV replication. The combination of 5 nM 17-AAG and 1 nM MG132 suppressed HCV RNA replication by 90% in NNC#2 HCV replicon cells (Figure 3B). The two drugs, 17-AAG and MG132, had synergistic inhibitory effects on HCV replicon (Table 1). Importantly, the combined use of these different groups of inhibitors showed strong synergistic inhibitory effects on HCV replication, indicating that combining these inhibitors might be a useful and efficient strategy for anti-HCV chemotherapy.
Given the absence of a single effective and proven antiviral agent against HCV, the combination of 17-AAG with agents that possess potential antiviral effects will continue to dominate novel therapeutic approaches. The present study demonstrated a strong synergistic effect of 17-AAG and MG132 on intracellular HCV replication, and these effects are attributable to the direct and specific inhibition of viral replication. Our results indicate that antiviral treatment with 17-AAG might be improved by combining 17-AAG with MG132, and that this combination therapy might be a feasible strategy for the treatment of HCV infection. Modifications of these inhibitors might also result in the development of more effective antiviral compounds.
Footnotes
Acknowledgements
We are grateful to M Sato and Y Katumura for excellent technical assistance. This work was supported by a Grant-in-Aid for HCV research from the Ministry of Health, Labor, and Welfare of Japan, and by a Grant-in-Aid for High Technology Research from the Ministry of Education, Science, Sports, and Culture of Japan.
The authors declare no competing interests.