
Abstract
Background:
Nucleoside analogues always require phosphorylation to be active. This appears to be a particular limitation for uridine-based nucleosides. Our ProTide method allows the direct use of masked membrane-soluble preformed nucleoside phosphates, bypassing the need for the initial phosphorylation step. We herein applied it to some novel 5-trimethylsilyl arabinosyl uridines.
Methods:
5-Trimethylsilyl-1-β-d-(arabinofuranosyl)uracil was prepared in six steps starting from uridine, and five phosphoramidate ProTide derivatives were synthesized. These compounds were investigated for activity against a range of DNA and RNA viruses, including herpes simplex virus type-1 and type-2, vaccinia virus and HIV.
Results:
Overall, these compounds did not show significant antiviral activity against any of the viruses tested.
Conclusions:
The inactivity of the ProTides of this nucleoside could correspond with poor ProTide activation in vitro, poor onward metabolism or low activity of the putative monophosphate metabolite.
Introduction
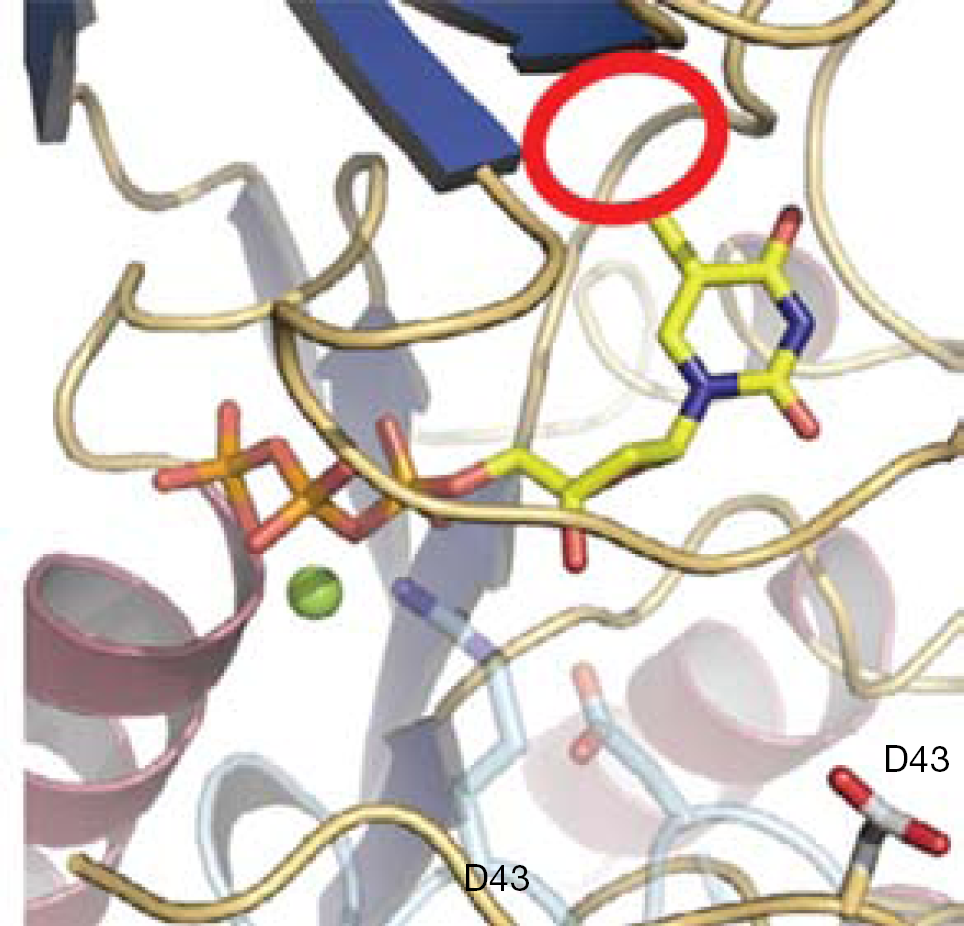
Although all the currently approved anti-HIV nucleoside-based drugs lack the 3′-hydroxyl group, giving them the ability to act as DNA chain terminators [1], recent reports suggested some ribo- and/or deoxyribonucleosides, which have the 3′-hydroxyl group present, to possess potent inhibitory effects on HIV [2–5]. Such compounds include 4′-ethynyl- or 4′-cyano-substituted 2′-deoxynucleosides, which (surprisingly) still afford their anti-HIV effects as a result of DNA chain termination and inhibition of reverse transcription [6]. Intrigued by this finding, we questioned whether having a β-hydroxyl group at the 2′-position, rather than a hydrogen atom would result in enough conformational change that might lead to a DNA chain termination effect. In addition, from the recently published crystal structure of vaccinia virus (VV) thymidine kinase [7], it was suggested that 1-β-d-(arabinofuranosyl)uracil nucleosides could result in potent agents against VV. Furthermore, it was proposed that bulky substitutions at C-5 of pyrimidines would make specific drugs against VV (Figure 1). To investigate these hypotheses, a series of 5-substituted-1-β-d-(arabinofuranosyl)uracils were synthesized and tested for antiviral activity, including herpes simplex virus type-1 and type-2, VV and HIV. Despite the various bulky substitutions that could be inserted at the 5-position of 1-β-d-(arabinofuranosyl) uracils, we focused our attention on the insertion of 5-trimethysilylethylene at position 5 making 5-trimethysilylethylene-1-β-d-(arabinofuranosyl)uracils (5-TMS-ethylene-AraU; Figure 2).
Docking image
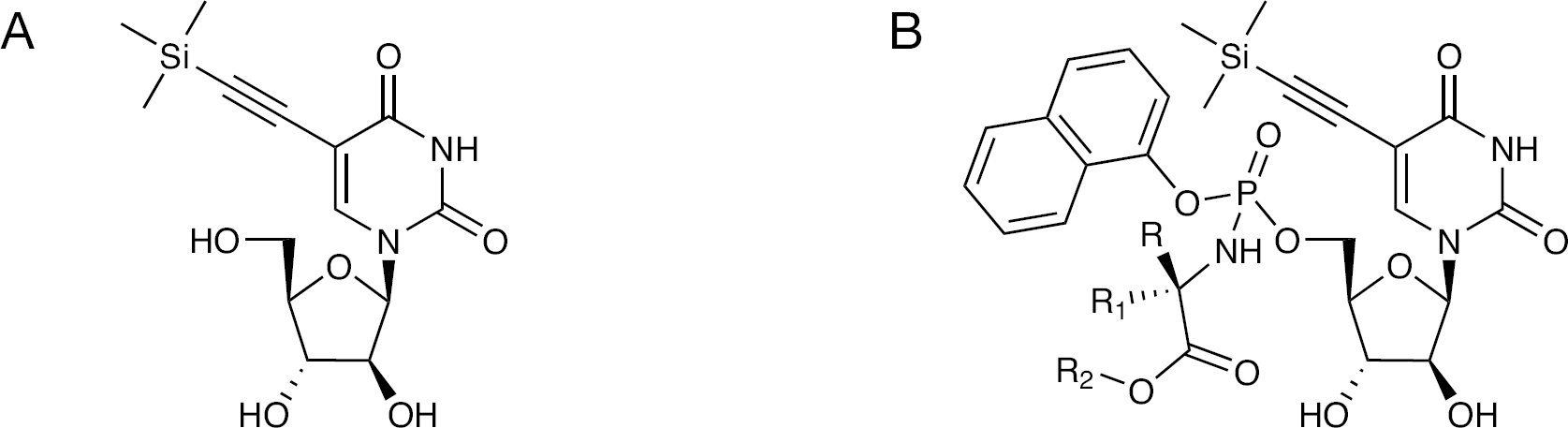
Structures of 5-TMS-ethylene-AraU and its phosphoramidate derivatives
Because uracil-based nucleoside analogues are often poorly phosphorylated to their triphosphate species, we also synthesized a series of phosphoramidate ProTides (Figure 2) to bypass the kinase-mediated first phosphorylation step [8,9]. In the phosphoramidate approach [10], the phosphate group is fully masked to provide membrane permeability. Upon entering the cell, phosphoramidates are metabolized by carboxypeptidases and phosphoramidase-type enzymes to release the nucleoside analogue monophosphate.
Methods
Chemistry
All experiments involving water-sensitive compounds were carried out under dry conditions. The solvents used were dry and used as purchased from Sigma-Aldrich (Gillingham, Dorset, UK). All the glassware was oven-dried at 130°C for several hours and allowed to cool under a steam of dry nitrogen. Thin-layer chromatography was performed using pre-coated aluminium-backed silica gel plates (60 F-54, 0.2 mm thickness; E Merck AG, Darmstad, Germany). Visualization of the plates was achieved using an UV lamp.
Glass columns were slurry-packed in the appropriate eluant under pressure with silica gel, 60 A, 40–60 μm, (Phase Separations Ltd, Deeside, Clwyd, UK). Samples were applied as a concentrated solution in the same eluant or pre-absorbed on silica gel. Fractions containing the product were identified by thinlayer chromatography (Merck KGaA, Darmstadt, Germany), pooled and the solvent was removed in vacuo.
1H, 13C NMR spectra were recorded on a Bruker Avance 500 MHz spectrometer (Bruker UK, Ltd., Coventry, UK) and autocalibrated to the deuterated solvent reference peak. The following abbreviations are used in the assignment of NMR signals: singlet (s), doublet (d), triplet (t), quartet (q) and multiplet (m).
Electrospray mass spectra were obtained using a Waters LCT time-of-flight (TOF) mass spectrometer coupled to a Waters M600 HPLC pump (Waters LCT, Milferd, MA, USA). Data was collected in the continuum mode over the mass range of 100–2,000 atomic mass units and processed using Masslynx 4.1 software.
1-((2R,4S,5R)-3,4-Anhydro-5-(hydroxymethyl) tetrahydrofuran-2-yl)pyrimidin-4(1H)-one (1)
Uridine (5.00 g, 20.47 mmol) and diphenyl carbonate (4.88 g, 22.80 mmol) were suspended in dimethylformamide (DMF; 40 ml). The slurry was heated to 100°C and sodium bicarbonate (150 mg) was then added and the reaction mixture was heated up to 137°C for 1.5 h. After completion, the reaction was cooled down to room temperature, filtered and washed with methanol (60 ml). Following drying under vacuum for 3 h, the title compound was obtained as a white solid in 78% yield (3.60 g). 1H NMR (d6-DMSO): δ 7.83 (1H, d, J=8.1 Hz, H-6), 6.31 (1H, m, H-1′), 5.90 (1H, d, J=8.1 Hz, H-5), 5.83 (1H, s, H-3′), 5.21 (1H, s, H-4′), 5.06 (1H, s, H-5′), 4.37 (1H, s, 5′-OH), 4.09 (1H, s, 3′-OH) and 3.15–3.24 (2H, m, H-2′). 13C NMR (d6-DMSO): δ 171.32 (CO), 159.79 (CO), 136.84 (C-6), 108.56 (C-5), 89.99 (C1′), 89.18 (C-2′), 88.74 (C-4′), 74.72 (C-3′) and 74.79 (C-5′).
1-((2R,3S,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)-tetrahydrofuran-2-yl)pyrimidines-2,4(1H,3H)-dione (2)
Compound
(2R,3R,4S,5R)-2-(acetoxymethyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydro-furan-3,4-diyl diacetate (3)
AraU (2.5 g, 10.23 mmol) and 4-dimethylaminopyridine (DMAP; 0.125 g, 1.02 mmol) were suspended in MeCN (80 ml). Triethylamine (5 ml, 35.80 mmol) was added to the reaction mixture and this was followed by the slow addition of acetic anhydride (3.2 ml, 33.76 mmol). After 1 h of stirring at room temperature, the reaction solvent was removed in vacuo. The residue was dissolved in methanol (20 ml) and diethylether (75 ml) was then added to form a precipitate, which was then filtered and dried to afford the desired compound as a white solid, 2.70 g (71%). 1H NMR (d6-DMSO): δ 11.43 (1H, bs, NH), 7.57 (1H, d, J=8.1 Hz, H-6), 6.22 (1H, t, J=4.6 Hz, H-1′), 5.65 (1H, d, J=8.1 Hz, H-5), 5.34 (1H, m, H-2′), 5.21 (1H, m, H-3′), 4.36 (1H, m, H-4′), 4.19–4.28 (2H, m, H-5′), 2.10 (6H, s, (CH3)2) and 1.99 (3H, s, CH3).
(2R,3R,4S,5R)-2-(acetoxymethyl)-5-(5-iodo-2,4-dioxo-3-,4-dihydropyrimidin-1(2H)-yl)-tetrahydrofuran-3,4-diyl diacetate (4)
Compound
1-((2R,3S,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)-tetrahydrofuran-2-yl)-5-iodopyrimidine-2,4(1H,3H)-dione (5)
Compound
1-((2R,3S,4S,5R)-3,4-Dihydroxy-5-(hydroxymethyl)-tetrahydrofuran-2-yl)-5-((trimethylsilyl)ethynyl)-pyrimidine-2,4(1H,3H)-dione (6)
Compound
(S)-Methyl 2-((((2R,3S,4S,5R)-5-(2,4-dioxo-5-((trimethylsilyl)ethynyl)-3,4-dihydropyrimidin-1(2H)-yl)-3-,4-dihydroxy-tetrahydrofuran-2-yl)methoxy)(naphthalen-1-yloxy)phosphorylamino)propanoate (7)
5-TMS-ethylene-AraU (200 mg, 0.58 mmol) was dissolved in 10 ml of pyridine/tetrahydrofuran (THF; 3:7) under N2. N-methylimidazole (NMI; 0.23 ml, 2.9 mmol) was then added followed by the addition of 1-naphthyl L-alanine methyl ester phosphorochloridate (570 mg, 1.74 mmol). The reaction mixture was stirred at room temperature for 15 h, after which the solvent was removed and the residue was purified by column chromatography using DCM: MeOH (94:6) as an eluant to afford 36 mg (10% yield) of the title compound as a white solid. 31P-NMR (d6-DMSO): δ 4.09, 4.37. 1H NMR (d6-DMSO): δ 11.67 (1H, bs, NH), 7.81 (1H, J=4.0 Hz, H-6), 7.43–7.65 (7H, m, Ar), 6.18 (1H, t, J=10.6 Hz, H-1′), 6.01 (1H, t, J=2.1 Hz, 2′-OH), 5.75 (1H, q, J=3.9 Hz, 3′-OH), 5.63 (1H, dd, J=4.1 Hz, J=7.4 Hz, 5′-OH), 4.28–4.34 (2H, m, H-2′ and H-3′), 3.98 (1H, m, H-4′), 4.91–4.96 (2H, m, H-5′), 3.65 (1H, s, CH), 3.62 (1H, s, CH), 3.58 (3H, s, OCH3), 1.26 (3H, t, J=1.9 Hz, CH3) and 0.3 (9H, s, 3xCH3). HRMS (ES+): Calculated mass for C28H34N3O10NaPSi: 654.1649. Found: 654.1653.
(S)-tert-Butyl 2-((((2R,3S,4S,5R)-5-(2,4-dioxo-5-((trimethylsilyl)ethynyl)-3,4-dihydropyrimidin-1(2H)-yl)-3-,4-dihydroxy-tetrahydrofuran-2-yl)methoxy)(naphthalen-1-yloxy)phosphorylamino)propanoate (8)
Compound
(S)-Benzyl 2-((((2R,3S,4S,5R)-5-(2,4-dioxo-5-((trimethylsilyl)ethynyl)-3,4-dihydropyrimidin-1(2H)-yl)-3-,4-dihydroxy-tetrahydrofuran-2-yl)methoxy)(naphthalen-1 yloxy)phosphorylamino)propanoate (9)
Compound
(R)-Methyl 2-((((2R,3S,4S,5R)-5-(2,4-dioxo-5-((trimethylsilyl)ethynyl)-3,4-dihydropyrimidin-1(2H)-yl)-3-,4-dihydroxy-tetrahydrofuran-2-yl)methoxy)(naphthalen-1-yloxy)phosphorylamino)propanoate (10)
Compound
(S)-Methyl 2-((((2R,3S,4S,5R)-5-(2,4-dioxo-5-((trimethylsilyl)ethynyl)-3,4-dihydropyrimidin-1(2H)-yl)-3-,4-dihydroxy-tetrahydrofuran-2-yl)methoxy)(naphthalen-1-yloxy)phosphorylamino)-3-methylbutanoate (11)
Compound
Virology
The antiviral assays, other than the anti-HIV assays, were based on inhibition of virus-induced cytopathic effect in human lung fibroblast (herpes simplex virus type-1 [strain KOS], herpes simplex virus type-2 [strain G], VV and vesicular stomatitis virus), African green monkey kidney (parainfluenza-3, reovirus-1, Sindbis, Coxsackie B4 and Punta Toro virus), human cervix carcinoma (vesicular stomatitis virus, Coxsackie virus B4 and respiratory syncytial virus) or Madin–Darby canine kidney (influenza A [H1N1; H3N2] and influenza B) cell cultures. Confluent cell cultures in microtitre 96-well plates were inoculated with 100 CCID50 of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures). After a 1 h virus adsorption period, residual virus was removed, and the cell cultures were incubated in the presence of varying concentrations (200, 40, 8, 1.6 and 0.32 μM) of the test compounds. Viral cytopathicity was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. The methodology of the anti-HIV assays was as follows: human T-lymphocyte CEM (approximately 3×105 cells/cm3) cells were infected with 100 CCID50 of HIV-1(IIIB) or HIV-2(ROD)/ml and seeded in 200 μl wells of a microtitre plate containing appropriate dilutions of the test compounds. After 4 days of incubation at 37°C, HIV-induced CEM giant cell formation was examined microscopically.
Results
Synthesis of 5-TMS-ethylene-AraU
The key intermediate for the synthesis of 5-TMS-ethylene AraU was AraU, which was obtained in two steps. Uridine and diphenyl carbonate were firstly suspended in dry DMF and the slurry was heated to 100°C. At this point, potassium carbonate (K2CO3) was added and the reaction mixture was heated to 137°C. After 1.5 h, the reaction mixture was allowed to cool to room temperature and then filtered. The white solid obtained was washed with methanol to afford the desired 2,2′-anhydrouridine (
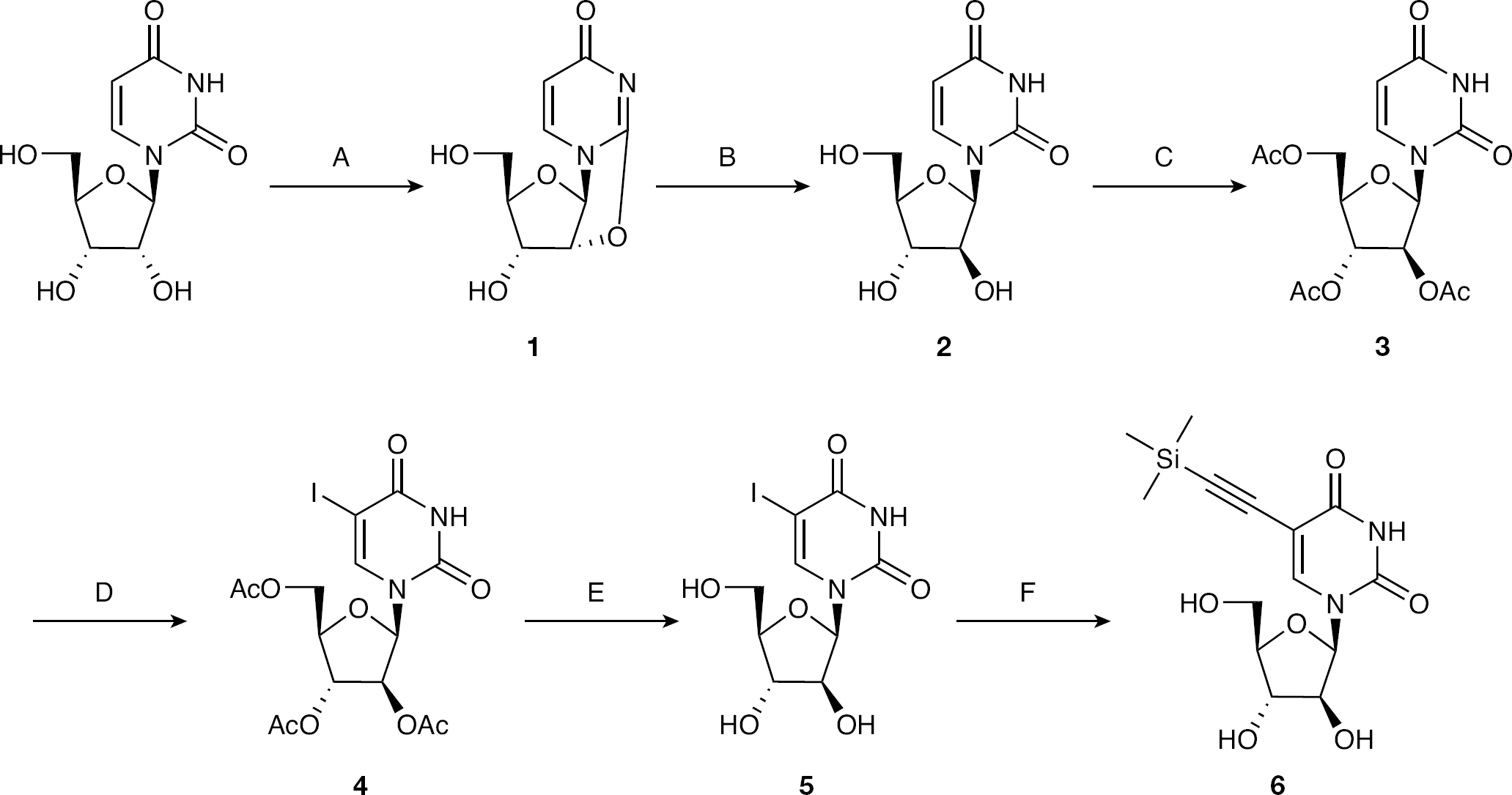
Synthesis of 5-trimethylsilyl-1-β-d-(arabinofuranosyl)uracil
Once AraU was obtained, 5-TMS-ethylene-AraU (
Synthesis of 5-TMS-ethylene-AraU phosphoramidates
The target phosphoramidates were synthesized according to the previously reported synthetic routes developed by McGuigan et al. [15–17] (Figure 4). Aryl phosphorochloridates were prepared by the reaction of phenyl/naphthyl dichlorophosphates with the appropriate amino acid ester hydrochloride. The obtained phosphorochloridates were allowed to react with 5-TMS-ethylene-araU in THF:pyridine (1:1) and NMI to give desired phosphoramidates in moderate yields. 31P NMR investigations of the phosphoramidates displayed two closely spaced signals, corresponding to two diastereoisomers resulting from mixed phosphate stereochemistry. All the phosphoramidate samples presented in this work were further investigated as a mixture of phosphate diastereoisomers.
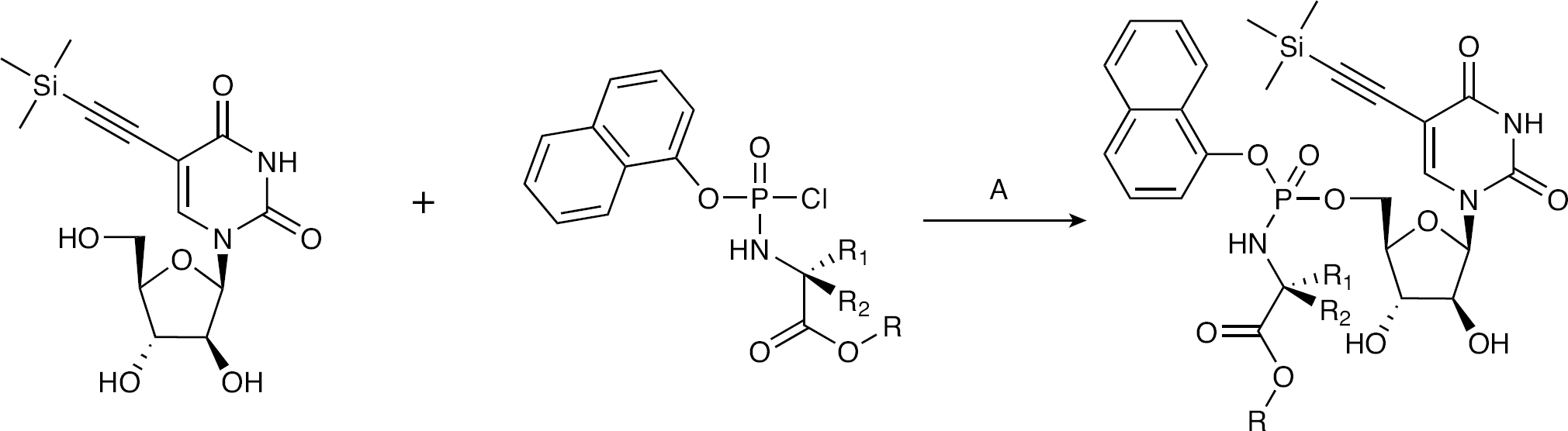
General synthetic scheme for the synthesis of 5-trimethylsilyl-1-β-d-(arabinofuranosyl) uracil
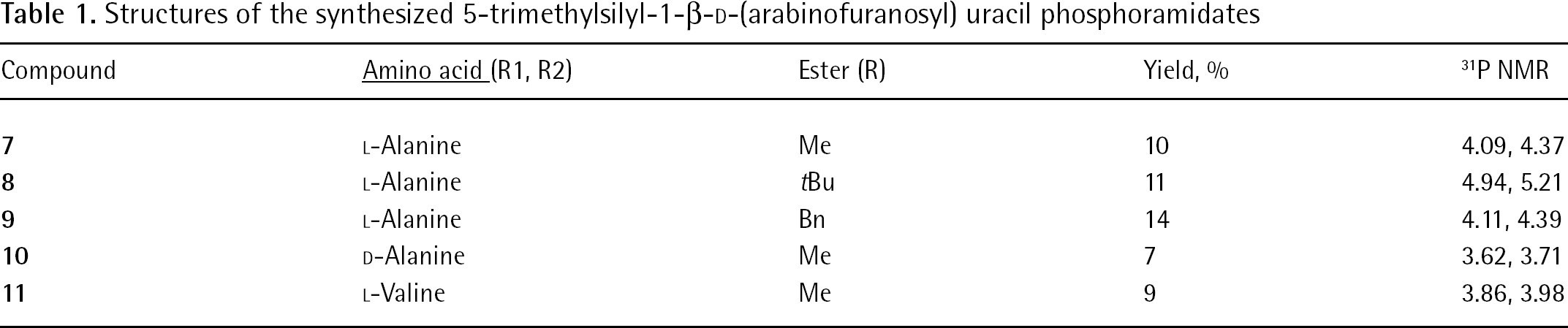
A series of 5-TMS-ethylene-AraU phosphoramidates bearing different ester and amino acid moieties were designed and synthesized (Table 1).
Structures of the synthesized 5-trimethylsilyl-1-β-D-(arabinofuranosyl) uracil phosphoramidates
Biological activity
The antiviral data of the various 5-TMS-ethylene-AraU nucleosides, which is summarized in Table 2, shows that none of the compounds possessed significant anti-HIV, anti-HSV or anti-VV activities. Interestingly, compounds
Anti-HIV, anti-HSV and anti-VV data of 5-trimethylsilyl-1-β-D-(arabinofuranosyl)uracil phosphoramidates
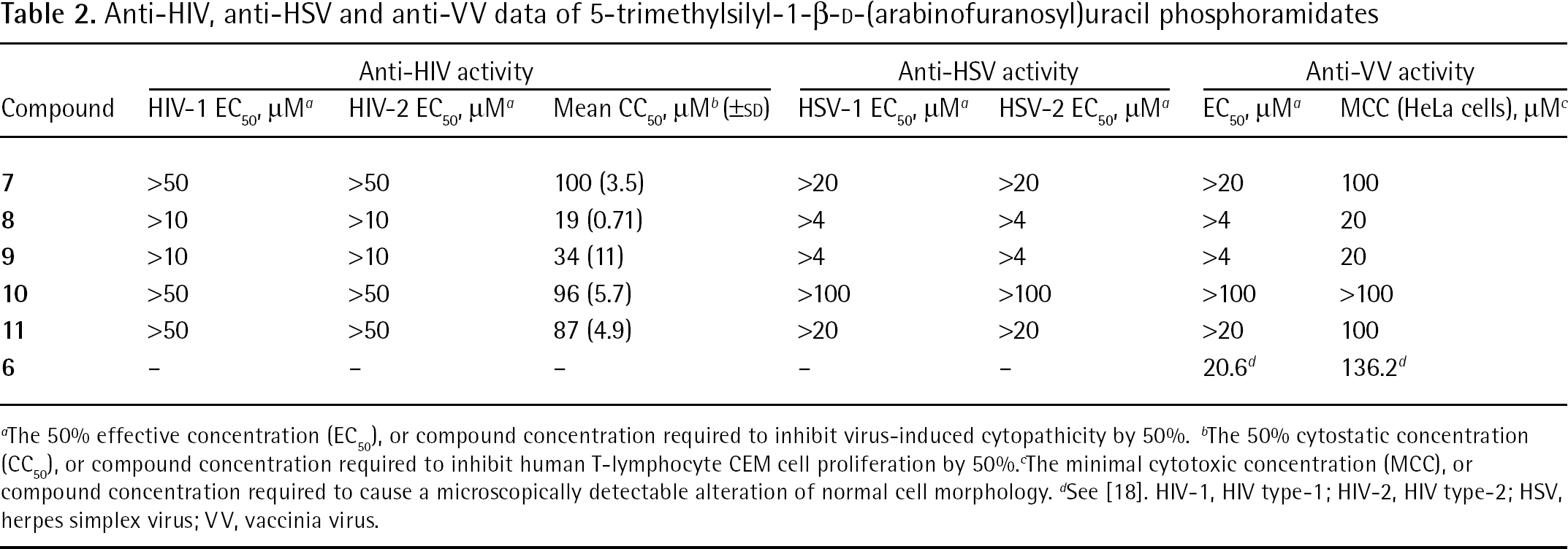
The 50% effective concentration (EC50), or compound concentration required to inhibit virus-induced cytopathicity by 50%.
The 50% cytostatic concentration (CC50), or compound concentration required to inhibit human T-lymphocyte CEM cell proliferation by 50%.
The minimal cytotoxic concentration (MCC), or compound concentration required to cause a microscopically detectable alteration of normal cell morphology.
See [18]. HIV-1, HIV type-1; HIV-2, HIV type-2; HSV, herpes simplex virus; VV, vaccinia virus.
The 5-TMS-ethylene-araU phosphoramidates were also tested against a wide variety of other viruses including vesicular stomatitis virus, Coxsackie virus B4, reovirus-1, Sindbis virus, Punta Toro virus, influenza virus A (H1N1; H3N2) and influenza virus B, parainfluenza-3 virus, respiratory syncytial virus, feline coronavirus and feline herpesvirus. In all cases, no significant antiviral activity at subtoxic concentrations was measured.
Discussion
As a result of the identification of some ribonucleosides bearing antiviral activities [2–6], and the suggestion that 5-substituted 1-β-d-(arabinofuranosyl)uracils might be selective agents versus VV [7], 5-TMS-ethylene-AraU (
The reason(s) why these phosphoramidates were devoid of any activity remains unclear. However, three reasons could be suggested as possible explanations for the lack of activity in this case. Firstly, the phosphoramidates were not converted to their corresponding monophosphates. This seems a very plausible explanation, given that in another study we found 1-β-d-(arabinofuranosyl)uracil phosphoramidates were hydrolyzed by carboxypeptidase Y, but they did not seem to be good substrates of the phosphoramidase-type enzyme (hint-1) that is believed to cleave the P-N bond (unpublished observations). Secondly, a poor second and/or third phosphorylation of the monophosphate to the corresponding triphosphate might be the reason for such a lack of antiviral activity as already found to be the case for 2′,3′-didehydro-2′,3′-dideoxyuridine and 2′,3′-dideoxyuridine monophosphates [8]. Thirdly, if the triphosphate of 5-TMS-ethylene-AraU is generated, it might be a poor substrate/inhibitor for the virally-encoded DNA or RNA polymerases.
Conclusion
A small series of 5-trimethylsilyl-1-β-d-(arabinofuranosyl)uracil phosphoramidate derivatives were designed, synthesized and investigated for antiviral activity. This was fulfilled by firstly synthesizing 5-TMS-ethylene-AraU from uridine in six steps, and secondly, the preparation of five different phosphoramidate derivatives. Although these compounds were tested against a wide range of viruses, including HIV and VV, no significant activity was observed at subtoxic concentrations. Such a lack of activity could be the result of a number of reasons that vary from the cleavage of the P–N bond of the phosphoramidates to the possibility of the triphosphate derivative of 5-trimethylsilyl-1-β-d-(arabinofuranosyl)uracil not being a (potent) DNA or RNA polymerase inhibitor. Overall, the antiviral data of 5-trimethylsilyl-1-β-d-(arabinofuranosyl)uracil phosphoramidates did not support further investigation, but we continue to study alternative 5-substituted uridine nucleoside analogue phosphoramidates as antiviral therapies.
Footnotes
Acknowledgements
The authors thank Leen Ingels, Leentje Persoons, Frieda De Meyer and Vicky Broeckx for excellent technical assistance. The research was supported by the Katholieke Universiteit Leuven (GOA number 05/19) to JB.
The authors declare no competing interests.