
Abstract
Bicyclic aryl furano pyrimidines represent the most potent anti-varicella zoster virus (VZV) agents reported to date. Lead compounds have 50% effective concentration (EC50) values in vitro that are in the subnanomolar range and selectivity index values that exceed 1 million. They have an absolute requirement for VZV thymidine kinase and most likely act as their phosphate forms. Some structural modification (such as aryl substitution in the base moiety) is tolerated, whereas little sugar modification is acceptable.
The Cf1743 compound has proved to be significantly more potent than all reference anti-VZV compounds, as measured either by inhibition of infectious virus particles and/or viral DNA production; however, the high lipophilicity and very low water solubility of this compound gives poor oral bioavailability (<14%). Use of the modified cyclodextrin captisol and the synthesis of the 5′-monophosphate prodrug of Cf1743 has significantly improved water solubility, but does not give any enhancement in oral bioavailability. By contrast, the synthesis of the ether series does not give any further improvement in terms of solubility. The most promising prodrug to emerge to date is the hydrochloric salt of the 5′-valyl-ester, designated as FV-100. Its uptake into cells has been studied using fluorescent microscopy and biological assays, which have indicated that the compound is efficiently taken up by the cells after a short period of incubation.
Introduction
Varicella zoster virus (VZV) is one of the eight members of the herpesviridae family. More specifically, it is classified within the α-herpesviruses (such as the herpes simplex viruses [HSV]). Although HSV replicates in the cells of numerous animal species, the host range of VZV is limited to humans. The VZV particle is 125–175 nm in diameter and has a lipid envelope bearing glycoprotein spikes [1]. VZV is the cause of both varicella (chickenpox) and zoster (shingles). The virus becomes permanently established in sensory ganglia, persists in latent form and recurs when reactivated as herpes zoster. VZV reactivation occurs in up to 15% of those who have had varicella, manifesting by a vesicular rash and affecting the region innervated by the sensory nerve concerned [2]. The drugs of first choice for the treatment of herpes zoster by oral administration are acyclovir and its prodrug valacyclovir, although vidarabine has also been used intravenously (Figure 1). These drugs have a beneficial effect on reducing acute pain caused from shingles and on speeding healing [3].
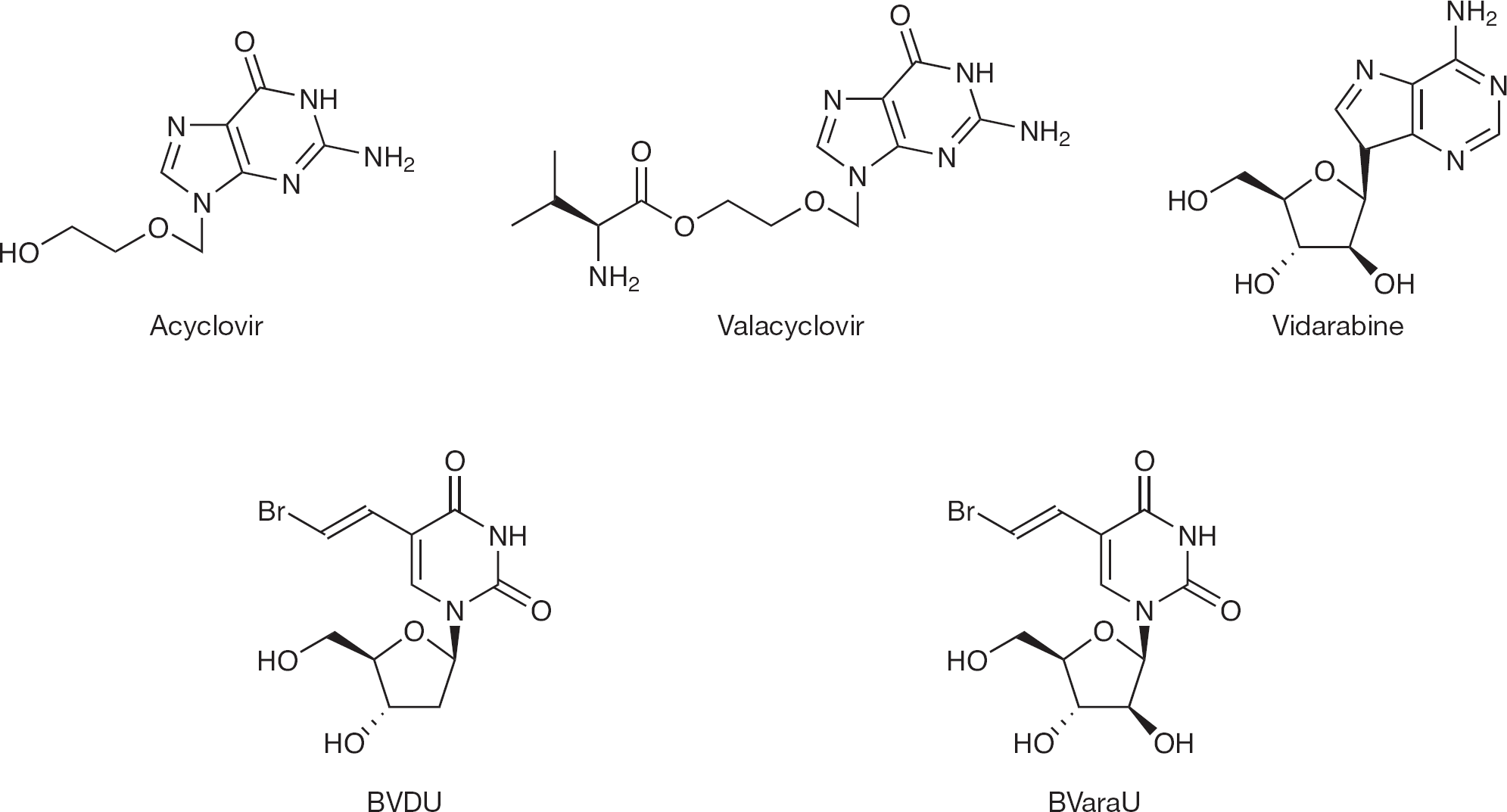
Drugs used for the treatment of varicella zoster virus
Valacyclovir and famciclovir have been licensed for the treatment of herpes zoster virus infections [4]. Their pharmacokinetic profile is improved and their half-life is longer, which makes these compounds better drugs for the treatment of VZV compared with their parent drugs acyclovir and penciclovir [5]. Also, brivudine (BVDU) and sorivudine (BVaraU) are very potent inhibitors of VZV, inhibiting virus replication in cell culture at a 50% effective concentration (EC50) of <10 nM [6,7].
Bicyclic nucleoside analogues (BCNAs) are a class of compounds that have been demonstrated, in the past 10 years, as very potent inhibitors of VZV. These nucleosides bear a furanopyrimidine structure, which is obtained by Sonogashira coupling of terminal alkynes and 5-iodopyrimidine derivatives in the presence of a base [8,9]. These bicyclic nucleosides were found to be inactive against HSV type-1 (HSV-1), HSV type-2 and cytomegalogvirus (CMV), but have showed considerable potency and selectivity against VZV, with the length of the side chain playing a crucial role in the activity. Indeed, the optimal length is between 8 and 10 carbon atoms, with an EC50 of 8–20 nM (Figure 2). These compounds are approximately 300-fold more active than acyclovir and are roughly equipotent to BVDU [10], and the low 50% cytotoxicity concentration (CC50; >50 μM) results in a high selectivity index of >5,000. The structure–activity relationships (SAR) regarding the long alkyl chain is clear: a short chain of ≤C6 leads to little antiviral activity and C7, C11 and C12 confers a moderate activity, whereas C8–10 gives a high potency [10] (Figure 2).
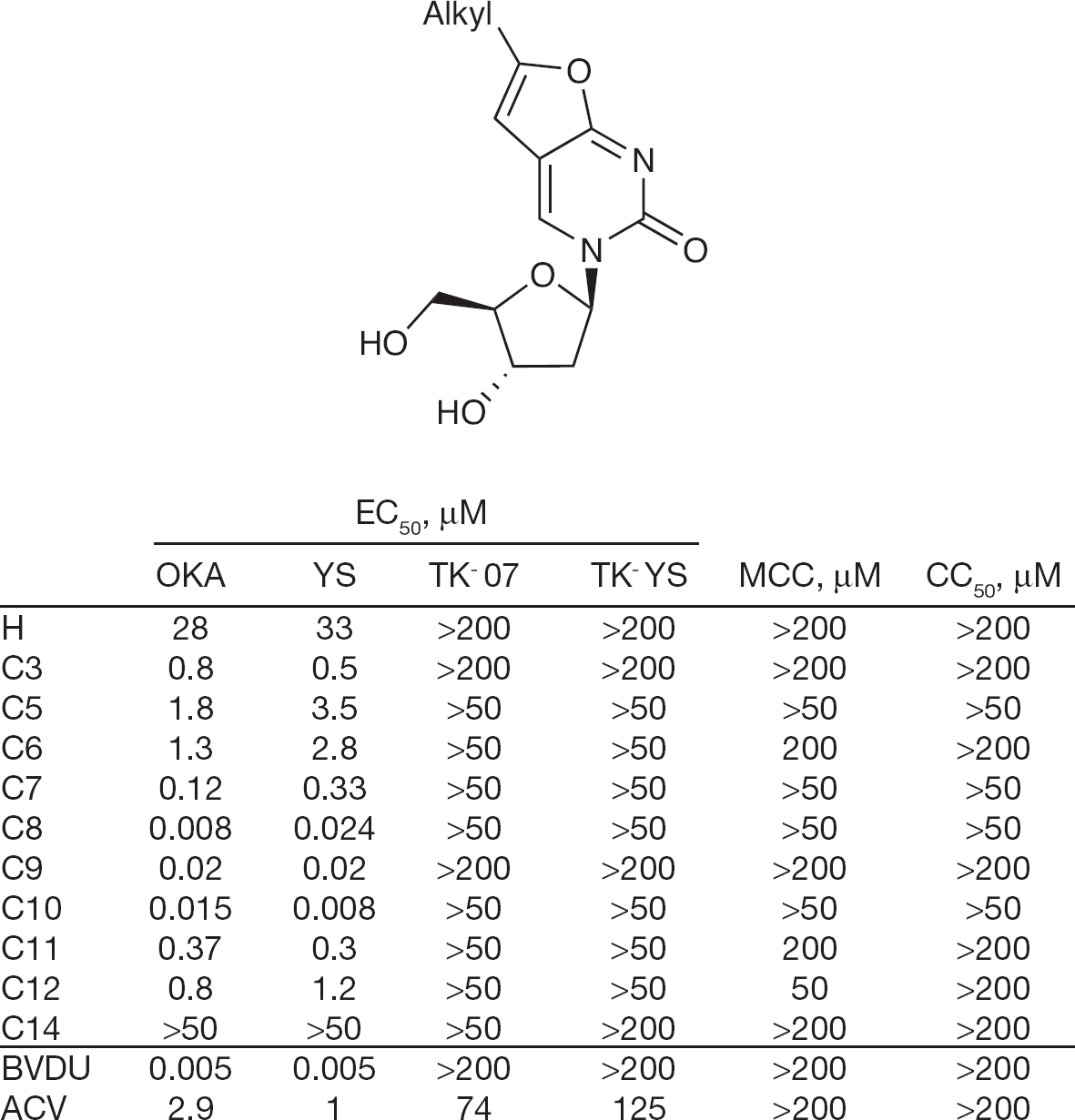
Anti-varicella zoster virus activity for alkyl-bicyclic nucleoside analogues
Alkylaryl modifications
In order to introduce a conformational constraint in the alkyl chain while retaining a high logP, some p-alkylaryl compounds have been synthesized. The introduction of a phenyl moiety between the base and the linear alkyl chain resulted in a significant increase of potency up to a subnanomolar concentration for the pentylphenyl derivative, also known as Cf1743 (Figure 3; C5 entry), which is 10,000-fold more active than acyclovir and approximately 20-fold more active than BVDU with no cytotoxicity [11]. The presence of a branched chain on the phenyl ring, instead, led to a reduction of activity, implying that lipophilicity or a more flexible chain is required for an extreme potency against VZV [12].
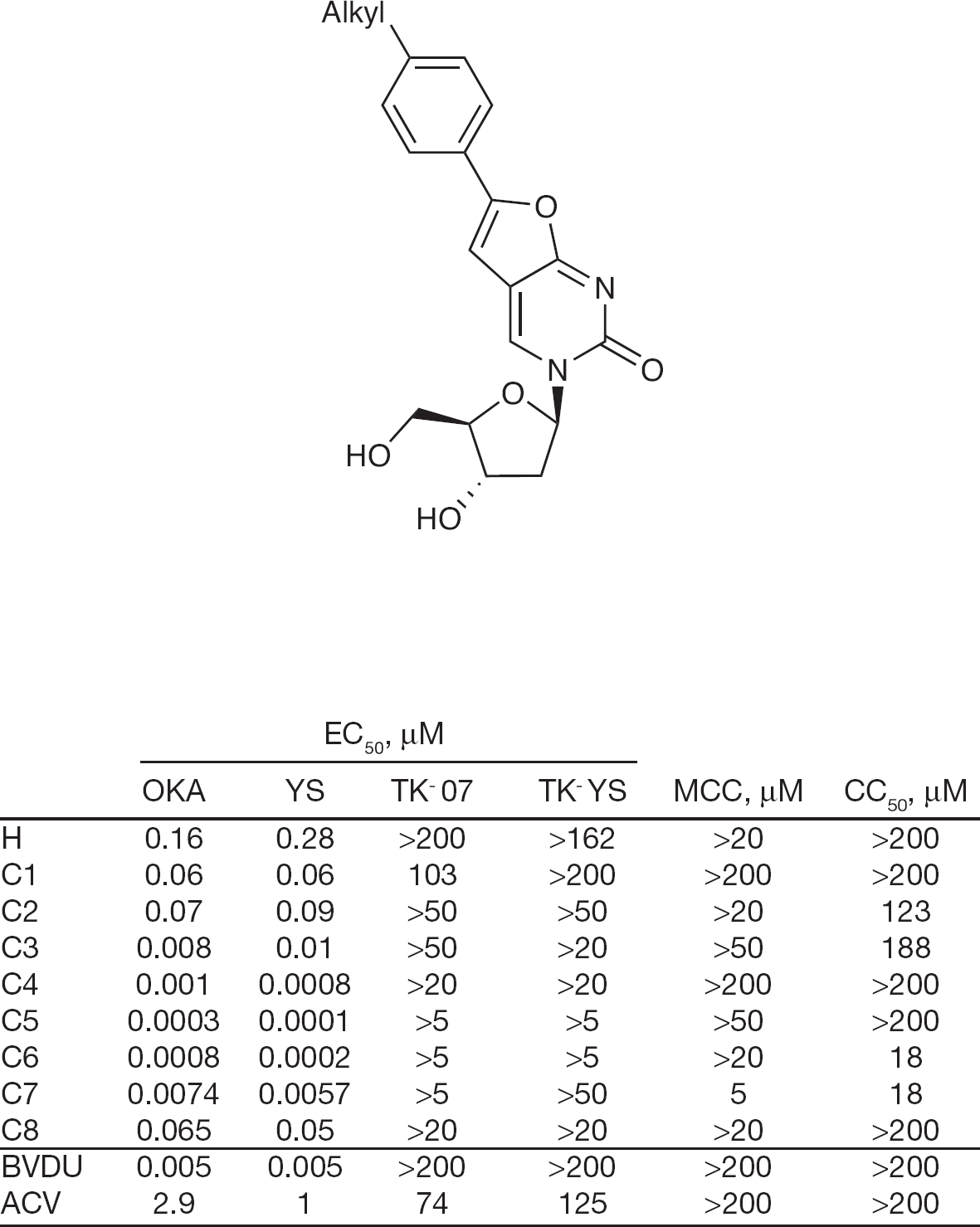
Anti-varicella zoster virus activity for aryl-bicyclic nucleoside analogues
Sugar modifications
Because the antiviral effect is strictly dependent on the VZV-encoded kinase activity, a deoxyribose nucleoside is the best substrate for the enzyme and only small modifications are tolerated in order to have a good antiviral activity [10]; thus, different sugar derivatives have been synthesized bearing different alkyl chains to probe the SARs on the sugar moiety. The introduction of a hydroxyl group in position 2′ resulted in a marked loss of activity (ara and ribo; Table 1) [10]. The removal of the 3′-OH from the sugar led to a loss of activity for the didehydro and didehydrodideoxy derivatives [13], but the latter surprisingly gained a modest anti-human CMV activity [13]. The corresponding analogue of acyclovir has been synthesized, showing that this compound has antiviral activity comparable to acyclovir and that the presence of a deoxyribose motif is beneficial for activity [14]. However, the 3′-OH has to be in a ribo position, as the dXylo derivative is less active than the corresponding dRibo analogue [15]. Even the carbocycle analogue showed a marked reduction (1,000-fold) in activity, probably because of poor phosphorylation [16]. From this data, it seems that even very small modifications in this region of the molecule are not very well tolerated and that the dRibo is the best substrate for the VZV-encoded kinase.
Anti-varicella zoster virus activity for sugar-modified bicyclic nucleoside analogues
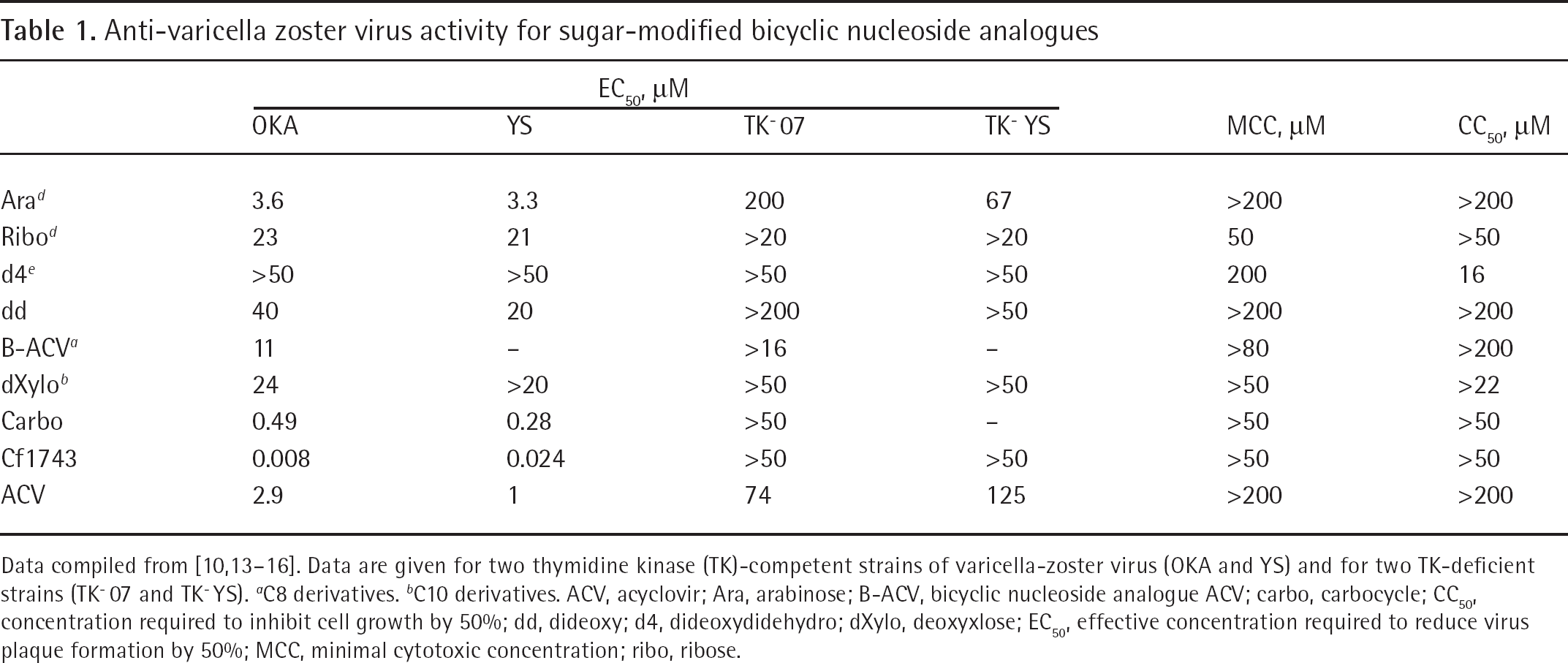
Data compiled from [10,13–16]. Data are given for two thymidine kinase (TK)-competent strains of varicella-zoster virus (OKA and YS) and for two TK-deficient strains (TK-07 and TK-YS).
C8 derivatives.
C10 derivatives. ACV, acyclovir; Ara, arabinose; B-ACV, bicyclic nucleoside analogue ACV; carbo, carbocycle; CC50, concentration required to inhibit cell growth by 50%; dd, dideoxy; d4, dideoxydidehydro; dXylo, deoxyxlose; EC50, effective concentration required to reduce virus plaque formation by 50%; MCC, minimal cytotoxic concentration; ribo, ribose.
Mechanism of action
Unlike the BCNAs, all other anti-VZV drugs known so far have also been shown to have antiviral activity against other herpesviruses. Pharmacological studies have revealed that the BCNAs are recognized as substrates for phosphorylation by VZV thymidine kinase (TK; Figure 4) [6]. However, in contrast to BVDU, HSV-1 TK does not recognize the BCNA or the BCNA 5′-monophosphates (MPs) as substrates for phosphorylation. Furthermore, the BCNAs are not recognized as substrates by the cellular TKs either, therefore, explaining the unusual specificity of the BCNAs for VZV [17]. This specificity excludes also the Simian varicella virus, which, despite being closely related to VZV, is not inhibited by BCNAs [18]. Interestingly, when nucleoside 5′-diphosphate (NDP) kinase was added to a mixture containing BCNAs and VZV TK, only the BCNA-diphosphate (DP) formation was observed and no trace of BCNA-triphosphate (TP) could be detected [17]. Although it cannot be excluded that the BCNA-DPs can be recognized by other NDP kinase isoenzymes or by other cellular enzymes, the mechanism of anti-VZV activity of the novel class of BCNAs might be entirely different from that of BVDU (being a inhibitor of viral DNA polymerase and incorporated into viral DNA when it is converted into BVDU 5′-TP).
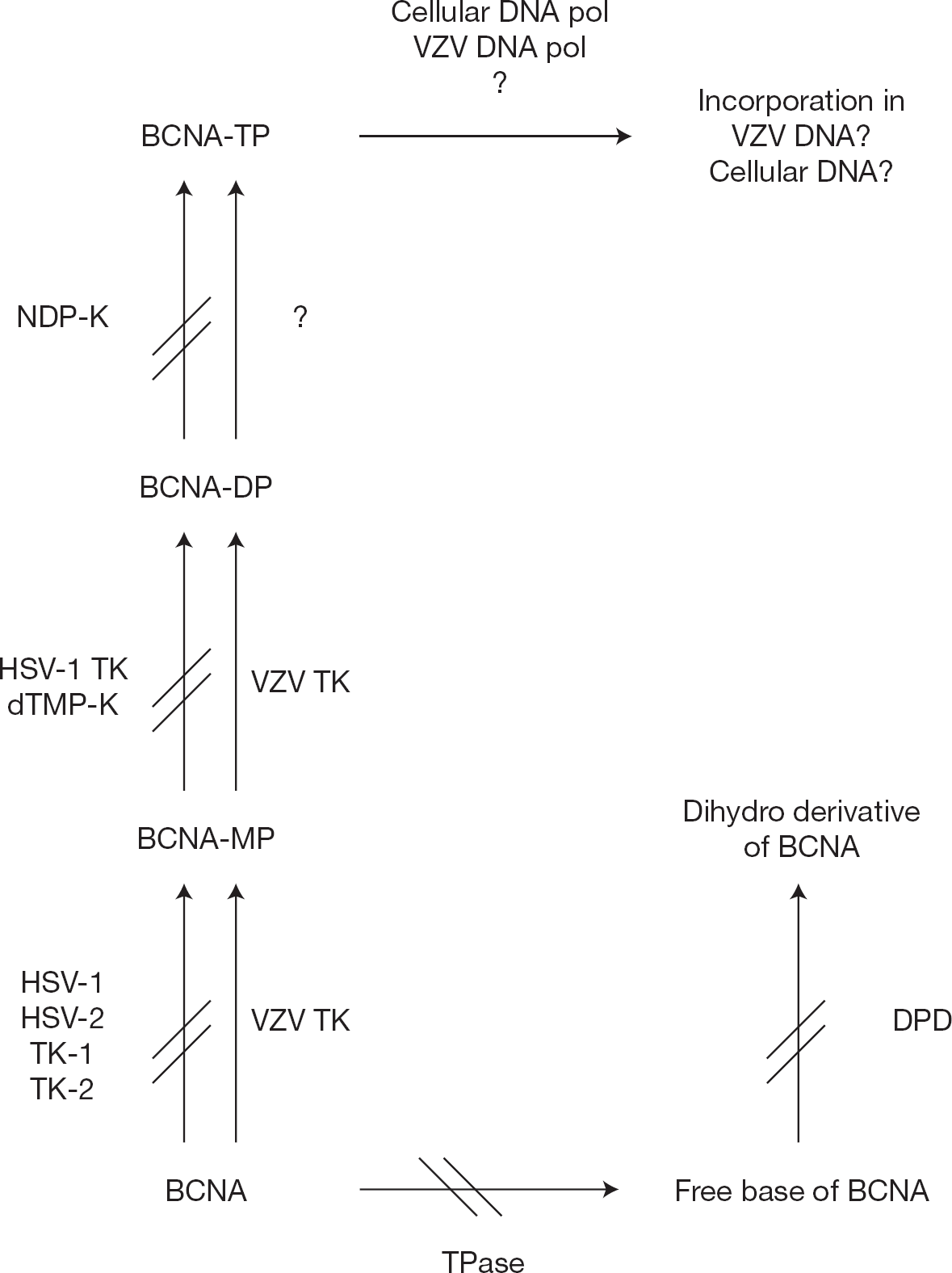
Metabolic fate of BCNA
Pyrimidine nucleoside analogues are susceptible to cleavage by the pyrimidine nucleoside catabolic enzyme uridine phosphorylase (UPase) or thymidine phosphorylase (TPase). The bases usually do not have any significant therapeutic activity; thus, the UPase and TPase enzymes often inactivate the antiviral pyrimidine nucleoside analogues, and BVDU is a well-known example of an anti-herpesvirus drug subjected to this kind of cleavage. The BCNAs were found to be resistant to the phosphorolytic cleavage of human erythrocytes and Escherichia coli TPase and, thus, are expected to be relatively stable in biological fluids [19]; however, it cannot be excluded that they can be targeted by purine phosphorylases. The free base (E)-5-(2-bromovinyl) uracil (BVU) and related compounds are known to be good inhibitors of human dihydropyrimidine dehydronase (DPD) [20], the catabolic enzyme responsible for the degradation of natural pyrimidines and pyrimidine analogues such as fluorouracil (5-FU). Given the potential complication of coadministration of BVDU and 5-FU, it is of clinical importance to know whether the free base of BCNAs can inhibit this enzyme. In contrast to BVU (50% inhibitory concentration 10 μM against human liver DPD), free BCNA bases are completely inactive in inhibiting DPD at 100–250 μM. In addition, plasma levels of 5-FU were not affected by the free BCNA bases [17].
Solubility enhancement
The high logP of these compounds helps with membrane permeability, but it might present a problem because of its low water solubility, which significantly reduces its concentration in plasma levels. Indeed, the solubility of Cf1743 in water is approximately 0.9 μg/ml [21] and the octyl derivative is only slightly more soluble [22]. A potential candidate could be found on the alkoxyphenyl series [23]; in particular, the propyloxy represents a good compromise between solubility and activity.
Preliminary studies of bioavailability on these compounds in rodents showed that the octyl exhibits the best bioavailability with an estimated value of <25%, followed by the pentylphenyl with a value <15% (Table 2). Instead, the more water soluble propyloxy shows a bioavailability value of 0%, probably caused by the rapid metabolism and excretion [23].
Solubility of bicyclic nucleoside analogues
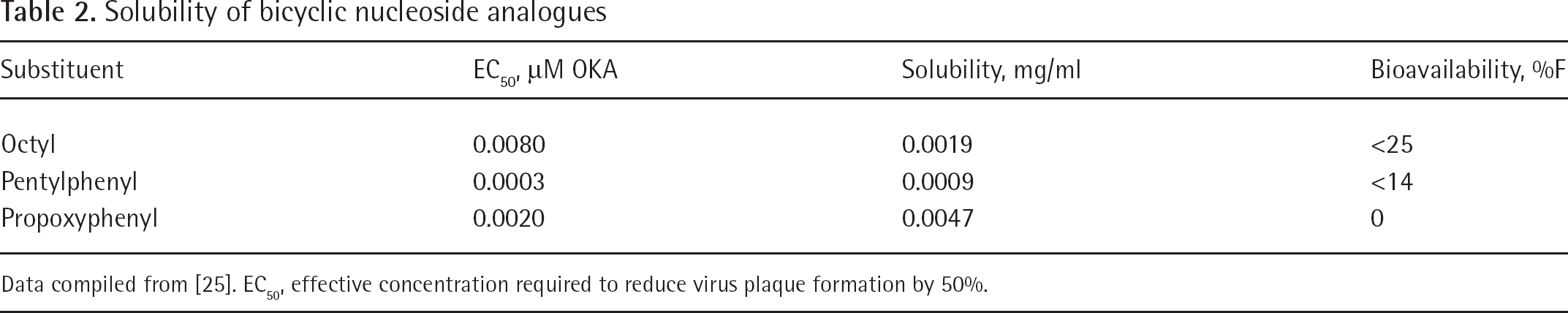
Data compiled from [25]. EC50, effective concentration required to reduce virus plaque formation by 50%.
Ethers
It is known that the intrinsic lipophilicity of the lead compounds has a negative effect on water solubility, affecting the bioavailability directly. The introduction of one or two atoms of oxygen in the alkyl chain has been reported, leading to the alkoxyphenyl series and glycol series [22,24]. The former retains a good antiviral activity but it is rapidly metabolized in vivo to a less active metabolite, whereas the latter shows a dramatic loss of activity [23,24].
The modification of the alkoxyphenyl series could be a good strategy in order to enhance the water solubility whilst keeping a high antiviral activity. The shifting of the oxygen through the side chain might retain the water solubility without affecting the antiviral activity, converting the aromatic ether into an aliphatic ether, making it harder to metabolize. Indeed, although these compounds retain full antiviral activity at non-toxic concentrations, the introduction of an atom of oxygen in the alkyl chain did not achieve significant water solubility enhancement [21].
Captisol
Captisol is a substituted cyclodextrin that has been successfully used in clinical settings; therefore, a captisol study was carried out in order to increase the solubility of Cf1743. As shown in Table 3, captisol led to a significant boost on the water solubility of the highly lipophilic drug. Solubility enhancements were found to be linear up to the highest concentration used (40%) with a 1,000-fold boost. The standard β- and γ-cyclodextrins showed a limited enhancement compared with captisol; however, when evaluated at the highest concentration for oral bioavailability in mice, it was observed only to have a limited (twofold) enhancement in the area under the curve [25].
Effect of captisol on water solubility of Cf1743
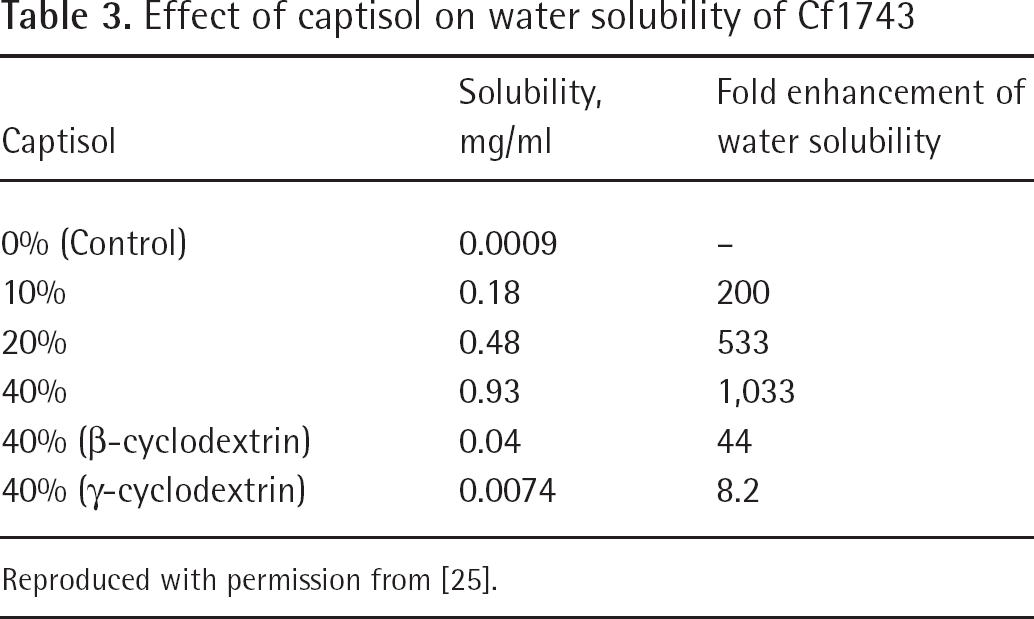
Reproduced with permission from [25].
FV-100
The high potency of the BCNA pentylphenyl makes FV-100 compound the most potent agent against VZV to date, with very low toxicity and a selectivity index of >100,000. These encouraging biological properties led to an extensive preclinical programme, where several issues were encountered. The major issue encountered was represented by low absorption resulting in a bioavailability value (<14%) [25]. The same problem of poor availability emerged also with acyclovir, which was solved by synthesizing amino acid ester prodrugs. In particular, valacyclovir increased the bioavailability of acyclovir from 20% to 55% [26].
Several prodrugs of Cf1743 were synthesized including the valinyl ester and the MP (Figure 5). After a preliminary study, it was found that the antiviral activity of these compounds did not show any significant variation from the parent drug. Because the above mentioned ether analogue results were not satisfactory, the MP of Cf1743 has been thought as a solution, as the presence of a charge should significantly increase the water solubility. This strategy has been used for fludarabine [27], an anticancer nucleoside used against chronic lymphocytic leukaemia. This drug is administered as an MP salt, which is rapidly dephosphorylated in human plasma and, once it is inside the cell, it is converted into a TP that inhibits the ribonucleotide reductase and DNA polymerase [27]. As expected, the MP is >100-fold more water soluble than its parent drug, but despite the solubility enhancement, the bioavailability did not show any significant improvement [25].
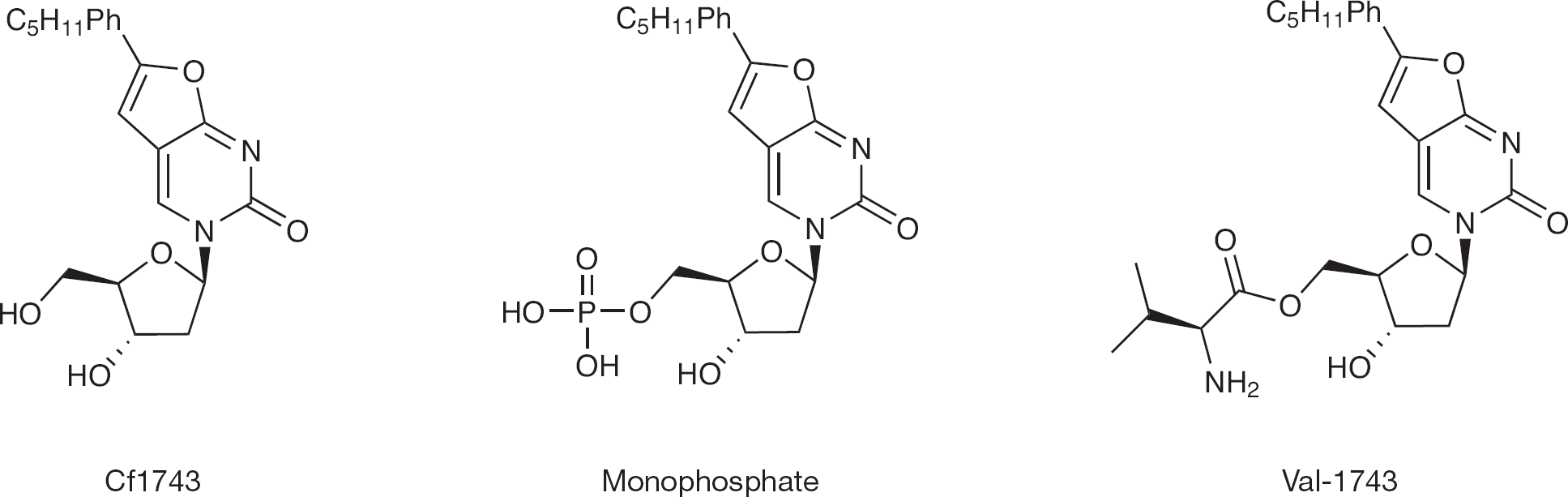
CF1743 and its prodrugs
The same strategy used to improve the bioavailability of acyclovir was used also on Cf1743, leading to the valinyl ester. This prodrug exhibited a significantly improved bioavailability, 10× higher than its parent drug and, along with increased water solubility, it qualified as a good candidate for preclinical studies [25] (Table 4 and Figure 6).
CF1743 levels in plasma

Reproduced with permission from [25].
Data presented as area under the curve ×106. An area under the curve of 1×106 corresponds to 40 ng/ml of Cf1743.
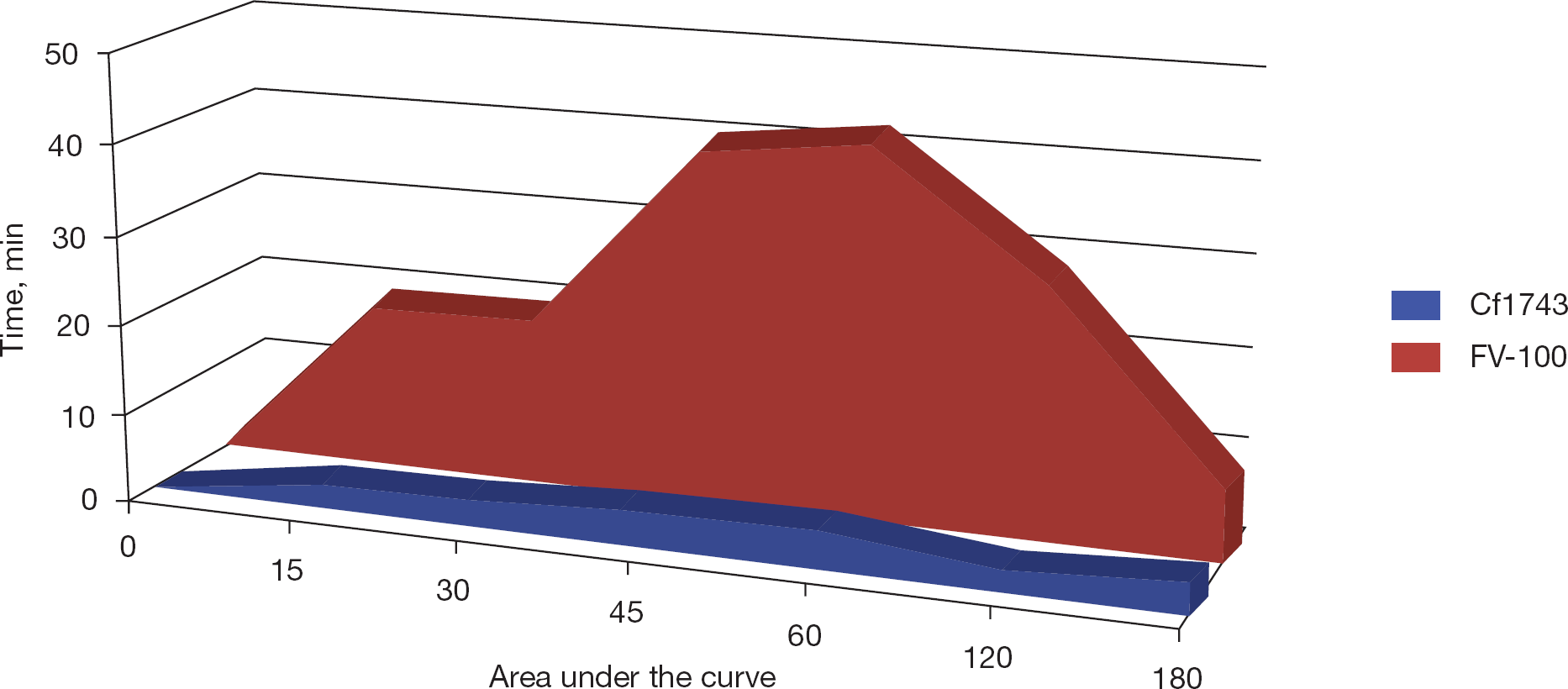
Area under the curve diagram versus time
Salts of Val-Cf1743
In order to further increase the water solubility of this prodrug, the hydrochloric and succinate salts were synthesized and their solubility evaluated. Val-Cf1743 was found to be 20× more soluble than the parent nucleoside. Its conversion to a hydrochloric salt increased the solubility by 25-fold, becoming 500× more soluble than the parent nucleoside, Cf1743. This form of Val-Cf1743 is also known as FV-100. The succinate salt showed an even greater boost in solubility, being 1,500× more soluble than Val-Cf1743 and 30,000× more soluble than Cf1743 (Table 5) [25].
Water solubility of Cf1743 and its derivatives
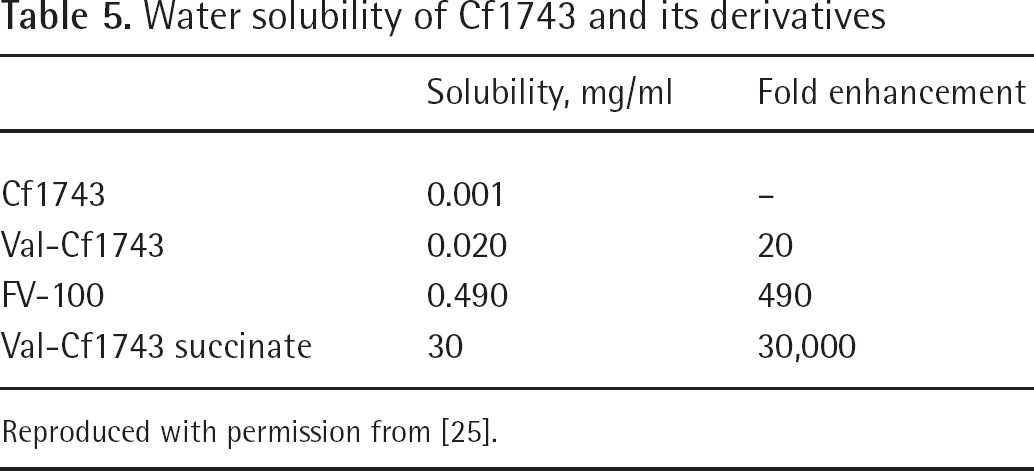
Reproduced with permission from [25].
Cell study
Cf1743 and FV-100 are intensely fluorescent when illuminated by wavelengths of 340–380 nm. This property has been used in a preliminary study to track the distribution of the compounds inside cells in order to investigate the mechanism of action of BCNAs. Untreated cells do not fluoresce in this range and thus they are virtually invisible. When HeLa cells (cervical carcinoma epithelial cells) are treated with an 8 μM solution of Cf1743 for 1 h, it can be seen that the compound is uniformly distributed in the cytoplasm. It was also noted that the compound did not diffuse inside the nucleus (Figure 7D). Treating cells with FV-100 for 1 h produced a different distribution (Figure 7B) wherein the compound was trapped inside small vesicles scattered around in the cell, which are probably lysosomes [28]. This effect might have been caused by the presence of the amino group in the compound that is protonated in the acidic environment of the lysosome and cannot recross the membrane by passive diffusion. Here, it remains trapped until it is slowly hydrolysed to Cf1743, which can cross the membrane by passive diffusion and then exhibit its antiviral activity. Administering FV-100 to HeLa cells and observing after 2 min, it can be seen that the vesicles are not yet formed and that FV-100 is uniformly distributed in the cytoplasm like the parent drug (Figure 7A). This implies that the accumulation in the vesicles is not an immediate process, but it requires some time. The dissipation of the pH gradient of these organelles with the antibiotic nigericin followed by treatment of the cells with FV-100 showed a very little evidence of sequestration (Figure 7C), suggesting that the trapping in these vesicles is a pH-dependent process. This preliminary study supports the notion that FV-100 is rapidly internalized, consistent with its promising bioavailability [25].
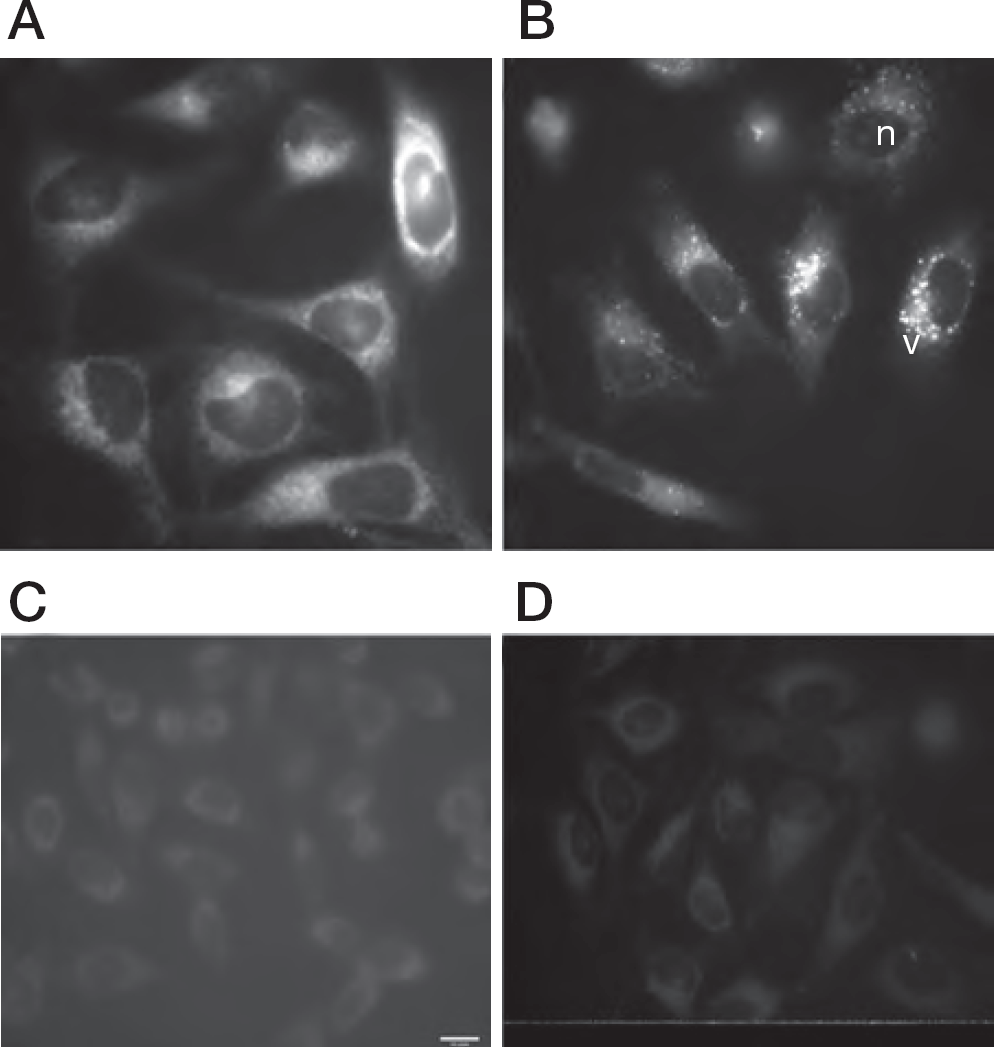
Subcellular distribution of CF1743 and FV-100
Conclusions
This brief SAR review has highlighted the high potency and selectivity of the lead aryl BCNA family, particularly for the p-pentylphenyl compound (Cf1743). Even minor structural modification of the sugar adversely affected potency, whereas modifications of the base were tolerated without any significant loss of activity. However, any modification applied to Cf1743 has led to an equipotent or less active compound. This compound was an excellent candidate for the preclinical phase, but its poor solubility in water required the use of a strategy to enhance its pharmacokinetic profile. Formulation with captisol increased the water solubility by 1,000-fold but not the bioavailability; the MP was 100-fold more soluble than Cf1743, but with a little improvement in bioavailability. The ether series kept the same potency but did not improve solubility. The 5′-valyl-ester of Cf1743 was more soluble and achieved a major boost in biovailability. The hydrochoric salt of the valyl-ester (FV-100) was chosen on the basis of its high solubility in water and resistance to decomposition. It has successfully passed the preclinical studies and has recently entered Phase II clinical trials for VZV shingles [29].
Footnotes
Acknowledgements
The author would like to thank Chris McGuigan (Welsh School of Pharmacy, Cardiff University, Cardiff, Wales), Geoffry Henson (Inhibitex, Inc., Alpharetta, GA, USA) and Jan Balzarini (Rega Institute for Medical Research, Universiteit Leuven, Leuven, Belgium) for their technical assistance and suggestions, and Oxford University Press for providing the images presented in this work.
The author declares no competing interests.