
Abstract
Background:
The development of antiviral drugs has provided crucial new means to mitigate or relieve the debilitating effects of many viral pathogens. New classes of inhibitors are essential to combat swine influenza viral infection.
Methods:
A series of isatine-sulfadimidine derivatives were screened for antiviral activity against swine influenza A/California/07/2009 (H1N1) virus in Madin–Darby canine kidney (MDCK) cell culture. Cytotoxicity of the synthesized compounds was also tested in uninfected MDCK cells.
Results:
All the compounds inhibit the influenza A (H1N1) in MDCK cells. The most active compounds, SPIII-5Br and SPIII-5H, inhibited virus-induced cytopathology by 50% at 27 and 30 μM, respectively, with 50% cytotoxicity occurring at a much higher dose (975–1,000 μM). The positive control compound ribavirin inhibits the replication of the virus at 18 μM and cytotoxic concentration was found to be >1,000 μM.
Conclusions:
SPIII-5Br and SPIII-5H exhibited potency in the same range as ribavirin and are suitable candidate molecules for further investigation.
Introduction
While the medical community is concerned about when the avian influenza A (H5N1) virus will adapt well enough in human to cause a pandemic, the emergence of a novel swine-origin influenza A (H1N1) virus (S-OIV) has shocked the world [1]. The S-OIV genome is made up of a unique combination of gene segments that had not been observed in previously known influenza A viruses as a result of genetic reassortment [2]. A major concern is the possibility that S-OIV will acquire more frequently encountered resistance to antivirals as the epidemic evolves. The two classes of antiviral agents against influenza viruses are the adamantanes, which include amantadine and rimantadine, and the neuraminidase inhibitors, which include oseltamivir and zanamivir. The M gene encodes the target of the adamantanes. So far, all strains of S-OIV from the current epidemic contain a serine asparagine mutation (S31N), which confers resistance to both amantadine and rimantadine. This mutation is also highly prevalent among circulating subtypes of European swine influenza A viruses [3]. Although rare, there were cases of oseltamivir-resistant S-OIV [4] – most of the strains tested were susceptible to zanamavir. At present, these two compounds are the mainstay of influenza virus treatment. This is in contrast to the situation in the 2008–2009 influenza season, in which most circulating influenza A (H1N1) viruses were apparently resistant to oseltamivir while still being susceptible to zanamivir [5]. However, massive use of these antiviral agents for treatment and prophylaxis might lead to development of resistance, which would pose a challenge to the management of patients severely compromised by these infections and result in increased mortality. For this reason the medical community is in need of newer therapeutic targets and antiviral agents to combat swine influenza.
Isatin (2,3-dioxoindole), a versatile lead molecule for synthesis of potential bioactive agents and its derivatives were reported to possess wide spectrum of activity. Methisazone (N-methylisatin-β-thiosemicarbazone) was one of the first clinically used synthetic antiviral agents [6]. The drug was not significantly effective in treating smallpox infections in humans and induced vomiting following oral administration of 1–6 g/day [7]. More recently, methisazone was tested for activity in mice infected with cowpox and vaccinia viruses, but was minimally beneficial in reducing disease severity [8]. N-methylisatin-β-4′:4′-diethylthiosemicarbazone was found to inhibit Moloney leukaemia virus replication and disease in mice [9]. N,N-disubstituted thiosemicarbazone derivatives of isatin were tested for inhibition and found to be active against HIV-1 replication in cell culture [10]. In earlier studies reported from our laboratories, some novel isatin derivatives were synthesized and evaluated for antiviral, anticancer and antibacterial activities [11–14]. These compounds showed significant inhibitory effects against HIV-1, orthopox and influenza viruses. In this study, we describe the antiviral activity of some of isatine-sulfadimidine derivatives (Figure 1) against influenza A/California/07/2009 (H1N1) infections in Madin–Darby canine kidney (MDCK) cells.
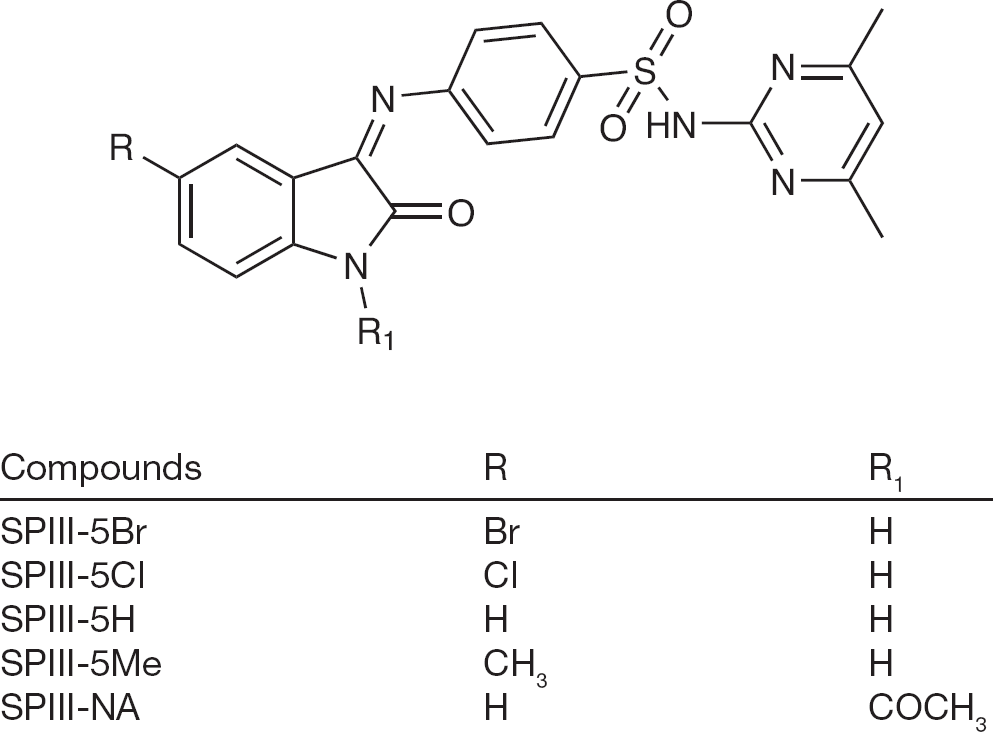
N-(4,6-dimethyl-pyrimidin-2-yl)-4-(2-oxo-1,2-dihydro-indol-3-ylideneamino)benzenesulfonamide and derivatives
Methods
Compounds
4-[(1,2-Dihydro-2-oxo-3H-indol-3-ylidene)amino]-N-(4,6-dimethyl-2-pyrimidin-2yl)benzene sulfonamide (SPIII-5H) and four derivatives (5-chloro [SPIII-Cl], 5-bromo [SPIIIBr], 5-methyl [SPIII-5ME] and N-acetyl [SPIII-NA]) were prepared by combining the isatin and the derivatives with sulfadimidine in the presence of glacial acetic acid [11].
Antiviral assays
The influenza A/California/07/2009 (H1N1) virus was obtained from the Centers for Disease Control and Prevention (Atlanta, GA, USA). The inhibitory effects of the compounds on virus replication were determined by cytopathic effect inhibition (CPE) assays in confluent MDCK cell monolayers conducted in 96-well microplates [14,15]. Compounds in half-log10 dilution increments were applied to cells 5–10 min before adding virus, using three wells for infection and two wells for uninfected toxicity controls. Fifty 50% cell culture infectious virus doses of virus were then added and the plates were incubated for 3 days, when inhibitor-free cell cultures were completely destroyed by virus. At this time, the mean percentage of cell viability in each set of three infected wells or set of two toxicity control wells was quantified by a neutral red dye uptake method, using 0.011% final concentration of the dye for 2 h. Eluted dye was quantified by optical density scanning at 460 nm. A Microsoft Excel® spreadsheet was developed for converting optical density readings to percentages of untreated control values. Concentrations of compounds reducing viral CPE by 50% (50% effective concentration [EC50] values) were calculated by linear regression plotting of concentration versus percent inhibition on semi-log graph paper. Antiviral activity and cytotoxicity of the positive control drug ribavirin were also performed in parallel.
Confirmatory virus yield reduction assays were performed as described previously [14], by replicating 50 50% cell culture infectious virus doses of A/California/07/2009 (H1N1) per well in 96-well microplates for 3 days in the presence of varying half-log10 concentrations of the active compounds. The same plates of cells used for the CPE assays above were processed for virus yield reduction analysis. The virus-containing samples were stored in the plates at −80°C prior to titration. Virus yields at each concentration were later determined by titration of samples in 10-fold increments from 10−1 to 10−8 dilutions on fresh monolayers of MDCK cells using four microwells per dilution. Virus titre in each sample was quantified by endpoint dilution method [16]. The concentration of compound reducing virus yield by 90% (1 log10) was determined in a similar manner as described for CPE inhibition assays.
Results
All the isatin compounds inhibited the replication of influenza A 2009 (H1N1) virus. Their 50% effective concentrations (EC50 values) ranged from 27 to 58 μM. SPIII-5Br and SPIII-5H had relatively the same antiviral potency, with the EC50 values of 27 and 30 μM, respectively, and similar selectivity index values. The other isatin derivatives were about half as potent as these two compounds. Ribavirin inhibited the replication of the virus at 18 μM (selectivity index >56). Toxicities of the isatin derivatives in confluent cells ranged from 183 to 1,000 μM. SPIII-5Me exhibited greater toxicity than the other four compounds.
The isatin compounds were also shown to inhibit virus yields at 31–125 μM (Table 1). SPIII-5H was more potent than SPIII-5Br in this assay. The most potent isatin in this assay was SPIII-5Me; however, this compound also exhibited the greatest cytotoxicity, resulting in a lower selectivity index. Ribavirin was inhibitory to virus yield at 23 μM.
Anti-influenza A/California/07/2009 (H1N1) activities of isatin derivatives in MDCK cells
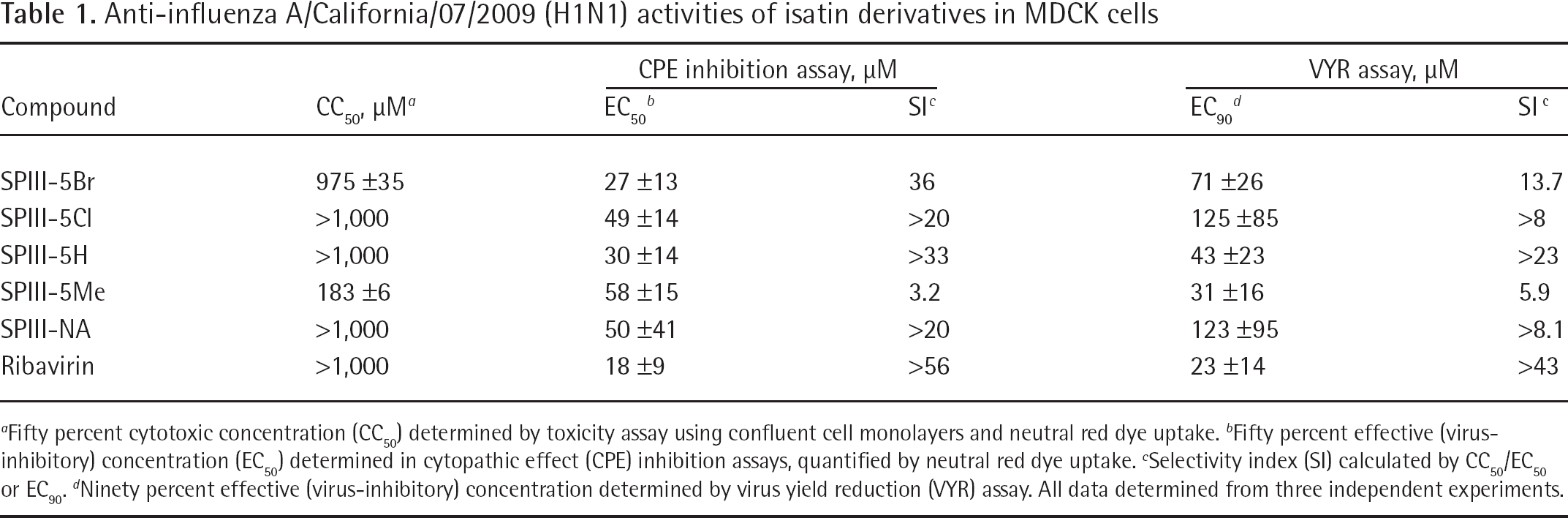
Fifty percent cytotoxic concentration (CC50) determined by toxicity assay using confluent cell monolayers and neutral red dye uptake.
Fifty percent effective (virus-inhibitory) concentration (EC50) determined in cytopathic effect (CPE) inhibition assays, quantified by neutral red dye uptake.
Selectivity index (SI) calculated by CC50/EC50 or EC90.
Ninety percent effective (virus-inhibitory) concentration determined by virus yield reduction (VYR) assay. All data determined from three independent experiments.
Discussion
The emergence of highly contagious influenza A virus strains, such as the new H1N1 swine influenza, represents a serious threat to global human health [1]. Efforts to control emerging influenza strains focus on surveillance and early diagnosis, as well as development of effective vaccines and novel antiviral drugs. Previously we investigated some novel heterocyclic compounds as potent inhibitors of influenza virus [17]. Herein we document the anti-influenza activity of five isatine-sulfadimidine derivatives effective against swine influenza A virus at 27–58 μM. These same compounds were also active against the influenza A/New Caledonia/20/99 (H1N1) seasonal influenza virus, as well as against influenza A (H3N2 and H5N1) and influenza B viruses [14]. Inhibition of the A/New Caledonia virus by the five compounds was reported at 2.7–5.2 μg/ml (6.4–12.8 μM) [14]. Ribavirin inhibited A/New Caledonia [14] and A/California (Table 1) viruses at 3.7 μg/ml (15 μM) and 18 μM, respectively. Thus, the isatin derivatives were less potent against the 2009 H1N1 virus compared with A/New Caledonia, whereas ribavirin was similarly potent against both viruses.
This class of compounds is suitable for further molecular modification for synthesis of potentially more potent inhibitors of swine influenza virus. Plans for synthesis of other molecules in this series are underway. Because of the antiviral activities reported here, SPIII-5Br and SPIII-5H merit consideration for evaluation in influenza mouse models. The mode of action of these compounds appears to be blocking of virus adsorption to cells [14]. Because of this, topical delivery of these compounds may be required for antiviral activity in vivo.
Footnotes
Acknowledgement
The anti-influenza experiments were performed at Utah State University under contract NO1-AI-30048 from the Virology Branch, NIAID, NIH (Bethesda, MD, USA).
The author declare no competing interests.