
Abstract
Integration is a distinctive and essential process in the HIV infection cycle and thus represents an attractive antiviral drug target. Integrase inhibitors combined with other classes of drug might contribute to long-lasting suppression of HIV type-1 (HIV-1) replication for many patients. Of the numerous potential integrase inhibitor leads that have been reported, few have reached clinical trials and only one, raltegravir, has been approved (in late 2007) for the treatment of HIV-1-infected patients. Another integrase inhibitor, elvitegravir, is currently showing promise in Phase III clinical studies. Once-daily administration of elvitegravir has a comparable antiviral activity to twice-daily of raltegravir in HIV-1-infected patients. Here, we highlight the salient features of elvitegravir: its chemical structure compared with representative integrase inhibitors, mechanism of action, in vitro and in vivo activity against HIV and other retroviruses, and the effect of integrase polymorphisms and resistance mutations on its anti-HIV activity.
Introduction
Combinational use of anti-HIV drugs as part of highly active antiretroviral therapy (HAART) has opened a new era in the treatment of HIV infection. With HAART, HIV replication has been remarkably suppressed, and reduction of viral load to undetectable levels and decrease of HIV-associated mortality have been successfully maintained [1]. To date, nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), non-nucleoside/nucleotide reverse transcriptase inhibitors (NNRTIs) and protease inhibitors (PIs) have been widely used in HAART. However, long-term administration of the drugs to keep viral replication to minimum levels unfortunately induces the emergence of drug-resistant variants and leads to treatment failure [2]. Moreover, some mutations can confer cross- and/or multidrug resistance to one or more inhibitors in the same class of agents [3]. To cope with this issue, in addition to discovery of new reverse transcriptase (RT) and protease (PR) inhibitors without cross-resistance to existing drugs, the development of novel therapeutic agents targeting virus-specific molecules remains necessary.
Integrase (IN) is one of three enzymes (including PR and RT) encoded by the viral genome and, like PR and RT, is essential for the virus replication. IN consists of three functional domains: N- and C-terminal domains, and a catalytic core domain (CCD). The CCD conducts the intrinsic function of IN, integration, which is a ligation reaction between viral and host DNA [4]. The human genome lacks functional homologues of HIV IN [5], consequently IN is an attractive target for the development of new anti-HIV drugs.
Many different molecules have been reported to function as IN inhibitors and some of them have been evaluated for their in vivo potency in animal and human trials; however, until recently, no IN inhibitors were approved in clinical use. Raltegravir (RAL; Merck) was approved for the treatment of HIV infection in October 2007 [6]. RAL shows potent activity in patients infected with HIV type-1 (HIV-1) with resistance to other classes of drugs, including NRTIs, NNRTIs and PIs [7,8]. Elvitegravir (EVG), another IN inhibitor that is currently in late Phase III clinical trials, also shows potent antiviral activity in HIV-1-infected patients [9,10]. Here, we describe both in vitro and in vivo antiviral activity and resistance profiles of EVG.
Similarities and differences in chemical structures
Compounds containing diketo acids or derivatives thereof, such as γ-ketone, enolizable α-ketone and carboxylic acid, were postulated to be the pharmacophore of inhibitors for retroviral IN activity. Structures of diketotriazole (S-1360; Figure 1), diketotetrazole (5CITEP), diketo-carboxylic acid (L-708,906), 1,6-naphthyridine-7-carboxamide (L-870,810 and L-870,812) have been designed, synthesized and demonstrated to inhibit HIV-1 IN as bioisosteres of a diketo acid motif. Such bioisosteres can be replaced with similar moieties. For instance, the carboxylic acid could be replaced with not only acidic bioisosters, such as triazole and tetrazole, but also by a basic heterocycle bearing a lone-pair donor atom, such as a pyridine ring. The heteroaromatic nitrogen in the pyridine ring mimics the corresponding carboxyl oxygen in the diketo acid as a Lewis base equivalent. The enolizable ketone at the α-position of diketo acids can be replaced with a phenolic hydroxyl group, indicating that the α-enol form of each diketo acid is its biologically active coplanar conformation. All bioisosteres of the diketo acid motif consist of the three functional groups that mimic a ketone, an enolizable ketone and a carboxyl oxygen, and can have a coplanar conformation (Figure 1). According to available information, we designed and synthesized a series of compounds that have 4-quinolone-3-carbocyclic acid, including EVG [11]. The 4-quinolone-3-carbocyclic acid has two functional groups, a β-ketone and a carbocyclic acid, which are coplanar. Therefore, a coplanar monoketo acid motif in 4-quinolone-3-carbocyclic acid could work as an alternative diketo acid motif, providing novel insight into new structural designs and the binding mode of this type of inhibitor. RAL (Figure 1) also has a structurally similar moiety, with a pyrimidinone carboxamide as a diketo acid bioisostere [12].
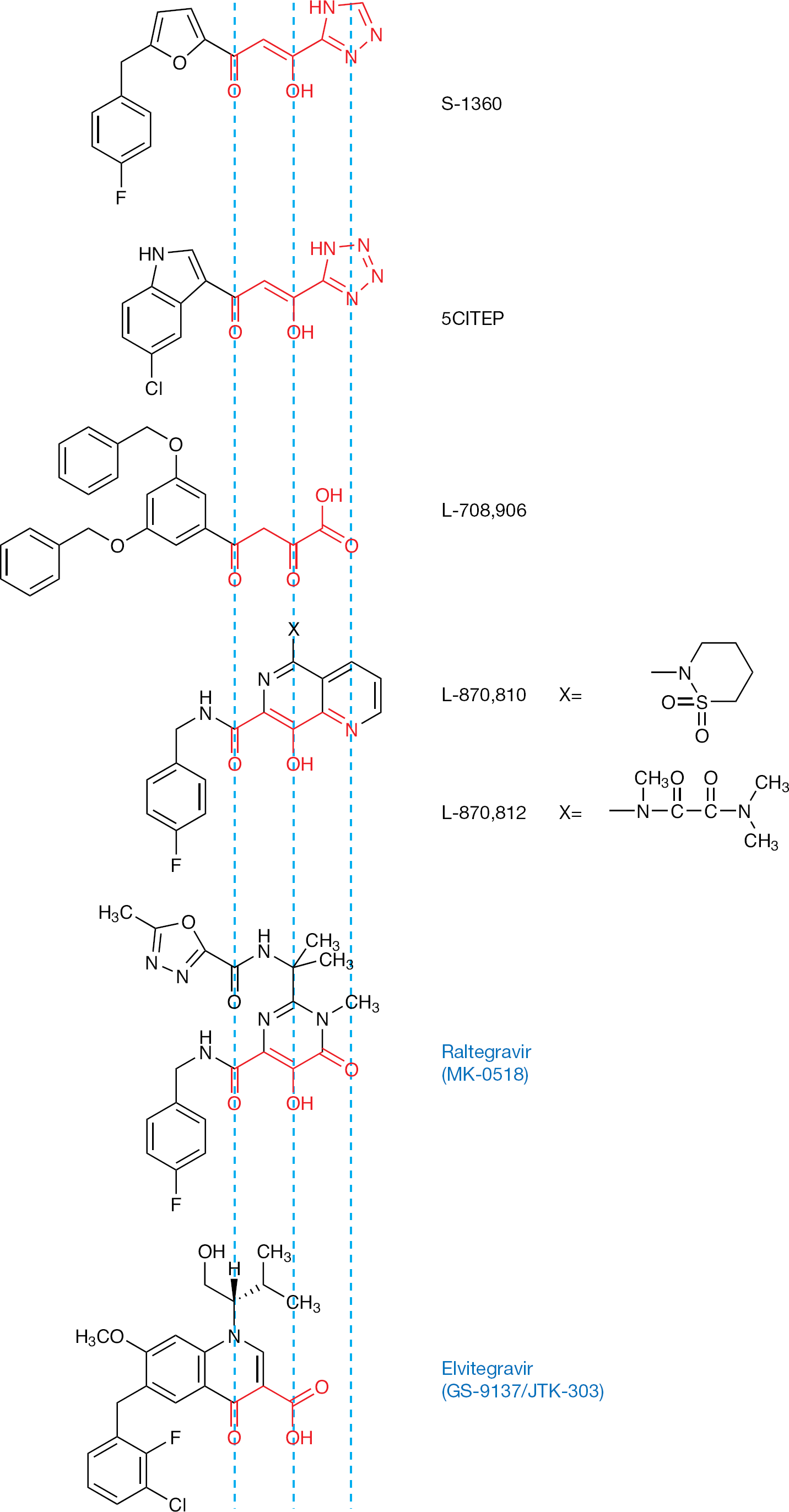
Structures of integrase inhibitors
Anti-HIV activity of EVG
EVG shows potent anti-HIV activity to various HIV strains that is comparable to other IN inhibitors, such as L-870,810 and RAL [13]. EVG suppresses replication of various HIV-1 subtypes with 50% effective concentration (EC50) values in the nanomolar to subnanomolar range, including clinical isolates that have NRTI, NNRTI and/or PI resistance mutations [14]. EVG also shows antiviral activity against HIV type-2 (HIV-2), such as ROD and EHO (subtypes A and B, respectively) strains with similar EC50 values to that observed for HIV-1 [14]. All 14 HIV-2 clinical isolates derived from IN-inhibitor-naive patients were susceptible to EVG with EC50 values ranging from 0.3 to 0.9 nM [15]. It has been described that the fusion inhibitor, T-20 (enfuvirtide), as well as most NNRTIs, minimally suppress replication of HIV-2 [16,17]. In contrast to these inhibitors, EVG is substantially active against HIV-2, indicating that EVG and other IN inhibitors are ideal for the treatment of both HIV-1 and HIV-2.
Mechanism of action
Integration is the crucial last step for the establishment of irreversible viral infection. The integration reaction consists of three sequential steps: firstly, two nucleotides located at the 3′-terminal end of the reverse transcribed HIV complementary DNA are removed (3′-processing); secondly, the processed HIV genome is integrated into the host chromosome (strand transfer); and lastly, the gap between virus and host DNA is thought to be filled by host repair machinery [18].
Using an enzymatic strand transfer assay in vitro, EVG has an enzymatic 50% inhibitory concentration (IC50) value of 54 nM [14]. EVG also inhibits the 3′-processing reaction, but the IC50 value of EVG for this reaction is much higher than that of strand transfer (150–300-fold) [19]. These results indicate that EVG exerts antiviral activity primarily by selective blockage of the strand transfer reaction.
An in silico docking simulation of IN with EVG has been performed by Savarino [20]; however, the precise binding site of EVG for IN has not yet been identified as cocrystallization of IN with EVG has not been reported. Recently, Marinello et al. [19] estimated the binding mode of EVG using an in vitro oligonucleotide-based assay. The authors postulated that EVG preferentially inhibits the strand transfer step as EVG recognizes the IN–DNA complex and binds the interface between 3′-processed viral DNA and IN.
Most IN inhibitors, including EVG and RAL, preferentially block strand transfer reactions; however, there are several compounds that inhibit another IN-catalyzing reaction, 3′-processing. For example, INH-I001/VNIII-083, one of the recently developed IN inhibitors, is a conceptually new β-diketo acid derivative and inhibits 3′-processing in addition to strand transfer [21,22]. It is important to keep developing IN inhibitors that target not only strand transfer reaction, but also 3′-processing and other steps involving in integration, such as IN interacting molecules.
Clinical efficacy of EVG
Anti-HIV activity of EVG in HIV-1-infected patients was initially evaluated in a Phase Ib clinical trial. In this 10-day EVG monotherapy trial, 40 HIV-1-infected patients were administrated one of five dosage groups as follows: 200 mg twice daily, 400 mg twice daily, 800 mg twice daily, 800 mg once daily or 50 mg EVG plus 100 mg ritonavir once daily. Ritonavir was used for boosting the half-life of EVG. In the 400 mg twice daily group, all patients showed >1 log10 copies/ml reduction of viral load and, moreover, half of the patients experienced a >2 log10 copies/ml decrease (Table 1) [9]. In the Phase II clinical trial, >90% patients of both groups of 50 mg plus ritonavir and 125 mg plus ritonavir decreased viral load by >1 log10 copies/ml, and mean CD4+ T-cell counts were increased compared with the control comparator protease inhibitor (Table 1) [10]. Through these trials, the potent activity of EVG was demonstrated. Currently, a Phase III study of the safety and efficacy of ritonavir-boosted EVG (EVG/r) has been initiated. The study is a multicentre, double-blind and randomized 48-week investigation. Participants receive 150 mg EVG/r once daily or 400 mg RAL twice daily, each with a background regimen, and are evaluated by the primary endpoint, which is defined as the achievement and maintenance of confirmed HIV-1 RNA levels <50 copies/ml [23].
Clinical efficacy of elvitegravir
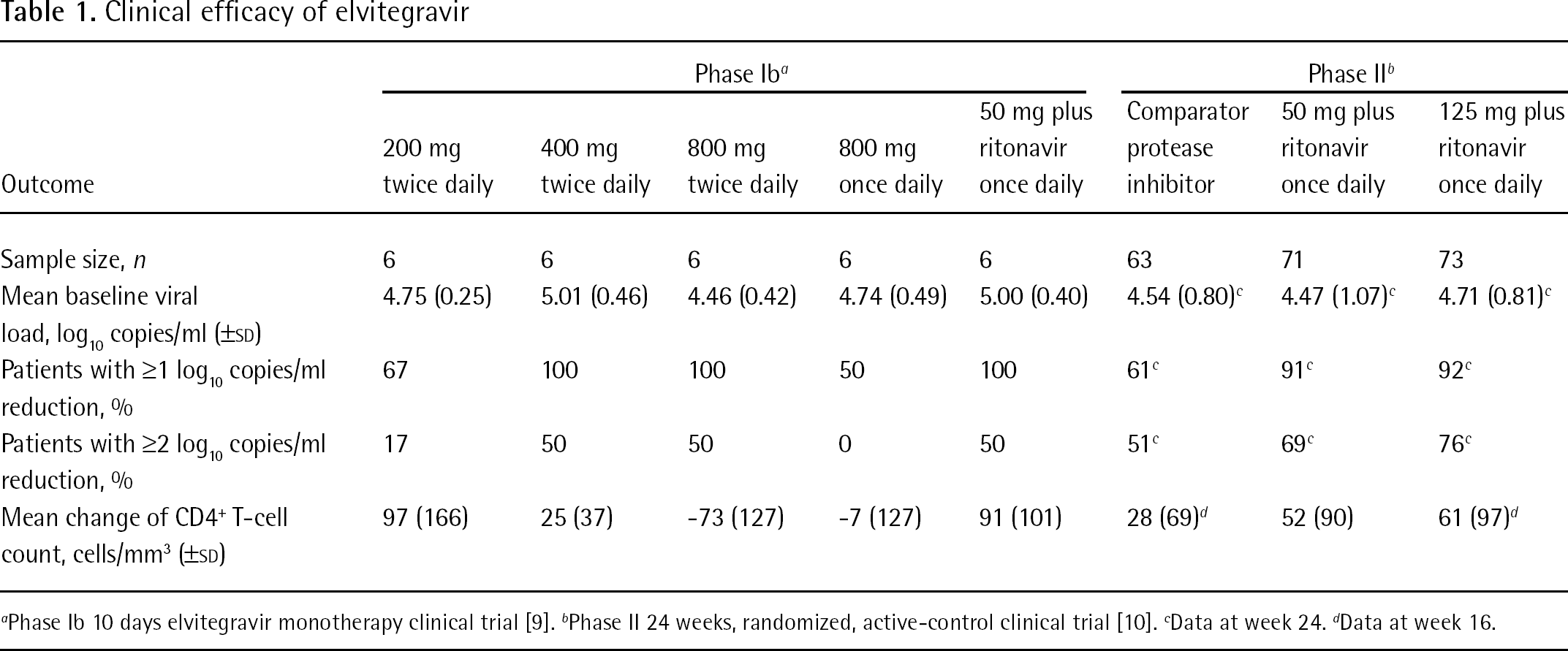
Phase Ib 10 days elvitegravir monotherapy clinical trial [9].
Phase II 24 weeks, randomized, active-control clinical trial [10].
Data at week 24.
Data at week 16.
Recently, Gilead Sciences (Foster City, CA, USA) announced that they were developing a CYP3A inhibitor, GS-9350, which has no antiviral activity but selectively inhibits CYP3A metabolism [24,25]. It has been reported that EVG is first oxidized by CYP3A4 and then glucuronidated by glucuronosyltransferase 1A1 and 1A3 in its metabolic pathway [26]; hence the rationale for combining EVG with ritonavir, which is known to be a CYP3A4 inhibitor (despite the fact that it was originally developed as a PI). It is expected that coadministration of EVG with GS-9350, instead of ritonavir, might reduce unfavourable adverse effects caused by ritonavir. Moreover, Gilead Sciences intend to use GS-9350 as part of a four-in-one single pill that includes EVG and two NRTIs (tenofovir disoproxil fumarate and emtricitabine) [24]. They plan to initiate a Phase II study in HIV-infected patients in the second quarter of 2009. This four-in-one single pill will contribute to the improvement in drug adherence.
In vitro resistance profile
Several experiments to select EVG-resistant variants have been performed by us and other groups. The EVG-selected mutations are summarized in Figure 2. We selected resistant variants to EVG that contained mutations in the IN-coding region (T66I/Q95K/E138K/Q146P/S147G and H51Y/E92Q/S147G/E157Q) by a dose-escalating method with HIV-1IIIB [14]. Among the selected mutations, T66I and E92Q, both of which were observed relatively early during the selection, mainly conferred EVG resistance with an EC50 change of 37-fold and 36-fold increase, respectively. Garvey et al. [27] selected T66(A/I/K)/V72A/E92(Q/V)/T124A/A128T/P145S/Q146L/Q148(K/R)/V151I mutations by increasing concentrations of EVG with HIV-1IIIB. Among these mutations, E92Q, P145S, Q148K/R and V151I mutations, which emerged at various time points of the selection, each showed >10-fold resistance to EVG. Goethals et al. [28] also induced the resistant variants and selected D10E/R20K/T66(A/I)/L74M/E92Q/H114Y/A128T/E138K/Q148R/S230R mutations by dose-escalating and automated selection experiments with HIV-1IIIB. Among these mutations, T66I and E92Q, both of which were observed in early passages of the in vitro selection, and Q148R mutations conferred resistance to EVG (33-fold, 57-fold and 96-fold, respectively). Kobayashi et al. [13] observed the emergence of T66(A/I/K)/V72A/E92(Q/V)/T124A/A128T/P145S/Q146L/Q148(K/R)/V151I mutations in the IN-coding region of HIV-1IIIB and phenotypic analysis revealed that T66K, E92Q, P145S and Q148K/R mutations, which were observed at various time points of the selection, showed high resistance to EVG (>10-fold). These results indicate that mutations at positions T66, E92 and Q148 were preferentially selected in these three in vitro inductions and confer high resistance to EVG as primary mutations.
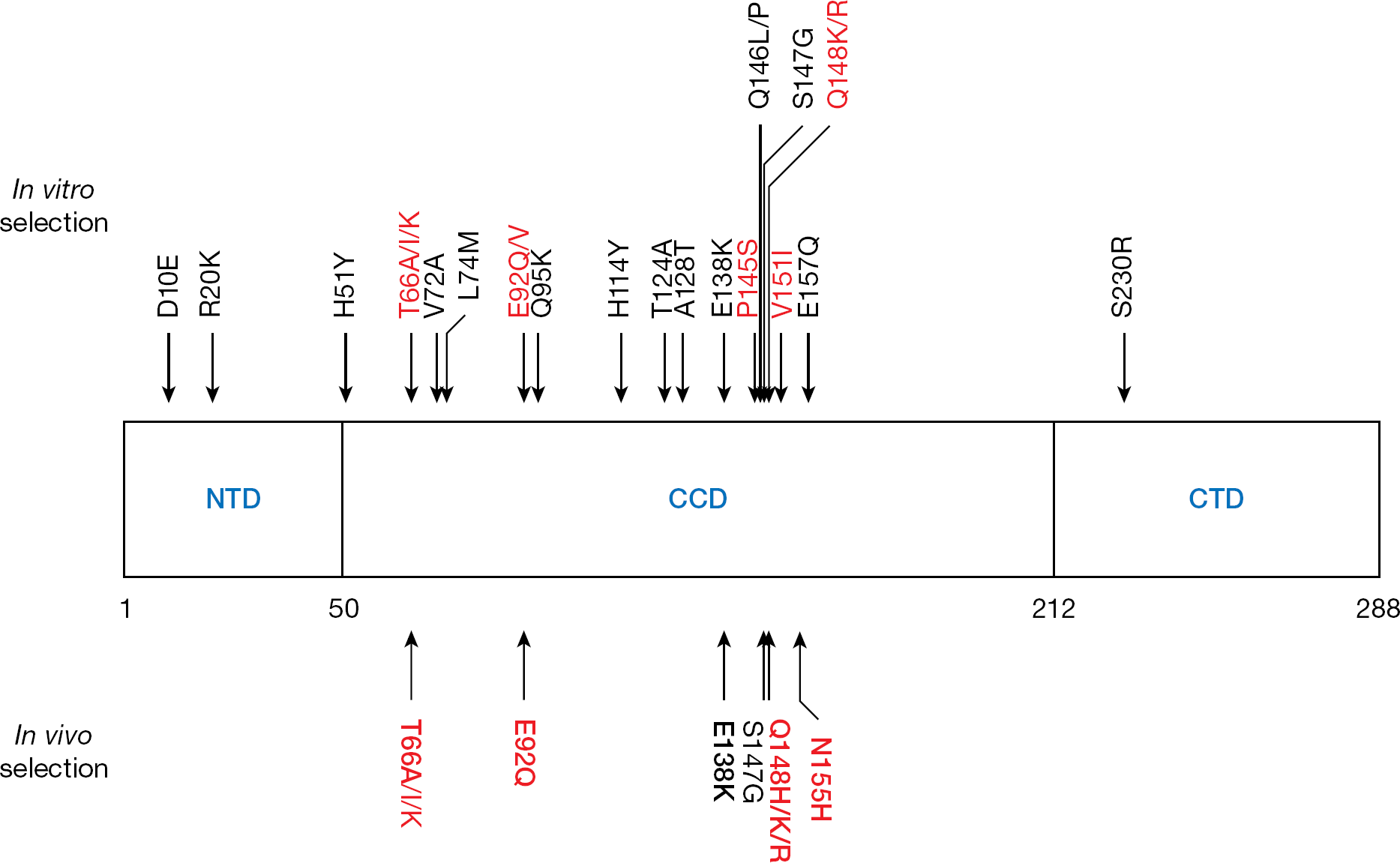
Mutation map for integrase inhibitors
In vivo resistance profile
Recently, the Phase II clinical trial of EVG (GS-US-183-0105) has been completed. From the data of virological failure (VF) in patients administrated with EVG, the in vivo resistance profile has been identified. T66I, E92Q and Q148K/R, which were confirmed as primary mutations for EVG resistance in vitro, were also observed among the VF patients (Figure 2) [29]. An amino acid substitution of glutamine to histidine at position 148 (Q148H) was newly observed in vivo. N155H, which was not induced during in vitro selection by EVG, was observed in vivo with a frequency of 39% of total VF [29]. Importantly, E92Q, Q148H/K/R and N155H mutations were also observed in HIV-infected patients experiencing VF on RAL (Figure 2) [8,30]. These results indicate a potential for cross-resistance between these IN inhibitors. Indeed, it has been reported that viruses from EVG VF that acquire a high resistance to EVG show cross-resistance to RAL (>28-fold resistance) in vitro [29]. The effect of resistant mutations on the replication kinetics has also been evaluated in vitro and in vivo. The in vitro fitness of HIV-1 harbouring EVG resistance mutations is relatively low: <20–86% reported by Shimura et al. [14] and 40–80% reported by Goethals et al. [28]. A similar result was observed in vivo: replication kinetics of viruses derived from EVG VF was 54% compared with wild type [29].
Effect of IN polymorphisms on the efficacy of EVG
One of the major obstacles to effective antiretroviral therapy is the emergence of resistant variants. To maximize the antiviral activities of inhibitors, genotyping assays are widely used to detect resistance mutations [31]. In addition, prior to initiation of antiretroviral therapy, the presence of natural polymorphisms are also a considerable factor for efficacious therapy [32,33]. The IN genes of a total of 243 HIV-1 clade B infected patients who had not received any IN inhibitors were analysed [34]. EVG-resistance-related polymorphisms at positions 66 and 92 were not observed, whereas Q148H and N155H polymorphisms were observed with a frequency of 0.4%, indicating that IN inhibitors (such as RAL and EVG) might show a reduced effect on patients who are infected with HIV-1 harbouring these polymorphisms. In contrast to HIV-1, no EVG-resistance-related polymorphism was observed in 11 HIV-2 isolates (10 subtype A and 1 subtype B) [35] and 52 HIV-2 clinical isolates [15], although the number of isolates tested was somewhat small.
Antiretroviral and lentiviral activity of EVG
The activity of EVG on viruses other than HIV has also been investigated. Simian immunodeficiency virus (SIV) is also susceptible to EVG and other IN inhibitors. EVG L-870,810 and prototype IN inhibitor L-708,906, which has weak antiviral activity compared with recently developed IN inhibitors, suppress SIV replication in a cell-based assay with similar effectiveness compared with that of HIV (Table 2) [14]. The anti-SIV effect of L-870,812 was further confirmed in a Simian–human chimeric immunodeficiency virus (SHIV)-infected rhesus macaque model. L-870,812 prevented viraemia and maintained levels of CD4+ T-cells in SHIV-infected monkeys [36]. Surprisingly, in addition to mammalian lentiviruses such as HIV and SIV, EVG shows antiviral activity against murine leukemia virus (MLV), although a higher concentration is necessary to suppress the replication of MLV than HIV (Table 2) [14]. These results suggest that IN inhibitors effectively inhibit various retroviruses and lentiviruses. Indeed, it has been reported that L-870,810 increases the 2-long terminal repeat product in feline immunodeficiency virus infection, which arises from defective integration (Table 2) [37]. Taken together, IN inhibitor is a promising therapeutic agent for a wide variety of both retroviruses and lentiviruses.
In vitro antiretroviral and lentiviral activity of integrase inhibitors
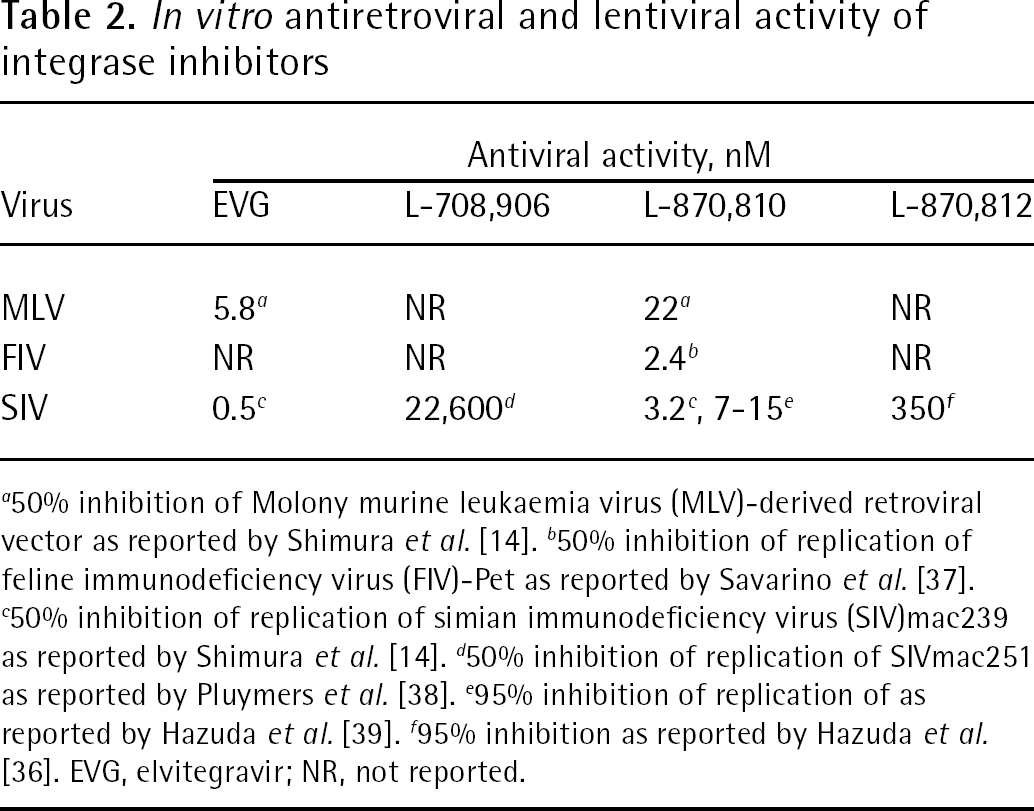
50% inhibition of Molony murine leukaemia virus (MLV)-derived retroviral vector as reported by Shimura et al. [14].
50% inhibition of replication of feline immunodeficiency virus (FIV)-Pet as reported by Savarino et al. [37].
50% inhibition of replication of simian immunodeficiency virus (SIV)mac239 as reported by Shimura et al. [14].
50% inhibition of replication of SIVmac251 as reported by Pluymers et al. [38].
95% inhibition of replication of as reported by Hazuda et al. [39].
95% inhibition as reported by Hazuda et al. [36]. EVG, elvitegravir; NR, not reported.
Conclusions
EVG is currently being studied in a Phase III clinical trial. Once-a-day administration of EVG potently suppresses HIV replication in HIV-infected patients. However, resistance mutations observed after VF in patients on EVG are also observed in patients who fail RAL, indicating cross-resistance between these inhibitors. EVG can inhibit the replication of several retroviruses and lentiviruses, suggesting that EVG is a promising inhibitor against IN-harbouring viruses.
Footnotes
Acknowledgements
This work was supported, in part, by a grant for the Promotion of AIDS Research from the Ministry of Health and Welfare of Japan (ENK) and a grant for Research for Health Science Focusing on Drug Innovation from the Japan Health Science Foundation (ENK). KS was supported by the 21st Century COE Program of the Ministry of Education, Culture, Sports, Science, and Technology (Japan). KS is a research fellow of the Japan Health Sciences Foundation.
The authors declare no competing interests.