
Abstract
Background:
A number of compounds were examined for their inhibitory effect on bovine viral diarrhoea virus (BVDV) replication in cell cultures and found that some cyclooxygenase (COX) inhibitors had antiviral activity against the virus.
Methods:
Determination of compounds for their anti-BVDV activity was on the basis of the inhibition of virus-induced cytopathogenicity in Mardin–Darby bovine kidney (MDBK) cells. Anti-hepatitis C virus (HCV) activity was assessed by the inhibition of viral RNA synthesis in the subgenomic HCV RNA replicon cells.
Results:
Among the test compounds, 5-(4-chlorophenyl)-1-(4-methoxyphenyl)-3-(trifluoromethyl)-1H-pyrazole (SC-560) was the most active against BVDV, and its 50% effective and cytotoxic concentrations were 10.9 ±2.8 and 93.9 ±24.5 μM in virus and mock-infected MDBK cells, respectively. The compound also suppressed BVDV RNA synthesis in a dose-dependent fashion. Studies on the mechanism of action revealed that SC-560 did not interfere with viral entry to the host cells. Furthermore, it was assumed that the antiviral activity of SC-560 was not associated with its inhibitory effect on COX. The combination of SC-560 and interferon-α was additive to synergistic in inhibiting BVDV replication. More importantly, the compound proved to be a selective inhibitor of HCV replication.
Conclusions:
SC-560 and its derivative might have potential as novel antiviral agents against HCV.
Introduction
Hepatitis C virus (HCV), a member of the hepacivirus genus from the family Flaviviridae, is a major aetiological agent of human liver diseases. The World Health Organization estimates that at least 170 million people in the world are chronically infected with HCV. Although HCV infection is often asymptomatic, it frequently causes chronic hepatitis, which progresses to end-stage liver diseases, such as liver cirrhosis and hepatocellular carcinoma [1]. No vaccine is currently available, and treatment with the nucleoside analogue ribavirin and pegylated interferon (PEG-IFN)-α has efficacy in a subset of patients. However, there is genetic heterogeneity in the HCV genome [2] and HCV genotype 1b cannot be eliminated from approximately half of the patients, even when treated with PEG-IFN-α and ribavirin [3]. In addition, side effects of these agents are sometimes serious and cannot be tolerated in some patients; therefore, alternative agents are highly desired for the treatment of HCV infection.
Because a lot of effort has been made to identify novel anti-HCV agents, several compounds have been developed and are currently under clinical trials [4]; however, progress is not rapid enough because of the inability of HCV replication in cell culture systems. The recent development of the subgenomic HCV RNA replicon system greatly accelerated the speed of anti-HCV drug discovery [5]; nevertheless, the system does not faithfully reproduce all steps in the HCV replication cycle. In addition, relatively high costs are required to use this technology for the screening and development of antiviral agents. Although a cell culture system of productive HCV infection with the replication-competent strain JFH-1 has been established in 2005, which makes it possible to identify inhibitors of every step in the virus replication cycle [6–8], this strain was isolated from a patient of fulminant hepatitis C and classified as genotype 2a with several mutations. Consequently, surrogate viruses are still widely used for the investigation of anti-HCV agents.
Bovine viral diarrhoea virus (BVDV) is considered as a surrogate model of HCV because HCV and BVDV share a significant degree of homology and common replication machinery [9]. BVDV, a member of the pestivirus genus of the family Flaviviridae, causes mucosal diseases in cattle. BVDV is easy to grow in cell culture. The genome consists of a positive-strand RNA approximately 12.5 kb in length that encodes a single open reading frame flanked by 5′ and 3′ untranslated regions. Similarly to the HCV genome, the 5′ terminus of the BVDV genome is not capped; instead, the initiation of translation is mediated by an internal ribosomal entry site [10]. Based on the effect on host cells, two biotypes of BVDV (cytopathogenic and non-cytopathogenic) have been recognized [11]. Common strains of BVDV, such as NADL, are cytopathogenic in cell culture, allowing the evaluation of compounds for their antiviral activity. Thus, BVDV has been most widely utilized in vitro as an HCV surrogate model for identification and characterization of antiviral agents against HCV [9].
We previously reported a simple and sensitive colorimetric assay of compounds for evaluation of their anti-BVDV activity in vitro [12]. Using this assay system, we have now found that some cyclooxygenase (COX) inhibitors are selective inhibitors of BVDV replication in cell culture. Among the compounds, 5-(4-chlorophenyl)-1-(4-methoxyphenyl)-3-(trifluoromethyl)-1H-pyrazole SC-560; Figure 1) was found to be the most potent inhibitor. Studies on its mechanism of action revealed that the compound did not interfere with viral entry to the host cells, but did suppress the viral RNA synthesis. Furthermore, HCV replication was also selectively inhibited by SC-560.
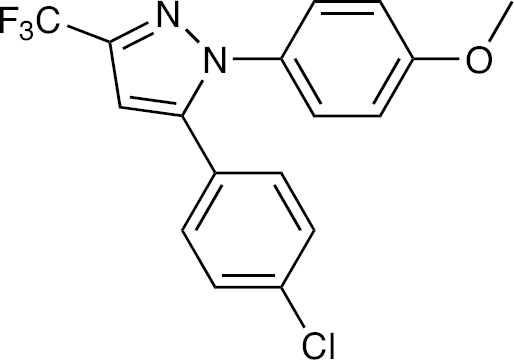
Structure of SC-560
Methods
Compounds
A total of 16 COX inhibitors, that is, aspirin, CAY10404, diclofenac, etodolac, ibuprofen, indomethacin, mefenamic acid, nabumetone, nimesulide, NS-398, phenylbutazone, piroxicam, resveratrol, SC-560, SC-58125 and vareoyl salicylate, and two reference compounds, cyclosporine A and ribavrin, were used for antiviral assays. Among the compounds, aspirin, CAY10404, NS-398, resveratrol, SC-560, SC-58125 and valeroyl salicylate were obtained from Cayman Chemical (Ann Arbor, MI, USA), and the other COX inhibitors and cyclosporine A were purchased from Sigma (Saint Louis, MO, USA). Ribavirin was synthesized by Asahi Kasei Pharma (Tokyo, Japan). All compounds were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 20 mM to avoid any antiviral and cytotoxic effects of DMSO. The stock solution was stored at −20°C until use. Human IFN-α2b was purchased from PBL Biochemical Laboratories (New Brunswick, NJ, USA) and stored at −80°C.
Cells and virus
Madin–Darby bovine kidney (MDBK) cells were purchased from Japan Health Sciences Foundation (Health Science Research Resources Bank, Osaka, Japan). MDBK cells were grown and maintained in Dulbecco's modified Eagle's medium (DMEM) with high glucose (Gibco/BRL, Grand Island, NY, USA). The medium was supplemented with 10% heat-inactivated horse serum (Gibco/BRL), 100 units/ml penicillin G and 100 μg/ml streptomycin. The cells were certified as BVDV contamination-negative. The cytopathogenic BVDV strain Nose was obtained from Kyoto Biken (Kyoto, Japan). BVDV was harvested from culture supernatants of virus-infected cells after a 3-day incubation. Virus stocks were stored at −80°C until use. The infectivity of the stocks was determined in MDBK cells and expressed as the 50% tissue culture infectious dose. The subgenomic HCV RNA replicon cells MH-14 were as described previously [13,14]. The replicon cells were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum (Gibco/BRL), 500 μg/ml G418 (Sigma) and the antibiotics listed earlier.
Anti-BVDV assays
Determination of compounds for their anti-BVDV activity was on the basis of the inhibition of virus-induced cytopathogenicity in MDBK cells, as described previously [12]. Briefly, the cells (2×105 cells/ml) were infected with BVDV at a multiplicity of infection (MOI) of 0.01 and 100 μl of the cell suspension was added into each well of a microtitre plate. The cells were incubated in the presence of various concentrations of test compounds (each compound alone or in combination with IFN-α) for 3 days at 37°C. After incubation, culture supernatants were collected to determine their lactate dehydrogenase (LDH) levels by an LDH detection kit according to the manufacturer's instructions (Takara Biochemicals, Otsu, Japan). The cytotoxicity of the test compounds was evaluated in parallel with their antiviral activity. The mock-infected MDBK cells (2×104 cells/well) were incubated in the presence of various concentrations of test compounds for 3 days. The cell viability was determined by a dye method using the water-soluble tetrazolium Tetracolor One® (Seikagaku Corporation, Tokyo, Japan).
The anti-BVDV activity of SC-560 was also determined by the inhibition of viral RNA synthesis in MDBK cells by real-time reverse transcription (RT)-PCR. The cells were infected with BVDV at a MOI of 1.0 and cultured in the presence of various concentrations of the test compound. After a 6 h incubation, the cells were extensively washed with phosphate-buffered saline (PBS), trypsinized and then washed again with PBS. Total RNA was extracted from the cells with a RNeasy Mini Kit® (Qiagen, Valencia, CA, USA) and subjected to real-time RT-PCR. The BVDV RNA level was determined using the sense primer 5′-TGGTCCGACGCCTTAGTATAAAGG-3′, the antisense primer 5′-GGCTGTATTCGTAACAGTTGGTTAAA-3′ and the fluorescence probe 5′-ACGAGGGCACGCCCAAAGCA-3′ (Applied Biosystems, Branchburg, NJ, USA). The primer pair amplifies the 5′ unsatulated region of BVDV RNA. The TaqMan® PCR reagent kit and TaqMan® Multiscribe™ RT reagent kit (Applied Biosystems) were used according to the manufacturer's instructions. Non-specific inhibition of host cellular messenger RNA synthesis by SC-560 was determined by amplification of a part of the bovine β-actin RNA using the sense primer 5′-GCCCTGAGGCTCTCTTCCA-3′, the antisense primer 5′-GCGGATGTCGACGTCACA-3′ and the fluorescence probe 5′-CATGGAATCCTGCGGCATTCACG-3′ (Applied Biosystems).
Anti-HCV assays
The anti-HCV activity of SC-560 was determined by the inhibition of viral RNA synthesis in subgenomic HCV RNA replicon cells by real-time RT-PCR as described previously [13,14]. Briefly, MH-14 cells (7×103 cells/12-well plate) were seeded and cultured in the presence of various concentrations of the test compound. Every 3 days, the culture medium was replaced by fresh culture medium containing an appropriate concentration of the compound. On day 7, total RNA was extracted from the cells and subjected to real-time RT-PCR. The 5′ untranslated region of HCV RNA was quantified using the sense primer 5′-CGGGAGAGCCATAGTGG-3′, the antisense primer 5′-AGTACCACAAGGCCTTTCG-3′ and the fluorescence probe 5′-CTGCGGAACCGGTGAGTACAC-3′ (Applied Biosystems). As an internal control, ribosomal RNA was also quantified using TaqMan® ribosomal RNA control reagents (Applied Biosystems).
Synergy calculation and statistical analyses
The multidrug effect was evaluated by the median-effect principle and the isobologram method as previously described [15,16]. The combination index (CI) was obtained by this method. CIs of <1, 1 and >1 indicate synergism, additive effect and antagonism, respectively. The statistical significance was determined by the Student's t-test. P-values <0.05 were considered to be significant.
Results
When 16 COX inhibitors were examined for their inhibitory effect on BVDV replication in MDBK cells, 8 compounds (diclofenac, indomethacin, mefenamic acid, nabumetone, NS-398, resveratrol, SC-560 and SC-58125) displayed selective inhibition of virus-induced cytopathogenicity (Table 1). With the exception of CAY10404, resveratrol and SC-560, these compounds did not reduce the viability of mock-infected MDBK cells at concentrations up to 100 μM. Among the active compounds, SC-560 (Figure 1), proved to be the most potent inhibitor of the virus. Its mean ±SD 50% effective concentration (EC50) and 50% cytotoxic concentration (CC50) were 10.9 ±2.8 μM and 93.9 ±24.5 μM, respectively, indicating that SC-560 is a selective inhibitor of BVDV. Cyclosporine A and ribavirin, both of which are known to inhibit HCV replication in cell cultures [13,14,17,18], also inhibited BVDV replication in this study. The mean ±SD EC50 values of cyclosporine A and ribavirin were 2.8 ±0.2 μM and 3.9 ±0.4 μM, respectively (Table 1); however, these compounds were more cytotoxic to the host cells than SC-560.
Anti-BVDV activity of COX inhibitors in MDBK cells
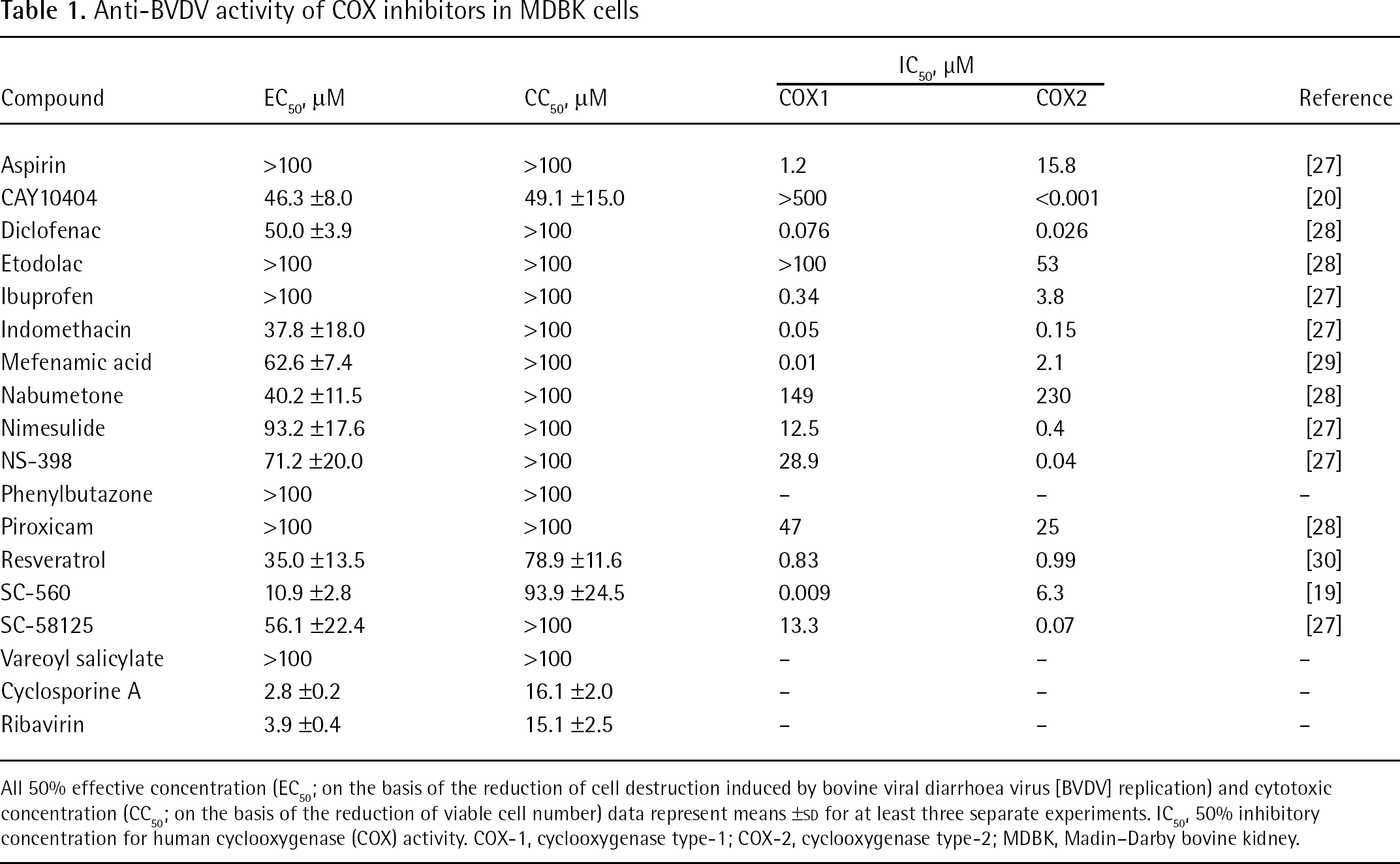
All 50% effective concentration (EC50; on the basis of the reduction of cell destruction induced by bovine viral diarrhoea virus [BVDV] replication) and cytotoxic concentration (CC50; on the basis of the reduction of viable cell number) data represent means ±SD for at least three separate experiments. IC50, 50% inhibitory concentration for human cyclooxygenase (COX) activity. COX-1, cyclooxygenase type-1; COX-2, cyclooxygenase type-2; MDBK, Madin–Darby bovine kidney.
SC-560 was found to almost completely inhibit BVDV replication at a concentration of 40 μM, and it did not show apparent cytotoxicity at this concentration (Figure 2A). The selective inhibition of BVDV replication by SC-560 was confirmed by dose-dependent inhibition of viral RNA synthesis in the infected cells, as determined by real-time RT-PCR (Figure 2B). In this experiment, almost complete inhibition of viral RNA synthesis was achieved by the compound without affecting the steady state level of β-actin RNA in MDBK cells at a concentration of 50 μM.
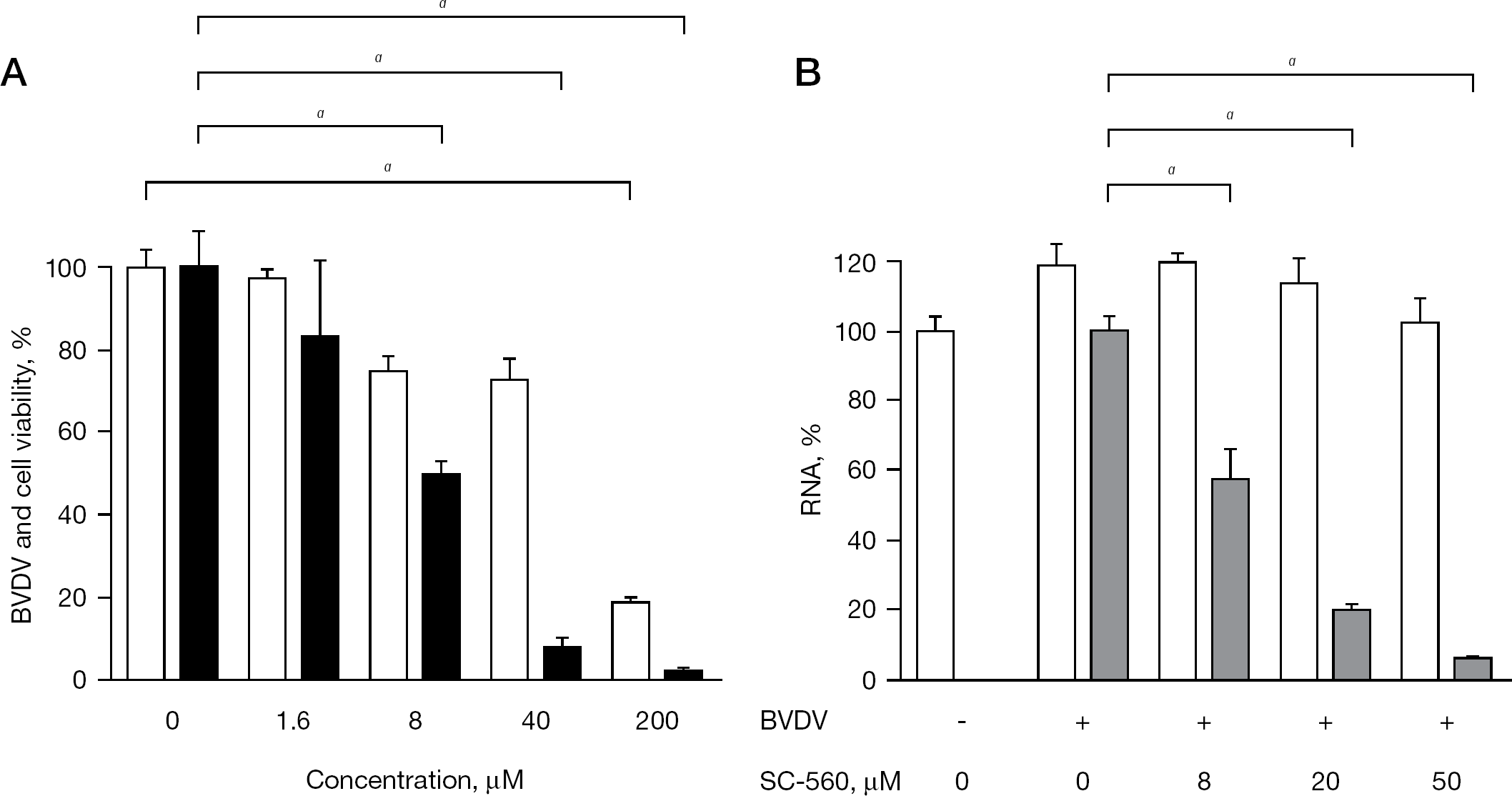
Inhibitory effect of SC-560 on BVDV replication in MDBK cells
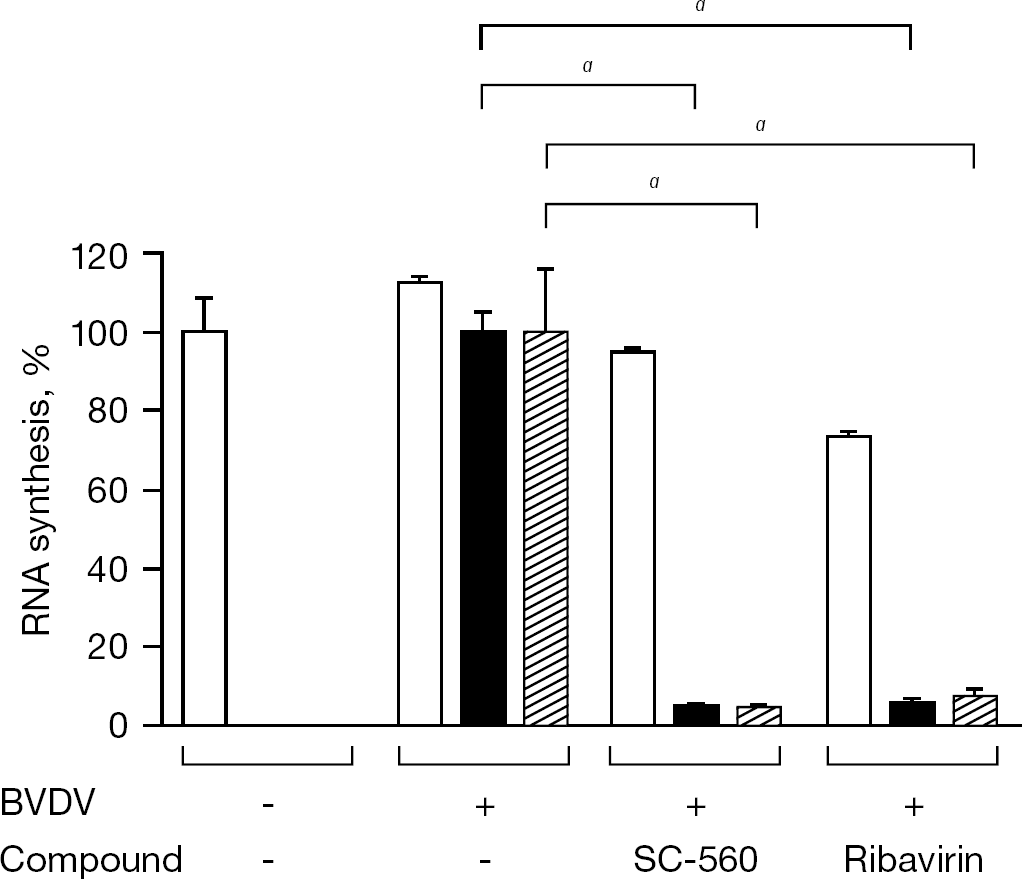
To gain insight into the mechanism of BVDV inhibition by SC-560, an experiment of delayed compound addition was conducted. When SC-560 (50 μM) was added to the cells simultaneously with the virus, almost complete inhibition of viral RNA synthesis was achieved (Figure 3A and 2B). A similar degree of the inhibition was observed, even when the compound was added at 2 h after virus infection (Figure 3B), indicating that SC-560 does not exert its anti-BVDV activity through the inhibition of viral entry to the host cells. In addition, like ribavirin, SC-560 could inhibit the negative-strand viral RNA synthesis (Figure 4).
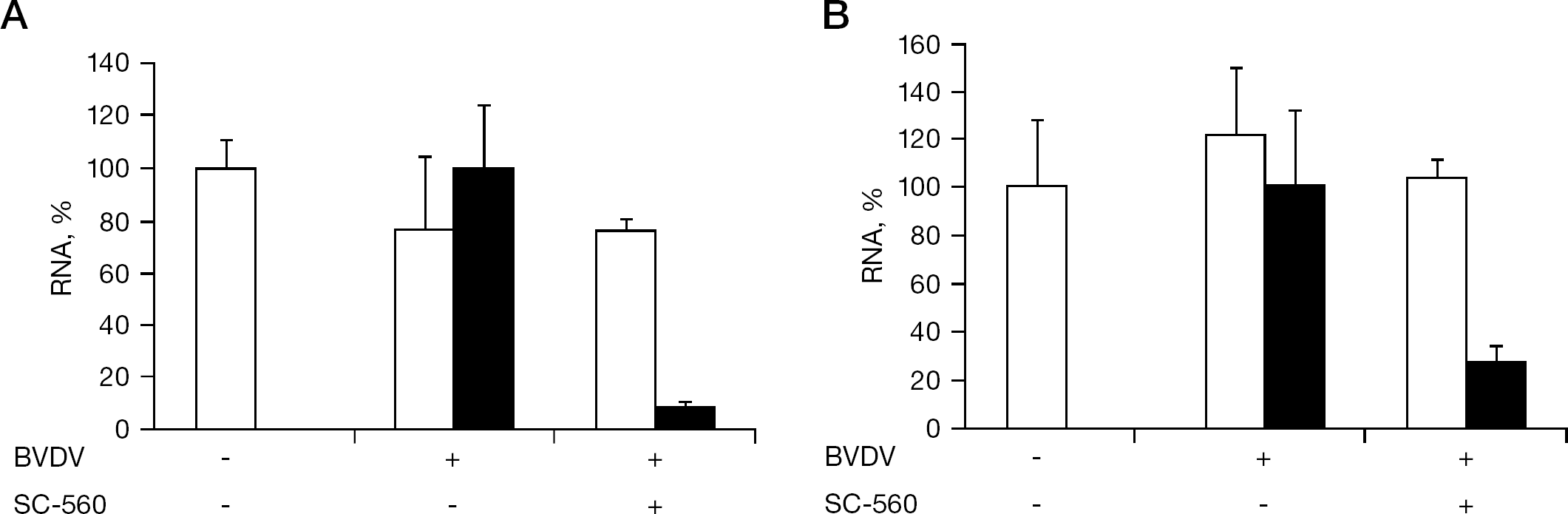
Inhibitory effect of SC-560 on BVDV replication after viral entry
Inhibitory effect of SC-560 on the BVDV negative-strand RNA synthesis
Combination antiviral activity of SC-560 and IFN-α was examined by a cytopathogenicity inhibition assay. Prior to the combination experiment, the optimal concentration ratio of the two compounds was determined (data not shown). Although SC-560 and IFN-α achieved 27.3% and 43.3% inhibition at a concentration of 6.25 μM and 0.625 IU/ml, respectively, their combination resulted in 63.6% inhibition (Figure 5). Furthermore, >90% inhibition of BVDV replication was recorded using the combination of 12.5 μM SC-560 and 1.25 IU/ml IFN-α. This combination did not generate any cytotoxicity to the host cells (data not shown). The CIs were 0.99, 0.73 and 0.46 at a level of 50%, 70% and 90% inhibition of BVDV replication, respectively, indicating synergism at higher inhibition levels.
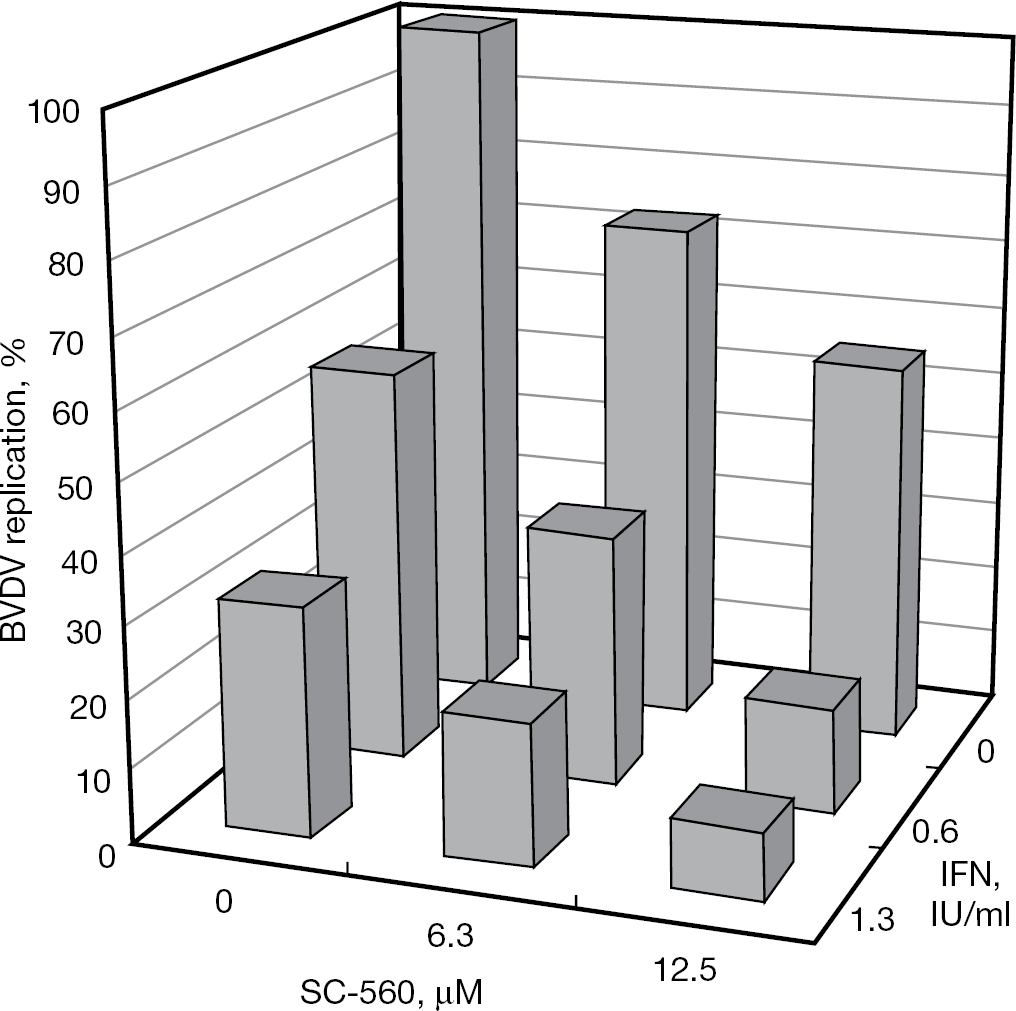
Inhibitory effect of SC-560 in combination with IFN-α on BVDV replication in MDBK cells
It seems important to know whether SC-560 is inhibitory to HCV replication. When the compound was examined for its inhibitory effect on HCV replication in MH-14 cells, dose-dependent suppression of HCV RNA was observed (Figure 6A). In particular, SC-560 significantly reduced the amount of intracellular HCV RNA at a concentration of 30 μM without apparent cytotoxicity to the replicon cells, indicating that the compound is a selective inhibitor of HCV replication. Furthermore, approximately 80% inhibition of HCV replication was achieved by 30 μM SC-560 in combination with 10 IU/ml IFN-α (Figure 6B).
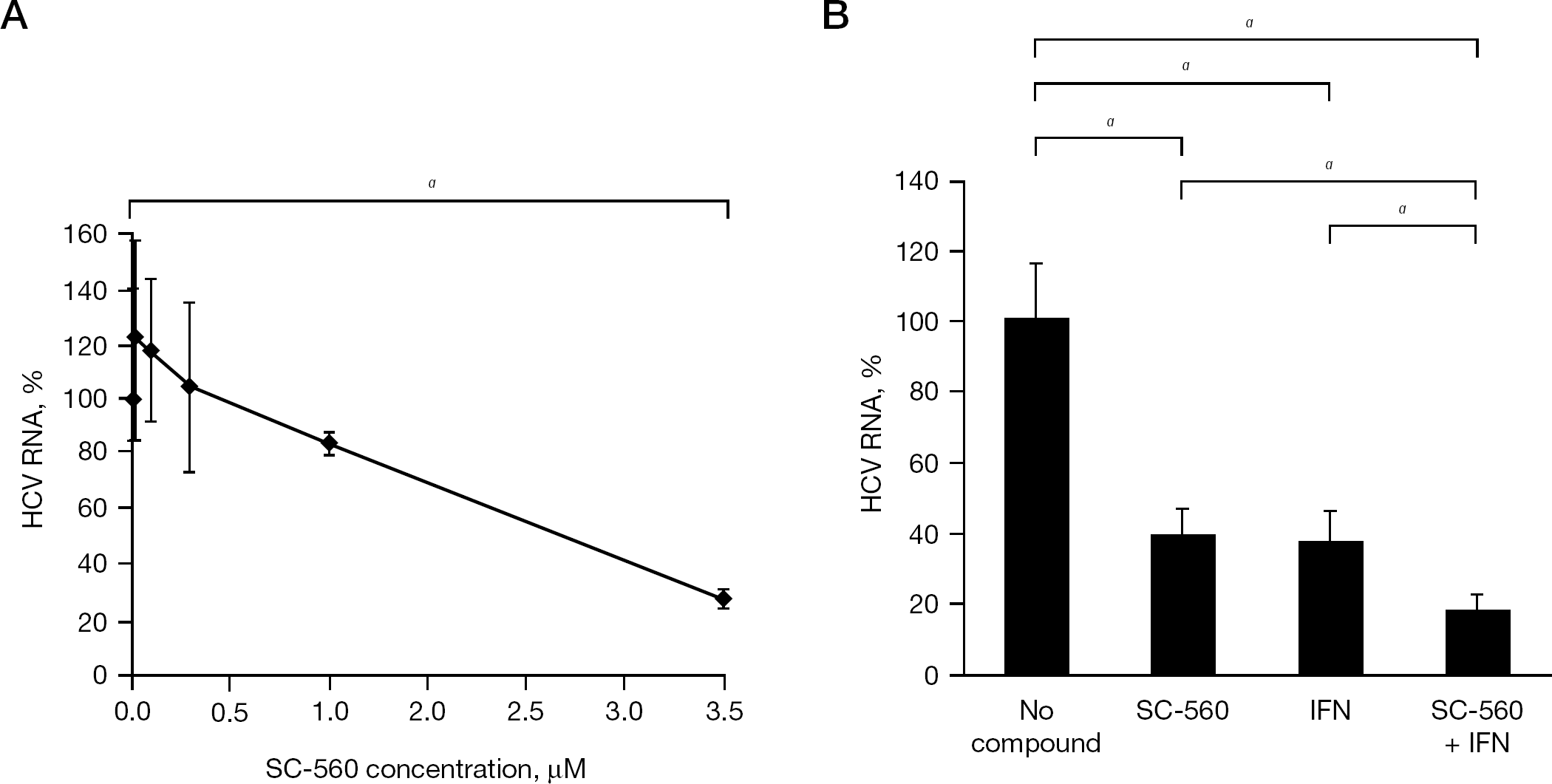
Inhibitory effect of SC-560 on HCV replication in subgenomic HCV RNA replicon cells
Discussion
SC-560 is a member of the diaryl heterocycle class of COX inhibitors, which include the selective COX type-2 (COX-2) inhibitors, celecoxib and rofecoxib. Unlike these inhibitors, SC-560 is a highly selective inhibitor of COX type-1 (COX-1). Using human recombinant enzymes, the 50% inhibitory concentrations (IC50) values of SC-560 for COX-1 and COX-2 were 0.009 μM and 6.3 μM, respectively [19]. We did not examine celecoxib and rofecoxib for their antiviral activity against BVDV and HCV; however, it is assumed that the antiviral activity of SC-560 is not associated with its inhibitory effect on COX-1 or COX-2 as there is no correlation between COX inhibition and antiviral activity (Table 1). For instance, CAY10404 is one of the most potent and selective COX-2 inhibitors reported to date. Its IC50 values for COX-1 and COX-2 were >500 μM and <1 nM, respectively [20], yet this compound did not show any selective inhibition of BVDV replication (Table 1). Furthermore, the COX-1-specific inhibitors ibuprofen and mefenamic acid were not active against BVDV.
It was recently reported that a triaryl pyrazoline compound, which has a similar chemical structure to SC-560, inhibited West Nile virus replication with an EC50 of 28 μM [21]. This compound did not inhibit viral entry or virion assembly but it did specifically suppress viral RNA synthesis. Although the compound was not tested for BVDV and HCV, it was active against other flaviviruses, such as dengue, yellow fever and Saint Louis encephalitis viruses. In our study, SC-560 did not inhibit viral entry but did inhibit BVDV and HCV RNA synthesis. As its anti-HCV activity was determined in HCV RNA replicon cells, the inhibition of virion assembly could also be excluded as the target of SC-560. Paeshuyse et al. [22] reported that two structurally unrelated compounds, BPIP (5-[(4-bromophenyl)methyl]-2-phenyl-5H-imidazo[4,5-c]pyridine) and VP32947, were highly potent and selective inhibitors of BVDV. Both compounds inhibited BVDV replication and RNA synthesis at nM concentrations; however, in contrast to SC-560, BPIP and VP32947 did not show any selective inhibition of HCV replication. Studies on the mechanism of action revealed that both BPIP and VP32947 targeted the same region of the BVDV RNA-dependent RNA polymerase.
The nonstructural protein 5B (NS5B) is the virus-encoded RNA-dependent RNA polymerase and is a major target for inhibition of anti-HCV agents [23,24]. Buckwold et al. [25] compared the complete amino acid sequences of NS5B between HCV and BVDV and found only 17.2% identity. Therefore, it is unsurprising that inhibitors such as BPIP and VP32947 did not show any activity against HCV replication. By contrast, ribavirin, an inosine mono-phosphate dehydrogenase inhibitor, has been shown to have antiviral activity against several RNA viruses [26]. It is hypothesized that ribavirin inhibits HCV replication by more than one mechanism, such as viral RNA transcription, elongation and cap formation and thus it might have broad-spectrum antiviral activity. Similar to ribavirin, SC-560 was found to be active against both BVDV and HCV in this study; however, because it is unlikely that SC-560 and ribavirin share the same target molecule for inhibition of these viruses, the target of SC-560 should be elucidated in future studies.
Another important issue to be elucidated is whether anti-COX activity of SC-560 can be dissociated from its antiviral activity by structural modification. A compound that inhibits COX-1 might generate serious side effects, even if it would have potent antiviral activity. Therefore, reducing COX activity as well as increasing antiviral potency seems indispensable for further development of this class of compounds as anti-HCV agents. Combination chemotherapy might be able to circumvent this problem by reducing a required dose of each compound.
In conclusion, the identification of SC-560 described herein represents the first step toward the future development of effective anti-HCV agents. Further studies are in progress to identify more potent and selective inhibitors of HCV replication.
Footnotes
Acknowledgements
We would like to thank Professors Tamotsu Kanzaki and Takuro Kanekura (Department of Dermatology, Graduate School of Medical and Dental Sciences, Kagoshima University, Kagoshima, Japan) for their valuable advice.
The authors declare no competing interests.