
Abstract
There are now 26 antiretroviral drugs and 6 fixed-dose combinations, including reverse transcriptase inhibitors, protease inhibitors, integrase inhibitors and fusion (or entry) inhibitors, approved by the US Food and Drug Administration for clinical use. Although they are clinically effective when used in combination, none of the existing drugs are considered ideal because of toxic side effects and the ascendance of inducing drug-resistant mutants. Development of new antiviral agents is essential. In the past decades, there has been great progress in understanding the structure of HIV type-1 (HIV-1) gp41 and the mechanism of HIV-1 entry into host cells. This opened up a promising avenue for rationally designed agents to interfere with this process. A number of fusion inhibitors have been developed to block HIV-1 replication. Enfuvirtide (T20) was one of those approved for clinical use. This signalled a new era in AIDS therapeutics. It is a synthetic polypeptide with potent inhibitory activity against HIV-1 infection. However, it is sensitive to proteolytic digestion and resistant virus strains are easily induced with multiple clinical use. One of the directions in designing new fusion inhibitors is to overcome these shortages. In the past years, large numbers of promising fusion inhibitory peptides have emerged. The antiviral activities are more potent or they can act differently from that of T20. Some of these new compounds have great potential to be further developed as therapeutic agents. This article reviewed some recent developments of these peptides and the possible role in anti-HIV-1 therapy.
Introduction
AIDS is caused by infection with HIV [1,2]. According to World Health Organization and Joint United Nations Programme on HIV/AIDS reports, by the end of the year 2007, more than 33.2 million people worldwide had been infected with HIV. Since the beginning of the epidemic, 25 million people have died of HIV-related complications. Above all, the dimensions of the epidemic remain staggering. In 2007 alone, approximately 33 million people were living with HIV; 2.7 million people were infected with the virus and 2 million people died of HIV-related complications [3]. This enormous human tragedy is also creating a serious impediment to the economic growth of many developing countries. Because of the lack of effective vaccines for the prevention of HIV infection, development of potent and affordable anti-HIV drugs is an international priority. Financial resources to support HIV projects have substantially increased in recent years. In the year 2007, US $10 billion were made available for HIV programmes from all sources.
To date, 26 antiretroviral drugs and 6 fixed-dose combinations have been approved by the US Food and Drug Administration (FDA) for the treatment of HIV type-1 (HIV-1) infection. These anti-HIV-1 drugs belong to five categories, including nucleoside reverse transcriptase inhibitors (NRTIs), non-NRTIs (NNRTIs), protease inhibitors (PIs), viral entry inhibitors and combinations of drugs [4] (Table 1).
Current anti-HIV type-1 drugs approved by the US Food and Drug Administration
NNRTIs, non-nucleoside reverse transcriptase inhibitors; NRTIs, nucleoside reverse transcriptase inhibitors; PIs, protease inhibitors.
In the past decade, development of a highly active antiretroviral therapy (HAART), with three to four anti-HIV-1 drugs used in combination, has markedly improved the life expectancy and greatly lengthened the progression course in HIV-1-infected patients. Antiretroviral therapy was also found to be a cost-effective or cost-saving intervention in high, middle and lower income countries [5]. It does not only support survival of the HIV-1 patient, but also enables them to be socially and economically active [6]. Despite their clinical effectiveness, none of the existing drugs can be considered as ideal because of toxic side effects and induction of drug-resistant mutants. The latter is of particular concern because once a viral mutant is found to be resistant to one drug, some level of cross-resistance resistance to other drugs within the same class is often developed [7,8].
Several steps in the HIV-1 replication cycle are potential targets for intervention. Anti-HIV-1 drugs are mainly designed to interfere with these targets. Currently, most approved anti-HIV-1 drugs focus on only two viral enzymes, namely reverse transcriptase and protease. Development of anti-HIV-1 drugs should not be restricted to these targets, and many potential targets should be explored. From a therapeutic perspective, viral entry is an attractive point for intervention in the viral life cycle because drug activity is independent of intracellular access. Although in the mucosal specificities of transmission and replications, where HIV-1 targets the cells that are usually CD4− but CCR5+, and where HIV-1 entry not only depends on gp120 but also on gp41, in this review, we will focus on the main HIV-1-targeted cells, CD4+ T-cells, and the entry process of HIV-1 into these cells. The HIV-1 entry into CD4+ T-cells has three discrete steps: HIV-1 Env glycoprotein gp120 attaches to the primary receptor CD4 on the host cell; gp120 engages with a coreceptor, CXCR4 or CCR5, on the host cell; and viral and cellular membrane fusion is mediated by HIV-1 transmembrane subunit gp41. These three steps are potential targets for the development of HIV-1 entry inhibitors. Based on these three steps, HIV-1 entry inhibitors can be divided into three categories: gp120-CD4 binding inhibitors, gp120-coreceptor binding inhibitors and fusion inhibitors. In this review, fusion inhibitor peptides blocking the gp41-mediated fusion will be discussed in detail [9–12].
In the early 1990s, the first anti-HIV-1 peptide SJ-2176 derived from the C-terminal heptad repeat (CHR or HR2) region was discovered by Jiang et al. [13,14]. Later, Wild et al. [15] identified another peptide, DP178 (later named as T20, enfuvirtide or Fuzeon®), that inhibits HIV-1-mediated membrane fusion at nM concentration. In March 2003, enfuvirtide was approved by the FDA and European Commission as a novel anti-HIV-1 drug for use in adults and in children aged 6 and older with advanced HIV-1 infection [16]. As the first agent in the class of fusion inhibitors, enfuvirtide binds to a region of the HIV-1 Env glycoprotein gp41 and prevents subsequent viral fusion with the target cell membrane. Enfuvirtide can be degraded by proteolytic enzymes, but not by the hepatic CYP450 isoenzyme systems [17,18]. There are no significant interactions between enfuvirtide and other medications. Enfuvirtide is particularly effective in treatment-experienced patients with detectable HIV-1 RNA, indicating an important role in subsequent therapy. In randomized controlled studies of PIs on patients infected with HIV that was resistant to NRTI, NNRTI and PI classes, the serum viral response was better in patients receiving enfuvirtide. It therefore provides a much needed fourth class drug for the treatment of HIV-1, which operates in the early viral life cycle.
Structure and function of HIV-1 gp41
HIV-1 envelope (Env) spikes are derived from gp160. It is a precursor protein enzymatically cleaved into gp120 and gp41 subunits by cellular furin protease before cell surface expression and incorporation into budding virus particles. Heterotrimeric Env spikes consist of three transmembrane (gp41) domains and three surface (gp120) domains [19]. The gp41 is a 345 amino acid glycoprotein corresponding to the Env sequence 512–856. It was subdivided into an ectodomain (ECTO; residues 512–683), a membrane-spanning domain (MSD; residues 684–704) and a long cytoplasmic tail (residues 705–856). ECTO locates outside the viral membrane and is considered to be the critical region of gp41. The sequence from N- to C-terminal of the ECTO: the fusion peptide (FP), together with the N-terminal heptad repeat (NHR, or HR1), and the CHR (or HR2) are the three important functional regions closely related to fusion activity [20].
FP is a hydrophobic glycine-rich peptide. It contains the amino acid residues 512–527. In the pre-fusion state, FP is surrounded in gp120–gp41 complex [21,22]. It is highly conserved with extensive homologies with those in the fusion sites on the Env glycoproteins of myxoviruses and paramyxoviruses [23]. There is evidence suggesting that FP plays a crucial role in the fusion process of HIV-1 and host cells in HeLa-T4 cells that expressed gp160: its fusion activity with T-cell lines decreased when three to four N-terminal amino acids were deleted from FP. Fusion activity was completely lost when the deletion reached five amino acids [24]. When the hydrophobic amino acids of FP were replaced by hydrophilic amino acids, cells that expressed gp160 also lost their fusion activities [25–27]. Synthetic FP peptide blocked cell-cell fusion [28], promoted lipid mixing and aggregation [27], inhibited HIV-1-induced syncytium formation [28] and induced leakage of lipid vesicles [29,30].
Following the FP region are the two heptad repeats (NHR and CHR). Both show periodic hydrophobicity predictive of α-helical structures. NHR-containing amino acid residues 542–592 are adjacent to FP, whereas CHR-containing amino acid residues 621–673 are adjacent to the membrane-proximal external region [31]. These two regions are crucial in the fusion process [32,33]. Point mutations in the conserved regions of NHR and CHR rendered greatly decreased fusion activity.
Very little is known about the structures of gp41 before fusion. In early studies, Lu et al. [34] expressed the ECTO of gp41 as a recombinant protein fragment. A stable and proteinase-resistant structure was found. It comprised two peptides, N-51 and C-43, derived from NHR and CHR. When isolated, N-51 was predominantly aggregated and C-43 was unfolded. When mixed, the peptides associated to form a discrete trimer of heterodimers, a 6-helix complex, which was stable in the aqueous solution. Proteolysis experiments indicated that the possible orientation of the N-51 and C-43 helices in the complex is antiparallel. A model was proposed that the N-51 formed an interior, parallel, homotrimeric, coiled–coil core, against which three C-43 helices pack in an antiparallel fashion surrounding the trimeric N-51 core, forming a 6-helix bindle. This 6-helix complex is the core of the fusion-competent state of the HIV-1 Env [34].
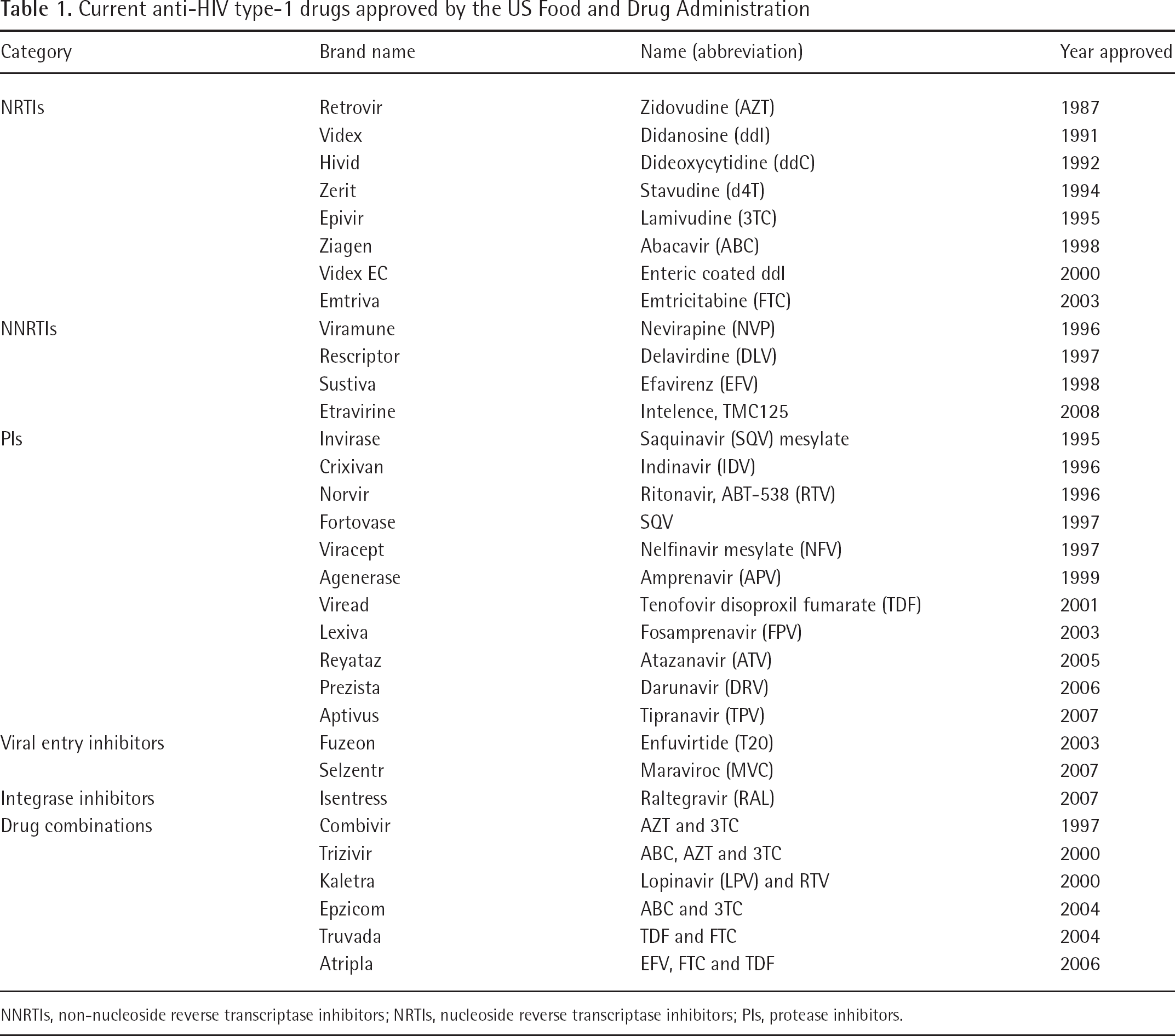
Later, a similar 6-helix structure was identified in the core of simian immunodeficiency virus (SIV) e-gp41. There were two peptides derived from NHR (N-peptide) and CHR (C-peptide). When the two peptides were mixed in a 1:1 molar ratio, there was a tendency to form a stable six-stranded helix bundle (6-HB) [35]. Based on the results of three independent research groups, the 6-helix structure has been resolved by nuclear magnetic resonance (NMR) and crystallography [36–38]. Three helices from the N-peptide region (the N-helices) associated to form the central trimeric coiled–coil. Another three helices from the C-peptide region (the C-helices) packed obliquely into the highly conserved hydrophobic grooves on the surface of the central coiled–coil. Residues of the heptad repeats were assigned to the a, b, c, d, e, f and g helix positions. During the process of 6-HB formation, residues at positions a and d of the C-helices helix packed against residues at the e and g positions of the N-helices coiled–coil trimer in an anti-parallel configuration. In each of the grooves, there was a highly conserved hydrophobic deep cavity (pocket) formed by the cavity-forming sequence (residues 565–581) in the NHR region. This is crucial for viral fusion and stability of the 6-HB (Figure 1A and 1B).
The mechanism of HIV-1 peptide fusion inhibitor blocking HIV-1 entering host cells
With better understanding of the structure, the next question relates to how HIV-1 enters the host cell. What is the role of gp41 in HIV-1 and host cell membrane fusion? When fusion occurs, FP is inserted into the target cell membrane. The conformation of NHR and CHR is altered to form a 6-helix structure. The process results in the formation of a fusion pore through which the HIV-1 capsid enters the CD4+ T-cell. The mechanism of gp41-mediated membrane fusion is analogous to the ‘spring-loaded’ model. Prior to formation of the 6-helix structure, there is an ‘intermediate state’ that the three CHR helices occupy only transiently, until they are associated with the trimeric NHR. After binding of gp120 to CD4 and a coreceptor (for example, CCR5 or CXCR4), gp41 changes its conformation to a pre-fusion (intermediate) state by inserting its FP into the target cell membrane. The gp41 N- and C-helices then associate to form the 6-HB, bringing the viral and target cell membranes to close proximity and eventually fusion of HIV-1 with the target cell. The intermediate state of gp41 provides a window in terms of time and space for synthetic peptides that mimic the N-peptides and C-peptides to form a heterogeneous 6-HB with CHR or NHR. This will block the formation of the fusion-active core and thereby inhibit fusion (Figure 1C). The understanding of this action of the fusion inhibitor has directed research towards identifying more antiviral peptides against those viruses with type-1 transmembrane Env glycoprotein [9,37,39,40].
The antiviral properties of peptides synthesized on the basis of the amino acid sequences of NHR and CHR of gp41 were originally recognized in the early 90s. DP107 mimicking a fragment of NHR was the first reported HIV-1 peptide inhibitor [41]. In 1993, the in vitro potency of another peptide, DP-178, synthesized on the basis of the amino acid sequence of CHR, was demonstrated [15]. During the past decade, peptides with potent HIV-1 fusion inhibitory activities have emerged.
The anti-HIV-1 peptides derived from the gp41 FP regions
Virus-induced membrane fusion is highly sensitive to single amino acid mutations in the FP domain [27,42]. This illustrates that FP is an important component in the fusion process. Indeed, several studies suggested that the peptides derived from the FP could inhibit cell fusion induced by HIV-1. The effects of the FPs are dependent on length but independent of chirality.
In 1990, a 6-amino-acid-long peptide with sequences corresponding to the N terminus of gp41 was shown to inhibit HIV-1 Env glycoprotein-induced syncytium formation [28]. However, this short peptide was only effective at a concentration of 1 mM. Similarly, Slepushkin et al. [43] found another 11-amino-acid-long peptide that could effectively block half of HIV-1 entry into host cell at concentration of 0.01 mM. In another study, Slepushkin et al. [44] showed that a 22-amino-acid-long FP and its conjugate with bovine serum albumin inhibited HIV-1 infection at a concentration of 1 μM. The effect of the peptide is certainly enhanced as its length increases. Kliger et al. [45] found another 33 amino acid FP that could inhibit fusion at a two-order magnitude lower concentration than that of the 22 amino acid peptide. Later, Gerber et al. [46] introduced D-amino acids into the peptides. A 33 amino acid peptide was synthesized in which four highly conserved amino acids, Ile4, Phe8, Phe11 and Ala14, were replaced by the respective D-enantiomers. This peptide was named as IFFA. They found that the wild-type (WT) peptide and its diastereomer exhibited similar inhibitory effects at concentrations <10 ng/ml. The ability of the diastereomer to maintain the inhibition properties of the WT FP makes it a promising candidate for designing new anti-HIV-1 inhibitors. However, neither the WT FP nor the IFFA could reach complete inhibition of cell-mediated fusion. The inhibitory activity of WT FP or IFFA increases quite rapidly with increasing dosage in the low nM range. The increase in activity slowed down, and even up to 1 μM it does not reach 75% inhibition. This observation might be accounted for by the nature of the FP itself. At low concentration FP promotes fusion. As the concentration increases, FP will enhance negative curvature leading to membrane destabilization. Therefore, FP has a dual role to promote and inhibit fusion. This property of the HIV-1 FP makes it unsuitable for further development of the FP peptides as a therapeutic agent [46].
Despite the limitation of HIV-1 FP for further development as a fusion inhibitor, it is worthwhile taking into consideration other aspects of FP. The sequence of HIV-1 FP is very conserved. Randomized change of one or two amino acid residues in FP can reduce infectivity of these HIV-1 variants by 21–483-fold [47]. As FP is performing an important function in HIV-1 and host cell fusion, it is still an ideal target for anti-HIV-1 drug development. In 2007, Münch et al. [47] extracted peptides from 10,000 l of human haemofiltrate derived from patients with chronic renal disease. It was fractionated by cation exchange followed by reversed phase chromatography to separate the peptides from human HF. A total of 322 different peptides were identified from the HF peptide library and screened for fractions that could block wild-type HIV-1 NL4-3 (NL4-3wt) replication in human peripheral blood mononuclear cells (PBMCs). A non-cytotoxic fraction with potent anti-HIV-1 activity was selected and further purified. The peptide had the sequence LEAIPMSIPPEVKFNKPFVF, with a molecular mass of 2,303 Da and was named VIRIP. The sequence matched residues 353–372 of human a1-AT (accession number P01009), which is the most abundant circulating serine PI. VIRIP is active against a broad spectrum of HIV-1 variants and targets at the highly conserved area of gp41 FP. Its inhibitory activity is independent of viral subtype or coreceptor usage. Both natural and synthetic VIRIP inhibit HIV-1 infection with the same potency. This peptide-inhibited infection of PBMC by X4- and R5-tropic HIV-1 with a 50% inhibitory concentration (IC50) of approximately 20 μM, and completely blocked HIV-1 NL4-3 replication in PBMCs at 100 μM. VIRIP also blocked HIV-1 variants resistant to PIs, RT inhibitors (X11 and ZT3-1) and fusion inhibitor T20 (DTV, L33S and I37Q/V38M). This suggests that VIRIP might be effective in patients failing current HAART regimens. VIRIP could also block HIV-1 transmission by monocyte-derived dendritic cells to T-cells at 100 μM. Even in the presence of an efficient virus, attachment factor did not overcome the ability of VIRIP to inhibit viral infection. However, in comparison with all HIV-1 variants analysed, VIRIP were hardly susceptible to inhibit the HIV type-2 (HIV-2) Rod, SIVmac239 and SIVmac251 strains by SIV particles carrying HIV-1 Env proteins (SHIV-89.6 and SHIV-RN). So far it is difficult to obtain VIRIP-resistant HIV-1 variants in cell culture. For several amino acids, change in FP enhanced the antiviral potency of VIRIP by approximately two orders of magnitude. Furthermore, it was found that many VIRIP derivatives were highly stable in human plasma. These compounds are not cytotoxic, even at exceedingly high concentrations. Together, these results showed that VIRIP is a broad-spectrum inhibitor of HIV-1 Env function and that the gp41 FP region is an attractive drug target [47].
The anti-HIV-1 peptides corresponding to the gp41 CHR regions
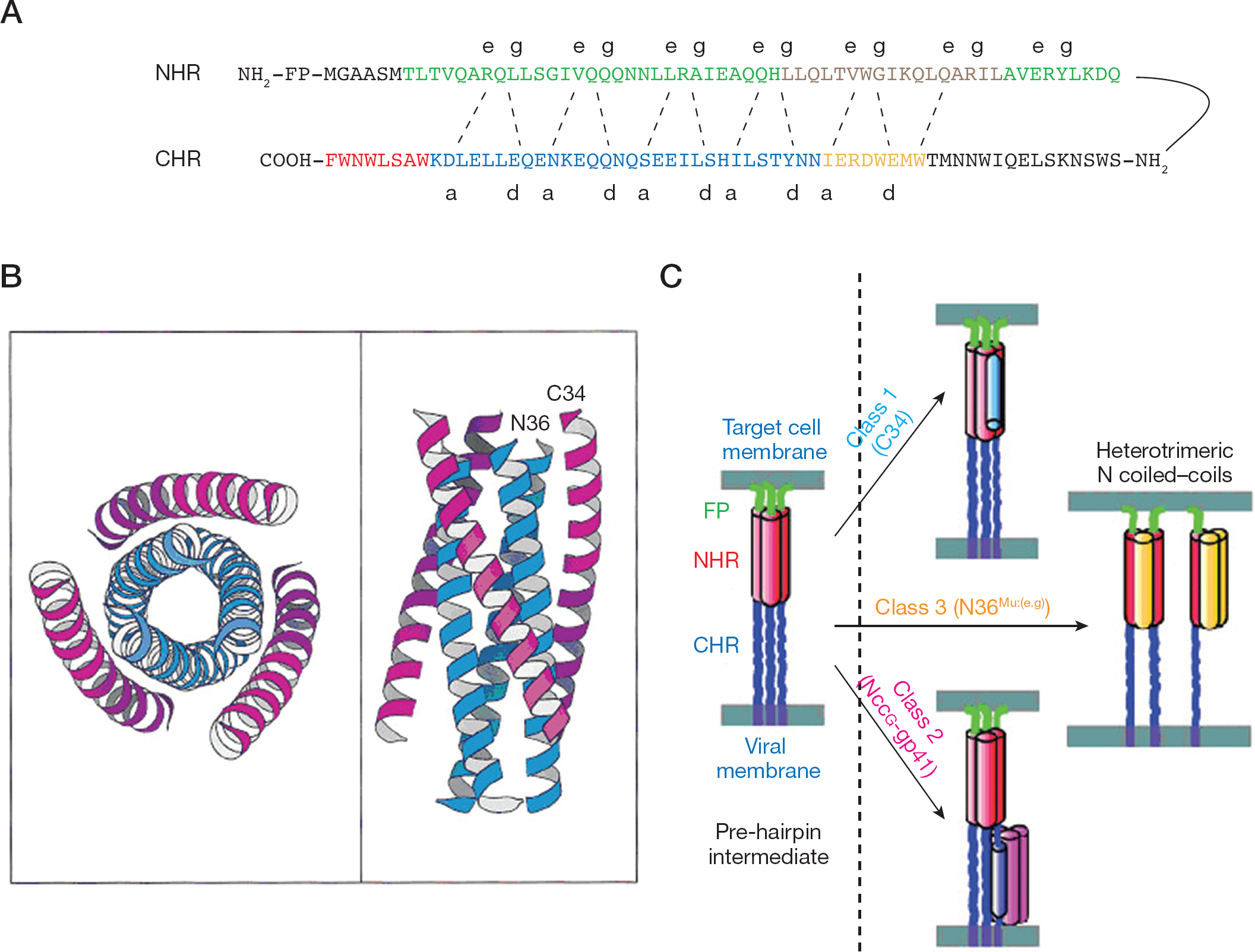
Currently, the gp41 CHR could be divided into four functional domains: upstream region (amino acids 621–627), NHR-binding domain (amino acids 628–666), pocket-binding domain (amino acids 628–635), and lipid-binding domain (amino acids 666–673) [48,49]. During the fusion process, the NHR-binding domain interacts with the NHR region (amino acids 536–581) to form a hairpin structure. This step can be blocked by C-peptide corresponding to the HIV-1 Env glycoprotein transmembrane subunit gp41 CHR region. These peptides interact with the gp41 NHR region and block the formation of the 6-HB (Figure 1A).
In 1993, Jiang et al. [13,14] found a synthetic peptide known as SJ-2176 (sequence: EWDREINNYTSLIHSLIEESQNQQEKNEQEGGC). This sequence corresponds to residues 637–666 of the HIV-1 glycoprotein gp41. It inhibits the replication of an array of HIV-1 strains at nM range. SJ-2176 selectively binds to the fusion domain at the N-terminus of gp41 and has no detectable cytotoxicity. In 1994, Wild et al. [15] identified another synthetic peptide, DP-178 (T20), corresponding to residues 643–678 of the HIV-1LAI isolate, as a potent antiviral agent. T20 exhibited remarkable potency in inhibiting both virus-mediated cell–cell fusion and infection by cell-free virus. It reduced syncytium formation in cells infected by cell-free HIV-1LAI at approximately 200 pM (1–2 ng/ml) and 18 nM (80 ng/ml), respectively. Similar results were obtained in infection with other prototypic HIV-1 isolates (MN, RF and SF2) and two primary isolates that had not been cultured in cell lines. The inhibitory activity of T20 was observed at peptide concentrations approximately 104–105× lower than the cytotoxic concentration. The peptide-mediated inhibition is HIV-1 specific in that approximately 102–103× more peptide was required for inhibition of HIV-2 isolates. Further experiments showed that T20 exhibited antiviral activity against both prototypic and primary HIV-1 isolates. As shown by PCR analysis of newly synthesized proviral DNA, T20 blocks the early step in the virus life cycle prior to reverse transcription. In the following years, it was developed by Trimeris, Inc. and Hoffmann–La Roche, Ltd. into a well known anti-HIV-1 drug, named enfuvirtide or FUZEON® [15].
In 1995, using proteolysis experiments, Lu et al. [34] identified a series of CHR-related C-peptides that are stable and process anti-HIV-1 activity (for example, C43 [residues at amino acids 624–666], C34 [residues at amino acids 628–661] and C28 [residues at amino acids 628–655]). The anti-HIV-1 activities of C-peptides are related to the number of residues, and the shortest active C-peptide is C28. C-peptides are believed to act by binding to the hydrophobic grooves that line the internal N-terminal trimeric coiled–coil core of the gp41 ECTO. As a result, the longer the C-peptides, the tighter the binding to the N-terminal core structure. Later, more C-peptides were identified. They are potent antiviral inhibitors with effective concentrations at nM range. The C-peptides block 6-HB formation and inhibit HIV-1 entry into cells in a dose-dependent manner (Figure 2A).
The amino acid sequences of some HIV-1 peptide fusion inhibitors
C-peptides derived from HIV-1, HIV-2 and SIV gp41 can generally inhibit HIV-1 Env-mediated fusion. However, inhibition of HIV-2 or SIV Env-mediated fusion is poor. For example, HIV-1 C34, HIV-2 C34 and SIV C34 inhibited HIV-1 Env-mediated fusion at an IC50<10 nM. By contrast, all three C34 peptides showed poor inhibition on HIV-2 or SIV Env-mediated fusion even at IC50>100 nM. This suggested that the fusion process of HIV-2 and SIV is different from HIV-1. In contrast to HIV-1, HIV-2 or SIV, Env do not expose their NHR region to C34 peptides following CD4 binding, but rapidly proceed to coreceptor engagement and 6-HB formation resulting in fusion. This data implied that several factors, including 6-HB stability and the ability of CD4 to destabilize the Env glycoprotein, might serve as determinants to the sensitivity of fusion inhibitors [50].
Amongst all of the C-peptides, T20 and C34 are favoured by researchers. Compared with T20, C34 is a more potent inhibitor and is weak in inducing resistant viruses. However, C34 is less soluble in aqueous media than T20. For this reason, C34 has a quite different destiny. T20 was used in clinical trials and became the first fusion inhibitor for AIDS therapy. C34 was used widely as a research tool [49,51–53].
It is widely believed that T20 and C34 shared the same mechanism of action in blocking the fusogenic core (6-HB) of gp41 [54–56]. However, new evidence casts doubt on the generality of the above model. Although the sequence of T20 partially overlaps the C34 peptide, T-20 does not contain the pocket-binding sequence (residues 628–635), which is important for gp41-mediated membrane fusion and the stability of the gp41 core conformation. Although there have been some reports on the direct interaction of T-20 with the NHR sequence [57,58], researchers believed that T20 targets at multiple sites in gp41 and gp120, and its anti-HIV-1 activity might be a combinatory effect mediated by different mechanisms of action. The major supporting evidence includes the following: first, C34 contains the pocket-binding sequence essential for inhibiting HIV-1 entry but not the lipid-binding sequence, whereas T20 contains the lipid-binding (or tryptophan-rich) sequence and partial NHR-binding domain but does not have the pocket-binding sequence [49,53]. Second, T20 inhibits phospholipid redistribution mediated by the HIV-1 Env glycoprotein at a concentration 8x greater than that of solute redistribution (the IC50 values are 43 and 335 nM, respectively). By contrast, C34, does not have this effect (11 and 25 nM, respectively) [52]. Third, in FN-PAGE experiments, ELISA assays and circular dichroism (CD) spectroscopy, T-20 does not form stable 6-HB with N36 like C34. Fourth, the determinant of T-20 resistance in the NHR region is not the critical site involved in the interaction between the NHR and CHR regions to form 6-HB. Fifth, T-20-mediated inhibitory activity is abrogated by peptides derived from the coreceptor binding region in gp120 and the MSD in gp41. Finally, the gp120 coreceptor binding region and gp41 MSD interact at different sites in T-20 [53]. These new findings implied that T-20 inhibited HIV-1 entry by targeting multiple sites in gp41 and gp120.
Recently, He et al. [59] identified a peptide CP621– 652 (residues at 621–652) with high anti-HIV-1 potency (IC50 value of 4.23 nM against HIV-1-mediated cell–cell fusion). As a control, the IC50 of C34 and T-20 under similar conditions is 3.65 nM and 26.43 nM, respectively. These results suggest that the heptad motif 621-QIWNNMT-627 is also a critical motif for anti-HIV-1 peptides [48]. Based on the parental peptide CP621–652, 11 of 32 (34.4%) residues in the peptide CP621–652 were mutated to enhance its hydrophobic interaction and salt bridges with the NHR target, a novel anti-HIV-1 peptide, CP32M, have been rationally designed [59]. CP32M is a potent HIV-1 fusion inhibitor against infection by both laboratory-adapted and primary HIV-1 strains with different genotypes and phenotypes at nM ranges. Of note, the peptide CP32M has potent inhibitory activity against enfuvirtide-resistant HIV-1 strains. These findings suggest that CP32M can be further developed as an antiviral therapeutic against multidrug-resistant HIV-1.
The anti-HIV-1 peptides corresponding to the gp41 NHR regions
The N-peptides represent another group of peptides with potential inhibitory effects against HIV-1 entry. These peptides interact with the CHR region blocking the 6-HB formation. Initial studies indicated that N-peptides are weaker inhibitors than the C-peptides, with IC50 values in the μM range. However, chimeric molecules composed of soluble trimeric coiled–coils have shown promising results.
In 1992, Wild et al. [41] synthesized a peptide named DP-107. It contained residues 558–595 of the Env glycoprotein gp41 of HIV-1LAI strain. Structurally, from CD spectroscopy studies, DP-107 is made up of approximately 85% α-helical structure. This suggests that the peptide can be self-associated and typical of a coiled–coil or leucine (Leu) zipper. In biological assays, this peptide efficiently blocked virus-mediated cell-cell fusion as well as infection of PBMCs by both prototypic and primary isolates of HIV-1. A single amino acid substitution in the peptide greatly destabilized its structure as measured by CD spectroscopy and abrogated its antiviral activity [41].
Later, Lu et al. [34] identified a series of N-peptides with anti-HIV-1 activity by proteolysis. The popular ones are N51 (540–590), N36 (546–581) and N34 (546–579; Figure 2B). The anti-HIV-1 activity of N-peptides is approximately 1,000-fold lower than that of C-peptides. These results suggest that N-peptides are more prone to oligomerization and cannot form ordered structure in the absence of C-peptides. The oligomerized and disordered N-peptides could hardly interact with the gp41 CHR region.
The anti-HIV-1 peptides derived from the gp41 CHR regions
SC34
C-peptides derived from the CHR regions are known to prevent formation of the fusogenic structure and inhibit HIV-1 replication. T20 was the first one used in clinical trials. The major drawback is induction of T20-resistant HIV-1 strains [60–62]. At the same time, C34 is less desirable because of its low solubility [39]. Otaka et al. [63] developed a highly soluble C34 variant (SC34) that is more potent in anti-HIV-1 activities. The design of SC34 was based on interaction of the inner coiled–coil strand formed by an N-region peptide (N36) with the C34 helices. First, amino acid residues crucial for interaction with the inner strand formed by N36 (a, d and e positions) were maintained. Second, non-conservative residues located at solvent-accessible sites in the 6-HB (b, c, f and g positions) by glutamic acid (Glu) or lysine (Lys) were replaced (Figure 2C). These arrangements allowed the Glu/Lys ion pairs to form between the i and i+4 positions along the C34 sequence. Such replacements with Glu or Lys could enhance solubility as well as α-helicity through intrahelical salt bridges, which were expected to stabilize the 6-HB crucial for anti-HIV-1 activity. The results showed that solubility of SC34 in aqueous solution increased by >1,000-fold over that of C34 without affecting the anti-HIV-1 activities. Furthermore, SC34 exhibited strong anti-HIV-1 effects against a T20 resistant strain, and NL4-3SIM. Although T20 showed anti-HIV-1 activity against wild-type HIV-1 clone NL4-3, its anti-HIV-1 effect decreased sevenfold against NL4-3SIM. By contrast, SC34 effectively inhibited the NL4-3SIM strain as well as the NL4-3 strain. To further improve SC34, more Glu and Lys residues were introduced in the non-conservative sites in order to stabilize the α-helix conformation and increase solubility. Two new SC34-derived peptides, SC34EK and SC35EK, were obtained. The solubility of both was similar to SC34, and both were 3x higher in anti-HIV-1 potency [63].
Sifuvirtide
Enfuvirtide (T20) is the first and only HIV-1 fusion inhibitor approved for clinical use. However, it can easily induce drug resistance and the plasma half-life is short (approximately 2 h) [17,18]. Large amounts are required to maintain the in vivo anti-HIV-1 efficacy in humans (approximately 200 mg/day). At present, T20 is only used as an auxiliary drug in HAART therapy. Another novel anti-HIV-1 peptide called sifuvirtide (SFT) was designed to address the limitations of T20. The prototypic peptide is derived from the gp41 CHR sequence of HIV-1 subtype E C34 (628-WIEWEREIS NYTNQIYEILTESQNQQDRNEKDLLE-661). Based on the three-dimensional structural information of HIV-1 gp41 and computer modelling analysis, SFT was designed similarly to SC34. The residues that are important for activity or stability were maintained. Charged Glu and Lys residues were introduced into SFT to help the formation of ion pairs (salt bridges) at the i and i+4 positions of the helical conformation. Glu 630 was substituted by threonine to cover the hydrophobic pocket. Furthermore, to increase its stability, serine was added to the N-terminal and Glu was added to the C-terminal of the peptide (Figure 2C). After a series of the abovementioned alterations, the resulting peptide was SFT, composed of 36 residues in length and shares some sequence and structural features with the native CHR peptide. Out of these 36 residues, SFT is different from C34 in 16 residues and from T20 in 22 residues [64].
The CD spectrum for SFT was typical of peptides with a random coil conformation. Unlike T20, SFT was able to form a 6-HB with N36, and could not interact with the lipid membrane [64]. Compared with T20, SFT had higher binding activity to gp41 [65,66]. The biophysical characterization of SFT was similar to that of C34. However, the solubility of SFT was quite different from C34. Because of the aromatic amino acid residues of the peptide, no significant aggregation of SFT was observed in aqueous solution [67]. SFT could inhibit HIV-1IIIB-mediated cell–cell fusion with an IC50 of 3.60 nM. By comparison, T20 inhibited the fusion with an IC50 of 21.39 nM [64]. In another research project done by our group, SFT inhibited HIV-1IIIB-mediated cell–cell fusion at an IC50 of 0.1 nM, whereas the IC50 of T20 was 1.5 nM [65,66]. Thus, SFT is approximately 6–15-fold more potent than T20 in inhibiting fusion of viral and cellular membranes. Consistently, SFT was also more potent to inhibit HIV-1IIIB-mediated infection of MT-2 cells than T20. Furthermore, SFT was highly active in inhibiting primary HIV-1 isolates with distinct genotypes and phenotypes (subtypes A to F), in particular those X4R5 viruses. More importantly, SFT showed strong inhibitory activities against all T20-resistant strains with IC50 ranging from 2.68 to 47.78 nM, whereas T20 failed to do so even with higher concentrations (40–700-fold). These results suggested that SFT could be used as an alternative fusion inhibitor for treatment of patients with HIV-1/AIDS, in particular those infected by T20-resistant variants [64].
To study the pharmacokinetics of SFT, it was injected into monkeys intravenously or subcutaneously (1.2 mg/kg), the terminal elimination half-lives (T1/2) were 6.3 ±0.9 h and 5.5 ±1.0 h, respectively [68]. A Phase Ia clinical study of SFT in 60 healthy individuals was performed, which demonstrated good safety, tolerability, and reasonable pharmacokinetic profiles. A single-dose regimen (5, 10, 20, 30 and 40 mg) by subcutaneous injection once daily at abdominal sites was well tolerated without serious adverse events [64]. Pharmacokinetic studies of single and multiple administration of SFT showed that its decay half-lives were 20.0 ±8.6 h and 26.0 ±7.9 h, respectively. SFT appeared to be an ideal fusion inhibitor with high potency, and a reasonable plasma half-life and resistance profile. It can be used for the treatment of HIV-1/AIDS patients, including those who are infected by HIV-1 strains resistant to T20. SFT is now being evaluated in Phase II clinical trails [64].
T-2410 and its derivatives
Treatment-acquired resistance to enfuvirtide highlights the need to create fusion inhibitor therapeutics with activity against enfuvirtide-resistant viruses. Using a rational design, Dwyer et al. [69] synthesized a series of C-peptides with enhanced helical structures that can self-associate into stable oligomeric structures. Such peptides were designed on the basis of C34. To increase the stability, a methionine and a threonine were added to the N-terminal and a Glu and a Leu were added to the C-terminal of C34. This new peptide was named as T-2410. This was further modified by introducing charged Glu and arginine residues into the non-core positions, so that the spacing (i, i+4) favoured the formation of an ion pair in the helical conformation. The N-terminal methionine was also changed to threonine to eliminate potential complications caused by oxidation. The resulting peptide was named as T-2429. Afterwards, based on T-2429, eight non-crucial residues that were not part of an ion pair were substituted with alanine. The resulting peptide was named T-290676. Finally, based on T-290676, two critical residues that were not part of an ion pair were substituted with alanine. The resulting peptide was T2635. From T-2410 to T2635, the proportion of α-helicity increased, and stabilities of peptides were enhanced (Figure 2D). There was little gain in anti-HIV-1 activity from the change, but a significant increase in potency against the resistant strain viruses. Remarkably, T2635 had potent antiviral activity against all tested viruses and it was 3,600-fold more active than enfuvirtide. Passage experiments using one of these peptides could not generate resistant virus even after >70 days in culture, suggesting superior durability as compared with enfuvirtide. In addition, the pharmacokinetic properties of T2635 were improved up to 100-fold than that of enfuvirtide. The potent antiviral activity against resistant viruses, the difficulty in generating resistant virus and the extended half-life in vivo make this class of fusion inhibitor peptides attractive for further development [69].
The anti-HIV-1 peptides derived from the gp41 NHR regions
IQN17, IZN17 and (CCIZN17)3
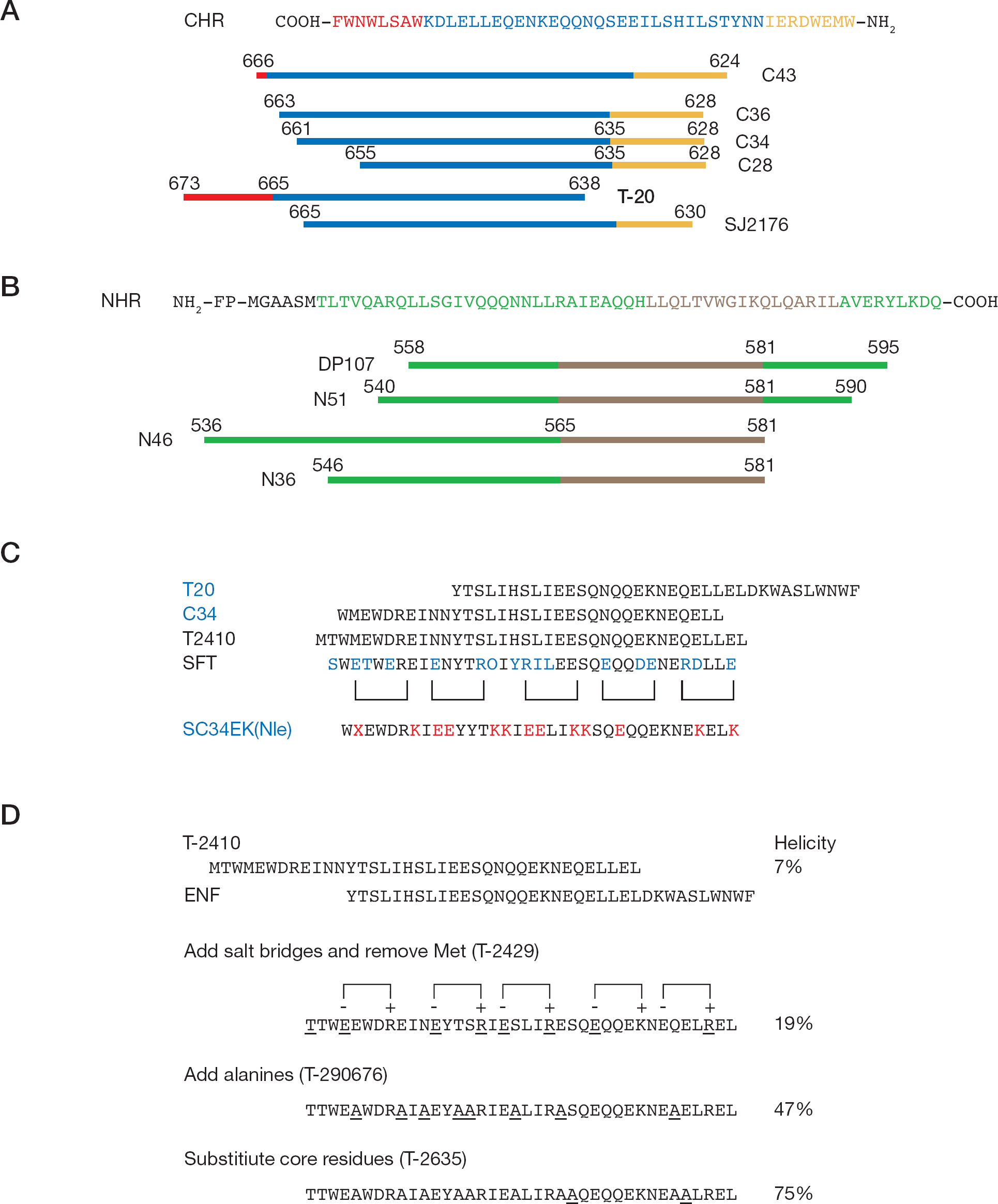
Eckert et al. [70] designed a molecule, denoted IQN17, in which a soluble trimeric coiled–coil was fused to the portion of the N-peptide that comprised the gp41 hydrophobic pocket. The crystal structures of the GCN4-pIQI and N36/C34 core of gp41 were analysed by X-ray. It indicated that the superhelical parameters of the two coiled–coils were similar. Thus, structural perturbations resulting from creation of the chimera were expected to be minimal. On the basis of this information, a peptide was constructed in which the first 29 residues of GCN4-pIQI were fused to the last 17 residues of N36. Three surface residue mutations in GCN4-pIQI were inserted in order to increase solubility. The resulting peptide, IQN17, was fully helical, and was a soluble trimeric species as determined by sedimentation equilibrium experiments (Figure 3B). IQN17 blocked cell-to-cell fusion induced by HIV-1 with an IC50 value of 190 nM [70]. Later, the number of residues used for fusion increased from 17 to 23 and 36, producing 2 peptides, IQN23 and IQN36, with IC50 values of 15 nM and 88 nM, respectively. Comparing the three, N36 aggregates in solution, and both N17 and N23, are extremely insoluble. These peptides were not very helical in isolation (Figure 3A). The inhibitory potencies of N17, N23 and N36 are low, with IC50 values of 13, 29 and 2 μM, respectively. The result indicates that the solubility, but not the length, of N-peptides is a crucial factor in the anti-HIV-1 activity of IQN peptides [71].
The structure models of IQN17, IZN17 and (CCIZN17)3
GCN4-pIQI was not the best soluble trimeric structure. Another soluble trimeric structure, IZN, was used to replace GCN4-pIQI. So the new peptides, IZN17 (Figure 3C), IZN23 and IZN36 were constructed. The IC50 values of IZN17, IZN23 and IZN36 in the HIV-1 infectivity assay were 22, 30 and 26 nM, respectively. IZN17 was close to an order of magnitude more potent than IQN17 in the viral infectivity assay. So the chimeric peptide IZN17 represented a good starting point for further improvement. In this molecule, the presence of a coiled–coil structure depended on the monomer-trimer equilibrium in solution and, thus, was strictly dependent on concentration. The observed antiviral potency of IZN17 should therefore result from the interplay between the binding constant of the trimer to the C peptide and the constant of oligomerization to form the coiled–coil. Rather than attempting to design more stable trimerization scaffolds to overcome this putative limitation, Bianchi et al. [72] designed and constructed CCIZN17, which can form a covalently stabilized coil–coil structure. They introduced two cysteine residues in the monomeric IZN17 precursor to form three interchain disulfide bridges upon spontaneous assembly of the monomers into a trimer. The resulting covalent trimer, (CCIZN17)3 (Figure 3C), had an extraordinary thermodynamic stability that translates into unprecedented antiviral potency. (CCIZN17)3 inhibits fusion in a cell–cell fusion assay (IC50 260 pM) and was the most potent fusion inhibitor described to date (IC50 40–380 pM) in a single-cycle infectivity assay against HIV-1HXB2, HIV-1NL4–3 and HIV-1MN-1. Because (CCIZN17)3 and T20 target at different regions of the pre-hairpin intermediate, and because T20 does not include the partial CHR region that binds to the hydrophobic pocket of N17, the two inhibitors should work independently and could be administered in combination [72].
N36 mutants
The crystal structure of the HIV-1 gp41 ECTO core consists of a complex of N36 and C34 peptides comprising residues 546–581 and 628–661 of HIV-1 Env. Internal interaction between the N helices will lead to formation of a trimeric structure. This interaction involves positions a and d of the N-helices. Next, two N-helices interact with each C-helix via positions e and g of the N-helices. To improve the antiviral activities of the N-peptides, Bewley et al. [73] designed two peptides: N36Mut (e, g), which could only undergo self-association but could not interact with C34, and N36Mut (a, d), which could no longer self-associate but could interact with C34. In the case of N36Mut (e, g), the residues at positions e and g of N36 have been replaced by residues at positions e and g of C34. Because the latter residues are located on the external surface of C34 in the context of the ECTO gp41 core and C34 is monomeric, this substitution will prevent any interaction between N36Mut (e, g) and the C-region of gp41 in its pre-hairpin intermediate state. The intermolecular interaction required to form the trimeric coiled–coil of N-helices is preserved. In the case of N36Mut (a, d), the residues at positions a and d of N36 have been substituted by residues at positions f and c of C34, which are located on the solvent-exposed face of the ECTO core of gp41, thereby removing the intermolecular interaction required to form the trimeric coiled–coil of N-helices.
CD spectra demonstrated that N36Mut (e, g) displayed characteristics of an α-helix (Figure 1C). N36Mut (a, d), by contrast, is largely a random coil. Although N36Mut (e, g) did not interact with the C-region of gp41 in its pre-hairpin intermediate state, it inhibited fusion with an IC50 308 nM, which is 50-fold more active in inhibiting fusion than N36. By contrast, N36Mut (a, d), which could not form the trimeric coiled–coil itself, failed to inhibit fusion even at concentrations as high as 0.1 mM. The result showed that the trimeric coil–coil of N-peptides was crucial for its anti-HIV-1 activity. Because N36Mut (e, g) forms a well defined trimeric species that does not interact with C34, it must target the N-region of the pre-hairpin intermediate by forming fusion-incompetent heterotrimers [73]. A model that explains inhibition of HIV-1 Env-mediated cell fusion by N36Mut (e, g) was established. In the presence of a dynamic equilibrium between monomeric and trimeric forms of membrane-bound gp41, it allows subunit exchange to take place in the pre-hairpin intermediate state. Thus, trimers of N36Mut (e, g) interact with monomeric or dimeric N-region of gp41, forming heterotrimetric N coiled–coils that inhibit 6-HB of gp41 formation [73,74].
The anti-HIV-1 peptides derived from the gp41 NHR and CHR regions
NCCG-gp41, N35CCG-N13 and N34CCG
N34 and C28 of HIV-1 gp41 can arrange in a minimal 6-HB. It is a thermo-stable and soluble trimer of hairpins [38]. Using the N34-(L6)-C28 core as a scaffold, Louis et al. [75] designed a more potent ‘N35 peptide’ against HIV-1. They grafted a 35-residue sequence (N35) comprising residues 546–580 of HIV-I Env onto the N terminus of the scaffold. To further stabilize the trimeric coiled–coil of N35 helices, and to ensure that the entire molecule remains trimeric at very low concentrations, they then substituted the sequence Leu-Gln-Ala located at the C-terminal end of N35 (residues 31–33 of the construct) to Cys-Cys-Gly (CCG). The modification allows formation of three intermolecular disulfide bonds between the three subunits of N35. The peptide designed in this way was named as NCCG-gp41 (Figure 4B).
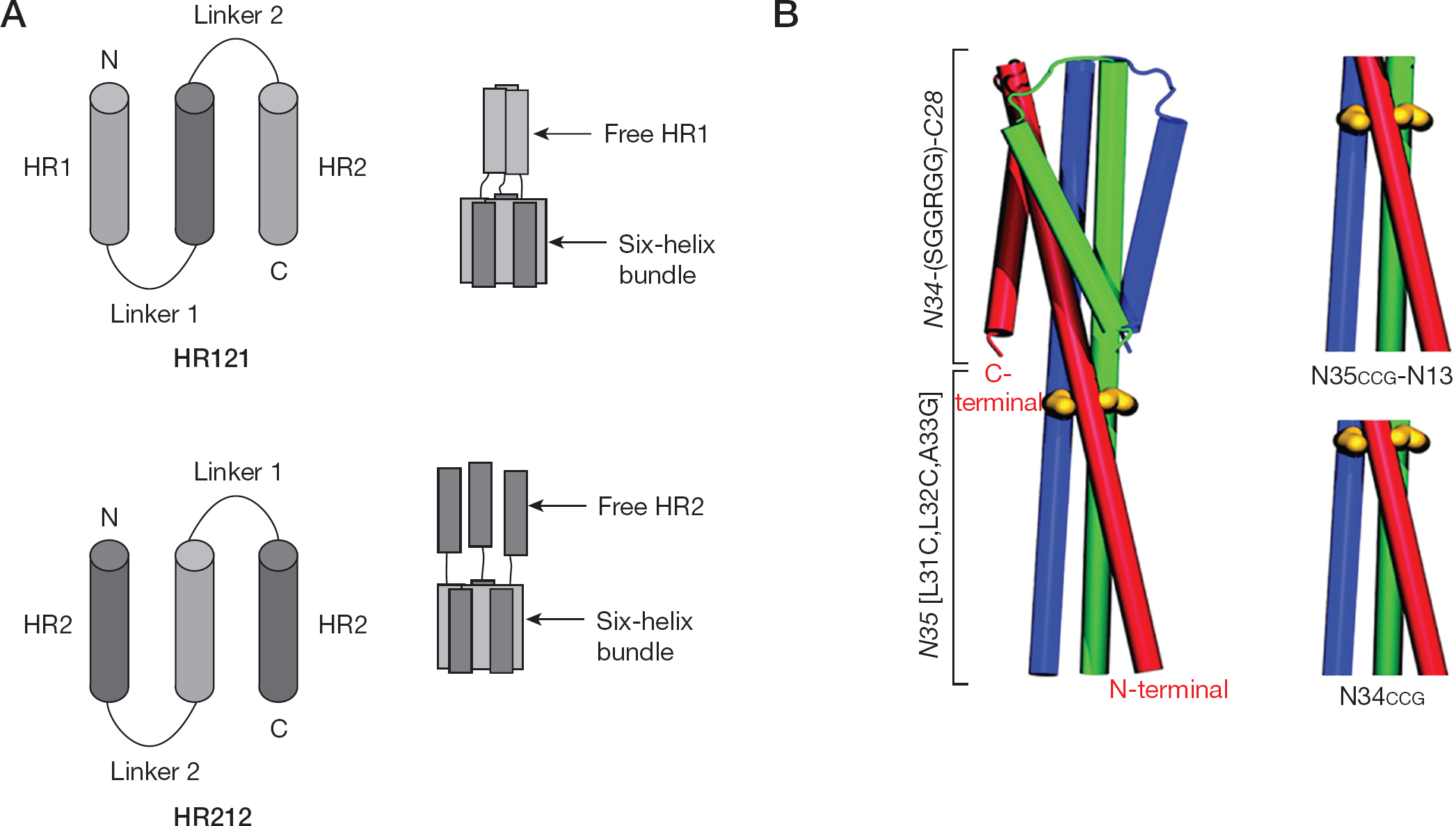
The structure models of HR121, HR212, NCCG-gp41, N35CCG-N13 and N34CCG
Different from other synthetic peptides, NCCG-gp41 was expressed and purified in Escherichia coli. It inhibited HIV-I Env-mediated cell fusion at nM concentrations with an IC50 of 16 nM, which was 1,000-fold lower than that of N35 [76]. This must be a result of the fact that the N-peptides easily aggregate and do not form a trimeric coiled–coil structure in the absence of C34 peptides [34].
From the perspective of a potential therapeutic, NCCG-gp41 trimer is too large (approximately 35 kDa), and the N34-(L6)-C28 core is useless in blocking HIV-1 entry. Louis et al. [77] therefore designed two smaller, covalently linked soluble peptides, N34CCG and N35CCG-N13, consisting solely of N-peptides of the gp41, and throwing the N34-(L6)-C28 core away. N34CCG and N35CCG-N13 blocked HIV-1 Env-mediated cell fusion with IC50 values of 96 and 15.5 nM, respectively (Figure 4B).
HR121 and HR212
N34 (residues 546–579) and C34 (residues 628–661) of HIV-1 gp41 could also form a 6-HB core. Using this 6-HB core as a scaffold, our group designed two symmetrical peptides, HR121 and HR212. In HR121, one N34 (HR1, N-peptide) is linked to the C terminus of N34-C34 (HR1-HR2-HR1), and similarly, in HR212, one C34 (HR2, C-peptide) is linked to the N terminus of N34-C34 (HR2-HR1-HR2). We selected the C34 segment, but not the C28 segment, primarily because C34 peptide inhibited HIV-1 entry with the lowest IC50 values of approximately 2–5 nM. As a consequence, three molecules of the peptides can form a stable 6-HB with three exposed free heptad-repeats (HR1 or HR2; Figure 4A). These free HR1 or HR2 can bind to the counterpart regions in the viral gp41, thereby blocking gp41-mediated membrane fusion.
These two peptides were expressed in E. coli as soluble proteins. When tested in luciferase reporter virus, both HR212 and HR121 potently inhibited HIV-1 Env glycoprotein-mediated virus–cell fusion, with the IC50 values of 2.8 nM and 16.2 nM, respectively. Similar to N34, HR121 was able to form a 6-HB with C-peptide (C43). In parallel, HR212 could interact with the N-peptide (N51). These findings suggested that the antiviral mechanism of HR121 or HR212 was analogous to the monomers HR1 or HR2, and might bind to their counterpart regions in the viral gp41 to block fusion. However, the CD spectrums revealed that HR121 and HR212 were of typical salient α-helix character conformations, whereas the corresponding HR1 or HR2 were in unordered formation. Both HR121 and HR212 could easily dissolve in water and were more stable than isolated N- and C-peptides. These results suggested that biophysical characterizations of HR121 and HR212 were different from related monopeptides [78].
Compared with HR121, HR212 had been shown to be more soluble and easily purified with noticeably more potent inhibitory activity. Therefore, we further investigated its anti-HIV-1 activities in vitro, and found that HR212 significantly inhibited entry and replication of laboratory and clinical HIV-1 strains. HR212 was effective in blocking laboratory strain HIV-1IIIB entry and replication with EC50 values of 3.92 and 6.59 nM, respectively, and inhibiting infection by clinic isolate HIV-1KM018 with EC50 values of 44.44 nM, as well as suppressing the HIV-1-induced cytopathic effect with an EC50 value of 3.04 nM. It also inhibited HIV-2ROD and HIV-2CBL-20 entry and replication in the μM range. Notably, HR212 showed strong inhibitory activities against all tested T20-resistant strains with EC50 values in the nM range, By contrast, T20 failed to do so, even with concentrations at least 100-fold or higher. Unlike T20, HR212 showed stability sufficient to inhibit syncytia formation in a time-of-addition assay, and was insensitive to proteinase K digestion. These results suggested that HR212 had a similar anti-HIV-1 activity with T20 and was more stable than T20 in vitro. Thus, HR212 is a promising therapeutic candidate against HIV-1 strains, particularly the T20-resistant variants [79].
Membrane-anchored peptides
Hildinger et al. [80] fused T20 with the hinge region of immunoglobulin G (IgG) heavy chain and the MSD from low affinity nerve growth factor receptor (LNGFR). The fusion protein was then inserted into a retrovirus vector M87. This reconstructed vector was packaged by transfection of Phoenix packaging cells and the supernatants were used to transduce the T-helper cell line PM-1. T20 was expressed on the cellular membrane of PM-1 cells, which are HIV-1 host cells. Then, the PM-1 cell lines were transduced: PM-1/M87 was infected with HIV-1 produced from the proviral clone NL4–3AGFP. On day 6, when 100% of the control cells were green fluorescent protein (GFP)-positive, <1% of the PM-1/M87 cells expressed GFP. Therefore, replication of NL4–3-AGFP was reduced by >2 orders of magnitude by M87. Conditioned medium from cells expressing M87 did not inhibit HIV-1 infection. This indicates that the peptide that inhibits infection anchored to the target cell membrane was not shed into the supernatant [80].
The peptide was anchored to the target membrane by a very short linker of 13 amino acids. To interact with the membrane-anchored T20, the N-terminal coiled–coil of gp41 must come very close to the target cell membrane. Egelhofer et al. [81] used a longer and more antiviral peptide, C46, to replace T20, and they used another retrovirus vector M87oRRE, to substitute for M87. They next inserted C46 into M87oRRE to construct a recombinant vector M87oRRE/C46. Compared with the vector M87, M87oRRE had several modifications: to reduce potential immunogenicity, the neo gene was eliminated and the murine IgG2 hinge was replaced by the human IgG2 hinge. To improve vector safety, the MSD of LNGFR was exchanged for the MSD of human tCD34. To increase the expression level, the DNA sequence coding for the M87o protein was adapted for human codon usage and cloned into an optimized retroviral vector backbone. In addition, a 41 base pair sequence derived from the HIV-1 Rev responsive element was added in the 3-untranslated region of the vector. PM-1 cells transduced with M87oRRE/C46 and wild-type PM-1 cells were challenged with HIV-1-NL4-3. The result showed wild-type PM-1 had a strong HIV-1 induced cytopathic effect, and all cells were dead by day 12 post-infection. By contrast, PM-1 cells sorted for the expression of C46 showed no viral cytopathic effect, and the 17% C46-positive culture only showed a transient cytopathic effect near day 10 and then grew normally for the rest of the experiment. Maximum virus production was seen in the control cultures on day 12. A short small peak of virus production was observed in the C46-positive culture on day 16, but no virus production was observed thereafter. Interestingly, virus production dropped continuously to become undetectable on day 55. These data clearly indicated that the M87o-RRE/C46 vector could confer an effective selective advantage to gene-modified cells in the presence of HIV-1 replication [81].
In primary cells, HIV-1 replication was inhibited by 1–2 log10 in PBMCs expressing M87oRRE/C46, and TZM-bl cells expressing M87oRRE/C46 were not infected T-20-resistant viruses. Extensive preclinical toxicity studies have been completed in mice and rhesus macaques for M87oRRE without any detectable side effects or immunogenicity. The lack of toxicity and the high antiviral efficacy thus justified further development of M87oRRE/C46, and in particular benefited the initiation of clinical trials [82].
Antibodies
In early research, de Rosny et al. [83] generated rabbit antisera against peptides derived from NHR, CHR and mixtures of the two that can self-assemble into a 6-HB. These antisera were used to investigate the fusion-induced conformational changes in Env. Several of these sera were shown to precipitate the receptor-activated forms of gp41, but these antibodies were not neutralized under conventional infectivity conditions at 37°C [83]. Similarly, monoclonal antibodies specific for the 6-HB (NC-1) were not neutralized [84]. Therefore, it was believed that the antibody molecules might be too large to access the fusion intermediates at the interface of the effector-target (E/T) or the virus-target cell membranes (steric problem). Alternatively, fusion might occur too quickly once fusion intermediates were formed (kinetics problem) [85].
Golding et al. [86] were able to slow down the fusion process by using suboptimal temperature (31.5°C) in order to dissect the steps in HIV-1 entry. This enabled evaluation of the antibodies on targeting fusion intermediates to block HIV-1 entry. Under these conditions, antibodies were raised against the native N36 peptide, and the 6-HB blocked E/T cell fusion and viral entry. Confocal microscopy demonstrated that binding of antibundle antibodies to effector cells interacted with target cells (E/T conjugates) prior to fusion. These data indicated that the fusion intermediates were accessible to antibodies and the lack of neutralization at 37°C was probably related to the kinetics of conformational changes and membrane fusion [86]. By contrast, Louis et al. [77] suggested that the kinetics of conformational changes of gp41 were not mainly responsible for antiviral activities of antibodies because the antibodies were raised against either the native N36 peptide or the 6-HB. Both did not represent the structure of the pre-hairpin intermediate state in gp41, and these antibodies have weak binding ability. Louis et al. [77] subsequently constructed a trimeric coiled–coil peptide, N35CCG-N13, which was similar to the internal trimeric coiled–coil of the N region in gp41. It was used to immunize rabbits and the anti-N35CCG-N13-specific antibodies were purified by affinity chromatography on a N35CCG-N13 immobilized column. A potent fusion inhibitory fraction, comprising 2.5–5% of the total IgG, was obtained. The tight binding anti-N35CCG-N13-specific antibody fraction inhibited both T tropic and M tropic HIV-1 Env-mediated cell fusion at 37°C. In a vaccinia-virus-based reporter gene assay, this specific antibody inhibited fusion completely at a concentration as low as 10 μg/ml. This data did not support the kinetic barrier and the tight binding of anti-N35CCG-N13-specific antibodies as explanations for the high avidity for the folded internal trimeric coiled–coil.
Miller et al. [87] used phage display libraries to select human-derived scFvs. Phage supernatants expressed IZN36 and 5H, which were the antigens designed to mimic HR1 as it might exist in the pre-hairpin intermediate. As a source of antibodies, large diverse and well characterized libraries of bacteriophages bearing scFvs derived from normal human B cells were used. From the libraries, they obtained an antiviral IgG, D5-IgG1, which blocked infection of diverse HIV-1 isolates with IC50 values ranging from 546 nM to 2,333 nM. NMR studies and functional analyses mapped the D5-binding site to a previously identified hydrophobic pocket situated in the HR1 groove [87]. Kinetic analysis of D5 mutants with perturbed D5-gp41 interactions implied D5 persistence at the fusion intermediate of gp41. This finding suggested that the hydrophobic pocket was a ‘hot spot’ for fusion inhibition. It provided a structural framework for the design of new therapeutic antibodies with potent neutralization potential against fusion intermediate of gp41 [76].
D-peptides
C-peptides are constituted of L-amino acids. Like many natural peptides, they can be easily degraded by proteolysis. Thus the bioavailability issue needs to be addressed before they can be used as a therapeutic agent. An example that illustrates this point is T20, which is a useful anti-HIV-1 drug used in humans. However, daily injection of 200 mg is required to achieve the therapeutic effect. It is therefore highly desirable if an effective and stable oral peptide could be identified. Peptides constituted of D-amino acids are not degraded by proteases. But there are very few natural D-peptides. Even if D-peptides are synthesized, there is no appropriate method to screen for antiviral activities. Eckert et al. [70] solved this problem. A peptide chimera, IQN17, was constructed as described above. It is a soluble trimeric structure and the formation of three N17 in IQN17 is similar to the internal trimeric coiled–coil of NHR region in gp41. More importantly, IQN17 contains the gp41 pocket, which is critical for drug screening. A peptide that interacts with IQN17 is likely to have antiviral activities. Eckert et al. [70] chemically synthesized the mirror image of IQN17 (denoted D-IQN17) with D-amino acids. They used D-IQN17 to screen through phage-expressed L-peptide libraries for anti-HIV-1 peptides. Next, the mirror images of the L-peptide sequences were chemically synthesized with D-amino acids. By symmetry, these D-peptides should bind to the natural form (L-amino acid) of IQN17. Using this method, potential inhibitors in the form of D-peptides can be identified. Subsequently, ten randomly encoded amino acid residues flanked by either a cysteine or a serine on both sides were screened from the phage-expressed peptides libraries. A potent D-peptide, D10-P5-2K, which inhibited fusion with an IC50 value of 3.6 pM, was found [70].
Later, Welch et al. [88] improved the mirror image target to a better trimeric pocket mimic peptide, IZN17, and generated a member 8-mer phage library for peptide screening. They found a D-peptide, PIE7, which is the most potent inhibitor (IC50 620 nM) in the library. Based on the trimeric nature of gp41, they predicted that multimeric D-peptides would have a significantly higher affinity for the N-trimer. Antiviral potency would therefore be enhanced. Subsequently, bis(NHS ester)PEG crosslinker was used to dimerize and trimerize PIE7. The resulting dimeric (PIE7)2 and trimeric (PIE7)3 inhibitors had IC50 values of 1.9 nM and 250 pM against HXB2, respectively. This represented an improvement of potency ranging from 325- to 2,500-fold over the PIE7 monomer.
D-peptides have several theoretical advantages: they are resistant to proteases leading to substantial increase in serum half-life; short D-peptides can be taken orally, whereas L-peptides must be injected to avoid gastrointestinal digestion; and D-peptides are a rich source unexplored structural diversity because they can bind to targets with unique interface geometries not available to D-peptides. Therefore D-peptides associated with current D-peptide entry inhibitors and are promising leads for the prevention and treatment of HIV-1/AIDS [88].
Cyclic peptides
θ-Defensins contain 18 cyclic peptides and their genes exist in primates. However, in humans, these genes and transcripts harbour a premature stop codon so that humans do not produce θ-defensin peptides. Retrocyclin-1 (RC-1) is a θ-defensin peptide with broad spectrum of antiviral properties in vitro. It protects cells from HIV-1, herpes simplex and influenza A infection. Gallo et al. [89] found that RC-1 inhibited HIV-1 Env-mediated membrane fusion in a dose-dependent manner with an IC50 of 1.5 μM. Complete inhibition of fusion was achieved at 4 μM. Further research found that RC-1 selectively targeted at the CHR region of the HIV-1 gp41 fusion intermediate, leading to inhibition of 6-HB formation.
Conclusions
HAART therapy has opened up a new avenue in the battle against AIDS. In the last decade, HAART therapy has significantly improved the prognosis for individuals infected with HIV-1. However, the drugs currently used in HAART regimens have significant side effects and resistance profiles. Therefore, development of new classes of anti-HIV-1 agents with low toxicity and low resistance is needed. From a therapeutic perspective, fusion inhibitors are among the most attractive agents for intervention in the viral life cycle. These compounds act outside the cell and are therefore independent of cellular transporters that might lower the effective intracellular concentrations. In addition, cytotoxicity is low because they do not actually enter cells.
Enfuvirtide is the first HIV-1 fusion inhibitor approved by the FDA. It is currently used in combination with other anti-HIV-1 drugs. Results from clinical trials indicated that it had potent activity against HIV-1 strains that are resistant to many other anti-HIV-1 drugs. By contrast, a large quantity of T20 is required to maintain the in vivo anti-HIV-1 efficacy. Treatment with T-20 is expensive at a price of US $20,000 and US $25,000 per patient per year in the USA and Europe, respectively. It is a huge economic and mental burden for AIDS patients. Moreover, resistance to enfuvirtide has been reported. The search for new fusion inhibitors with more potent activity and longer half-lives is imminent and essential. This review described several new fusion inhibitors, such as SFT, PEG(PIE7)3 and (CCIZN17)3, as potent antiviral agents; others, such as, M87oRRE/C46 as having a progressive gene therapy perspective; D-peptides as having the potential of oral administration; and HIV-1 broadly neutralizing antibody that extracts its epitope from a gp41 ECTO region have important implications for structure-guided vaccine design [90]. In addition to this, some fusion inhibitors can be developed as potential microbial microbicides, such as C34 [91], HR212 and peptide P1 (residues 649–683) [92], and they can be expressed into a live microbe and be secreted into mucosal surfaces [93]. In the prospect of being developed into microbial microbicides, both the lipid environment and pH are crucial factors to be taken into consideration for the structural stability of these peptides [92]. In the near future, some of these compounds will be added to the antiviral armoury to combat AIDS.
Footnotes
Acknowledgements
This work was supported in part by grants from the Chinese Academy of Sciences (KSCX1-YW-R-15 and KSCX1-YW-R-24), Key Scientific and Technological projects of China (2008ZX10001-002, 2008ZX10001-015, 2008ZX10005-005 and 2009ZX09501-029) and Yunnan province (2007BC006 and 2009CD109), 973 program (2006CB504302, 2006CB504208 and 2009CB5223006), the China Postdoctoral Science Foundation (20090451433) and the National Natural Science Foundation of China (U0832601 and 30671960).
The authors declare no financial disclosure or conflicts of interest.