
Abstract
Background:
Hepatitis C virus (HCV) NS5B is an essential component of the viral replication machinery and an important target for antiviral intervention. Aurintricarboxylic acid (ATA), a broad-spectrum antiviral agent, was evaluated and characterized for its anti-NS5B activity in vitro and in HCV replicon cells.
Methods:
Recombinant NS5B, HCV replicase and Huh-7 cells harbouring the subgenomic HCV replicon of genotype 1b were employed for biochemical and mechanistic investigations.
Results:
Analysis of ATA activity in vitro yielded equipotent inhibition of recombinant NS5B and HCV replicase in the submicromolar range (50% inhibition concentration [IC50] approximately 150 nM). Biochemical and mechanistic studies revealed a bimodal mechanism of ATA inhibition with characteristics of pyrophosphate mimics and non-nucleoside inhibitors. Molecular modelling and competition displacement studies were consistent with these parameters, suggesting that ATA might bind to the benzothiadiazine allosteric pocket 3 of NS5B or at its catalytic centre. Kinetic studies revealed a mixed mode of ATA inhibition with respect to both RNA and UTP substrates. Under single-cycle assay conditions, ATA inhibited HCV NS5B initiation and elongation from pre-bound RNA, but with ≥fivefold decreased potency compared with continuous polymerization conditions. The IC50 value of ATA for the native replicase complex was 145 nM. In HCV replicon cells, ATA treatment ablated HCV RNA replication (50% effective concentration =75 nM) with concomitant decrease in NS5B expression and no apparent cytotoxic effects.
Conclusions:
This study identified ATA as a potent anti-NS5B inhibitor and suggests that its unique mode of action might be exploited for structural refinement and development of novel anti-NS5B agents.
Introduction
Hepatitis C virus (HCV) is a significant human blood-borne pathogen affecting an estimated 3% of the world's population, of whom 80% progress to chronic infection [1,2]. Sequelae include fibrosis, cirrhosis and hepatocellular carcinoma, making HCV the leading cause of liver transplantation in the USA [2,3]. The currently approved anti-HCV therapy of pegylated interferon-α (PEG-IFN-α) in combination with ribavirin exhibit sustained virological response (SVR) rates of 40–50% for genotype 1 and ≤80% for genotypes 2 and 3, and are associated with severe side effects resulting in limited patient compliance [4]. Additional challenges are posed by the high cost of therapy and the emergence of HCV quasispecies during treatment [5]; therefore, it is paramount to develop new and improved anti-HCV therapeutic agents. A prime area of focus is the development of small molecule inhibitors targeting the essential viral enzymes.
HCV is an enveloped, single-stranded RNA virus with approximately 9.6 kb genome of positive polarity that encodes three structural and seven non-structural (NS) proteins [6,7]. Among these, the NS5B RNA-dependent RNA polymerase (RdRp), a crucial and unique component of the viral replication machinery with no functional equivalent in the host, has emerged as an important and attractive target for the development of anti-HCV agents [8,9].
HCV NS5B is a 66 kDa phosphoprotein with predominant perinuclear localization [10]. Characterization of biochemical and enzymatic attributes of NS5B has been made feasible by purification of full-length and various C-terminally truncated forms of recombinant NS5B [9,11,12]. A significant advancement in unravelling structural features of NS5B has been achieved by solving three dimensional crystal structures of several truncated forms of NS5B and its complexes with ribonucleotides or non-nucleoside inhibitors (NNIs) [9,13,14]. These investigations have revealed that NS5B exhibits a classical ‘right hand’ topology of the polymerase family, with the characteristic fingers, palm and thumb subdomains, as well as several unique structural features, which have substantially aided medicinal chemistry efforts for developing anti-NS5B compounds.
Employing biochemical or cell-based replicon assays, several distinct classes of NS5B RdRp inhibitors have been identified in recent years. Based on their chemical structure and putative mode of action, these have been broadly categorized as nucleoside analogue inhibitors (NIs) or NNIs. The NI, which resemble nucleoside substrates, are first metabolized to the corresponding triphosphate analogues by the host cell machinery and compete with the natural ribonucleoside triphosphates (rNTPs) of NS5B at its catalytic centre, causing chain termination upon incorporation into the RNA strand [9,13,14]. This group of inhibitors has been shown to suppress HCV replication in both subgenomic replicons and HCV-infected chimpanzees. The NNIs include a number of structurally diverse scaffolds, which interfere with the initiation step of RNA synthesis through allosteric inhibition of NS5B [9,13,14]. At least three distinct NNI binding sites have been mapped to date, all of which, interestingly, are located exclusively in the palm and thumb subdomains of NS5B [9,13,14]. Pyrophosphate mimetics, sometimes categorized within the NNI class, represents a third group of NS5B inhibitors. Representative members of this group include derivatives of dihydroxypyrimidine carboxylic acid and diketo acid (DKA) [14–17]. These compounds function as product-like analogues and are presumed to inhibit NS5B through an interaction with the catalytic metal ion at the enzyme's active site. Studies with the aforementioned inhibitor classes have been instrumental in providing substantial insights into the mechanism of NS5B inhibition and have opened new possibilities for antiviral drug design.
Aurintricarboxylic acid (ATA) is a carboxylated triphenylmethane derivative with versatile biological activities and modes of action. These include its inhibitory effects on enzymes that bind nucleic acid, such as DNA and RNA polymerases [18], nucleases [19], DNA topoisomerase [20] and reverse transcriptase (RT) [21]. In addition, ATA has also been reported to inhibit protein synthesis by blocking mRNA association with ribosomes [22], disrupt cellular signalling pathways by diverse mechanisms [23–26] and promote cell survival in response to variety of insults in various cell types [23,26,27]. The therapeutic potential of ATA is highlighted by its powerful antiviral activity against HIV [18,21], vesicular stomatitis virus [28], severe acute respiratory syndrome coronavirus (SARS-CoV) [29], Rauscher leukaemia virus [18,21] and vaccinia virus [30].
The effect of ATA on any RdRp family of enzymes, including HCV NS5B, has not been investigated to date. In this study, we describe the identification and characterization of ATA as a highly potent anti-NS5B agent both in vitro and in the HCV replicon system. Moreover, ATA inhibits the native HCV replicase complex in cell-free replication assay. We have extended our studies to understand the ATA-mediated NS5B inhibition mechanism and provide evidence for its dual mode of action by biochemical, kinetic and competition displacement assays. Furthermore, we also report the computationally predicted binding energy of ATA within the different allosteric pockets of NS5B and at its active site through a molecular docking approach.
Methods
Compound
ATA was obtained from Sigma-Aldrich (Saint Louis, MO, USA).
Reagents, antibodies and cell culture medium
The source of materials used in this work were as follows: nickel-nitrilotriacetic acid agarose (Ni-NTA) and NAP-10 columns from GE Healthcare (Piscataway, NJ, USA); radiolabelled [α-32P] rNTPs from Perkin Elmer (Waltham, MA, USA); [14C]-foscarnet from Moravek Biochemicals (Brea, CA, USA); HPLC-grade nucleoside triphosphates, RNase Out, glycogen and Complete™ protease inhibitor cocktail from Roche (Mannheim, Germany); GF-B filters from Whatman (Piscataway, NJ, USA); T4 polynucleotide kinase from Invitrogen (Carlsbad, CA, USA); western blotting luminol reagent from Santa Cruz Biotechnology (Santa Cruz, CA, USA); MEGAscript T7 transcription kit from Ambion (Austin, TX, USA); RNeasy kit from Qiagen (Valencia, CA, USA); rabbit polyclonal antibody against NS5B was developed at Covance (Denver, PA, USA) using recombinant NS5BCΔ21 as immunogen; anti-NS5A antibody was a kind gift from Dr Craig Cameron; monoclonal antibodies against NS3 and NS4B were purchased from Virogen (Watertown, MA, USA); GRP78 antibody was procured from BD Transduction Laboratories (San Jose, California, USA); anti-GAPDH antibody was purchased from Fitzgerald Industries International (Concord, MA, USA); and horseradish peroxidase-conjugated secondary antibodies were obtained from Rockland (Gilbertsville, PA, USA). Dulbecco's modified Eagle's medium was from Invitrogen; l-glutamine, non-essential amino acids, penicillin, streptomycin and G418 sulfate were purchased from Mediatech, Inc. (Manassas, VA, USA); and fetal bovine serum (FBS) was from Hyclone (Logan, UT, USA) or Mediatech, Inc. All other chemicals were of the highest available molecular biology grade and purchased from Fisher (Pittsburgh, PA, USA), Sigma (Saint Louis, MO, USA) or Bio-Rad (Hercules, CA, USA).
Cells
Three subgenomic genotype 1b HCV replicon cells, NNeo/3-5B (RG) [31], MH14 [32] and BB7 [33] obtained from Drs Stanley Lemon, Kunitada Shimotohno and Charles Rice, respectively, were employed in our studies. The replicon-bearing cell lines were cultured in Dulbecco's modified Eagle's medium, supplemented with 2 mM L-glutamine, 100 units/ml non-essential amino acids, 10% heat-inactivated FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 500 μg/ml G418. Huh-7, Huh-7.5 (provided by Dr Charles Rice) and cured MH14 cells (provided by Dr Kunitada Shimotohno) were maintained in the same media described above in the absence of G418.
Enzymes
Recombinant NS5BCΔ21 protein was purified from the plasmid pThNS5BCΔ21 expressed in Escherichia coli DH5α by Ni-NTA column chromatography as described previously [34,35]. Recombinant heterodimeric HIV type-1 RT was purified by Ni-NTA chromatography employing the pET-28a-RT66 and pET-28a-RT51 plasmids, carrying metal-binding His-Tag sequences at their N-terminal region as described before [36].
Primer-dependent and de novo RNA-dependent RNA polymerase assays
The standard primer-dependent NS5B RdRp assay was performed on poly rA/U12 template–primer (TP) as previously described [34,35]. Briefly, NS5B RdRp activity was determined in the absence or presence of ATA at 30°C for 60 min in a reaction buffer (25 μl) containing 20 mM Tris–HCl (pH 7.0), 100 mM NaCl, 100 mM sodium glutamate, 0.5 mM dithiothreitol (DTT), 0.01% bovine serum albumin (BSA), 0.01% Tween-20, 5% glycerol, 20 U/ml of RNase Out, 0.5 μM poly rA/U12 (pre-annealed), 25 μM UTP, 2–5 μCi [α-32P]UTP and 300 ng of NS5BΔΔ21. All reactions, except where specified, were initiated by the addition of 0.5 mM MnCl2 and terminated by the addition of ice-cold 10% (v/v) trichloroacetic acid (TCA) containing 0.5 mM pyrophosphate. The quenched reaction mixtures were spotted onto GF-B filters, washed with 5% (v/v) TCA-0.5 mM pyrophosphate to remove unincorporated rNTPs, rinsed and quantified on a liquid scintillation counter (Beckman Coulter Fullerton, CA, USA). Activity of NS5B in the absence of the inhibitor was set at 100%, and its presence was calculated relative to this control. The 50% inhibition value (IC50) for ATA was evaluated from the dose–response curve employing typically 9–12 concentrations of the compound and SigmaPlot 8.0 software (Aspire Software International, Ashburn, VA, USA). N,N-disubstituted phenylalanine derivative number 14, a well characterized NS5B inhibitor [37], was included as an internal reference standard.
The primer-independent de novo initiation activity of NS5B in the presence of ATA was reconstituted on HCV 5′-untranslated region (UTR) RNA template. To synthesize this transcript, we first generated the HCV 5′-UTR PCR product by using the HCV pCVJ4L6S template and the primers N1 5′-TAATACGACTCACTATAGCCAGCCCCCTGATGGGGGCG-3′, carrying the T7 promoter sequence (sequence in bold) and N2 5′-GGGTGCACGGTCTACGAGACC-3′. The resulting 341 base-pair PCR product was gel-purified and directly used as a template for synthesizing the HCV 5′-UTR run-off RNA transcript in vitro using the MEGAscript® T7 transcription kit. Reaction conditions for the primer-independent assays were similar to the primer-dependent assays except that incubations were carried out at 30°C for 90 min at increased concentrations of NS5BCΔ21, HCV 5′-UTR RNA (500 ng each) and all four rNTPs at final concentrations of 0.5 mM each ATP, GTP, UTP and 10 μCi/20 μM [α-32P]CTP were included in the reaction.
Analyses of the reaction products of primer-dependent (poly rA/U12) and primer-independent (HCV 5′-UTR RNA) assays as a function of ATA concentrations were carried out on a 8% polyacrylamide gel containing 7 M urea, under conditions as described above except that the amount of NS5B and reaction time was reduced by one-third. Reactions were stopped by the addition of 25 mM EDTA-0.5% SDS, supplemented with glycogen (5 μg) as carrier, followed by phenol chloroform extraction and alcohol precipitation. The reconstituted RNA products in formamide gel-loading buffer were resolved on a denaturing gel and visualized by phosphorimaging (Molecular Dynamics, Piscataway, NJ, USA).
Experiments involving assessment of different pre-incubation conditions required modified order of addition of the reaction components and were carried out essentially as described previously [34,38,39]. For the single-cycle RdRp assays, the RNA TP was first pre-incubated with NS5B on ice for 30 min, after which heparin (at 1.2 μg/ml final concentration), ATA and substrates were successively added. To ensure effectiveness of the heparin trap, NS5B was pre-incubated with heparin (1.2 μg/ml) on ice for 30 min, prior to the addition of RNA TP and substrates. Reactions were quenched after 60 min of incubation, and percentage inhibition and IC50 values were evaluated as described above.
For comparison, the activity of ATA against HIV-1 RT was measured under conditions as previously described [34,35].
Determination of the inhibition constant and mode of inhibition
Two series of reactions were performed to determine the mode of inhibition [34,35,38]. In the first series, concentration of UTP was fixed at 25 μM, whereas the concentrations of ATA and poly rA/U12 (0.2–2.5 μM) varied. In the second series of experiments, reaction velocities were determined at a fixed concentration of poly rA/U12 (0.25 μM) and varying concentrations of ATA and UTP (ranging from 2.5 to 80 μM). For each set, aliquots were withdrawn at defined times, quenched with 10% (v/v) TCA-0.5 mM inorganic pyrophosphate and radiolabelled UTP incorporated into RNA product was determined by GF-B filter-binding assay as described above. To determine the mode of inhibition and inhibition constant (Ki), data were plotted according to the methods of Dixon and Cornish-Bowden [40]. The assays were performed at least twice and values represent an average of at least duplicate samples. All Ki SD were <10%.
Cross-linking of NS5B to TP
Cross-linking of NS5B to RNA was performed in accordance with previously standardized conditions at increasing ATA concentrations on a pre-annealed 32P-labelled U12/rA20 (15,000 cpm/pmol) TP as described earlier [34,35]. Reactions were incubated for 10 min on ice in the absence or increasing concentrations of ATA in 50 μl reaction volume containing 20 mM HEPES (pH 7.0), 50 mM NaCl, 0.5 mM DTT, 0.01% BSA, 5% (v/v) glycerol, 20 U/ml of RNasin, 200 nM of 32P-labelled U12/rA20 (15,000 cpm/pmol), 1.5 μg of NS5B and 0.5 mM MnCl2. For cross-linking, the reaction mixture was exposed to 254 nm UV radiation at a dose of 300 mJ/cm2 (Spectronic Corporation, Westbury, NY, USA); the cross-linked species were resolved by SDS-PAGE (8%) and visualized by phosphorimaging.
Molecular modelling
All computations were carried out on a Dell Precision 470n workstation with the RHEL 4.0 operating system using Glide 4.5 (Schrodinger LLC, New York, NY, USA). For docking experiments, ATA was constructed using the fragment dictionary of Maestro 8.0 and geometry optimized using the optimized potentials for liquid simulations-all atom force field [41], with the steepest descent followed by truncated Newton conjugate gradient protocol as implemented in Macromodel 9.5. Water molecules of crystallization were removed from the complex, and the protein was optimized for docking using the protein preparation wizard provided by Schrodinger LLC and the Impact Program (First Discovery v4.5). Partial atomic charges for ATA and protein were assigned according to the Optimized Potentials for Liquid Simulations-all atom force field. The extra precision Glide docking method was then applied to dock ATA into the HCV NS5B NNI binding sites for tetracyclic indole (Protein Data Bank indentification [PDB ID]: 2DXS) [42], N,N-disubstituted phenylalanine (PDB ID 1NHU) [37] and benzothiadiazine (PDB ID 2FVC) [43] inhibitors. The binding site, for which the various energy grids were calculated and stored, is defined in terms of two concentric cubes: the bounding box, which must contain the centre of any acceptable ligand pose, and the enclosing box, which must contain all ligand atoms of an acceptable pose. Cubes with an edge length of 12 Å and centred at the midpoint of the longest atom–atom distance in the respective cocrystallized ligand defined the bounding box in the protein. The larger enclosing box was also defined in terms of the cocrystallized ligand: an edge length of 30 Å was used. Poses with a root mean squared deviation of <0.5 Å and a maximum atomic displacement of <1.3 Å were eliminated as redundant in order to increase diversity in the retained ligand poses. The scale factor for van der Waals radii was applied to those atoms with absolute partial charges ≤0.15 (scale factor of 0.80) and 0.25 (scale factor of 1.0) electrons for ligand and protein, respectively. The maxkeep variable, which sets the maximum number of poses generated during the initial phase of the docking calculation, was set to 5,000 and the keep best variable, which sets the number of poses per ligand that enters the energy minimization, was set to 1,000. Energy minimization protocol includes dielectric constant of 4 and 1,000 steps of conjugate gradient. Upon completion of each docking calculation, 100 poses per ligand, at most, were generated. The best docked structure was chosen using a Glidescore (Gscore) function. The Gscore is a modified and extended version of the empirically based Chemscore function [44].
Competition displacement assays
Binding of ATA to NS5B was examined by competition displacement assay employing [14C]-foscarnet as a probe and nitrocellulose membrane filter assembly [17]. Briefly, 9 μg of NS5BCΔ21 and 0.2 μCi [14C]-foscarnet (54 mCi/mmol) were incubated for 15 min at 30°C in the absence or increasing concentrations of ATA in NS5B-binding buffer (20 μl) containing 20 mM Tris–HCl (pH 7.0), 100 mM NaCl, 100 mM sodium glutamate, 0.5 mM DTT, 0.01% Tween-20 and 2 mM MgCl2. As a negative control, equivalent amount of BSA was incubated with [14C]-foscarnet. The nitrocellulose membrane circles (2.5 cm) were pre-washed with 0.5 N NaOH, rinsed with water and equilibrated with NS5B-binding buffer prior to use. An aliquot of the binding mix (18 μl) was spotted on the nitrocellulose membrane circle, washed with NS5B-binding buffer containing 0.1% triton to remove unbound [14C]-foscarnet, rinsed and quantified on a liquid scintillation counter (Packard). The extent of [14C]-foscarnet bound to NS5B under these conditions was determined as the percentage of control (no ATA) and was plotted versus ATA concentration.
Isolation of HCV cytoplasmic replicase complex
HCV crude replication complexes were isolated and fractionated from NNeo/3-5B (RG) and MH14 cells as described previously [45,46]. Corresponding control cytoplasmic membrane fractions were processed from Huh-7 and cured MH14 cells in parallel. Briefly, replicon cells cultured in 100 mm diameter petri dishes to 80% confluency were washed with cold buffer A containing 150 mM sucrose, 30 mM HEPES (pH 7.4), 33 mM ammonium chloride, 7 mM KCl and 4.5 mM magnesium acetate, treated with 3 ml lysolecithin buffer (250 μg/ml lysolecithin in buffer A) and followed by two consecutive washes (3 ml each) with buffer A to remove traces of lysolecithin. The cells were collected by scraping in extraction buffer (20 mM HEPES [pH 7.5], 25 mM ammonium chloride, 10 mM KCl and 1.5 mM MgCl2), lysed gently by pipetting at least 15×, and the unbroken cell and nuclei were removed by centrifugation at 800×g for 5 min. Membrane-bound crude replication complexes were pelleted from the cytoplasmic extract by centrifugation at 30,000×g for 30 min, reconstituted in a buffer containing 10 mM HEPES (pH 7.5), 10 mM KCl, 1 mM DTT and 15% glycerol at 5×105 cell equivalent/ml, and stored in aliquots at −70°C.
Cell-free replication assays
Replicase assays with HCV cytoplasmic replicase complex (CRC; 10 μl) were performed in the absence or presence of ATA at 30°C for 120 min in a reaction buffer (25 μl) containing 50 mM HEPES (pH 7.5), 10 mM KCl, 10 mM DTT, 50 mM ammonium chloride, 7 mM KCl, 1 mM spermidine, 20 μg/ml actinomycin D, 1 mM ATP, 1mM GTP, 1mM CTP, 30 μCi [α-32P]-UTP (20 μM), 20 U/ml of RNase Out and 10 mM MgCl2 as described previously [45,46]. Equivalent amounts of cytoplasmic membrane fraction isolated from Huh-7 or cured MH14 cells were employed as negative controls. Reactions were terminated by the addition of 50 mM EDTA and 0.5% SDS. RNA products were recovered by phenol–chloroform–isoamyl alcohol (25:24:1) extraction and ethanol precipitation, washed with 70% ethanol and resuspended in RNase-free water. Radiolabelled HCV subgenomic RNA, transcribed in vitro from the XbaI-linearized MH14 plasmid using a MEGAscript T7 kit according to the manufacturer's instructions, was loaded as a molecular marker. Samples were resolved on a denaturing formaldehyde agarose gel (0.8%), the gel was dried and products were visualized on a phosphoimager. RNA synthesis activity of the HCV CRC as a function of ATA concentration was quantified by ImageQuant software, expressed relative to the RNA synthesis obtained in the absence of the inhibitor, and IC50 values were evaluated using SigmaPlot 8.0 software.
Quantitative real-time PCR-based HCV replicon assay
The antiviral activity of ATA was determined in HCV subgenomic replicon cell line BB7 by quantitative real-time PCR. Briefly, BB7 cells seeded at a density of 2×105 cells/well in a 12-well plate were allowed to attach overnight in complete culture medium without G418. The next day, the culture medium was replaced with medium containing serially diluted concentrations of ATA in the presence of 5% FBS. The culture medium was replaced every day with fresh medium containing the specific concentration of ATA. After the cells were treated for 72 h with ATA, total RNA was extracted using an RNeasy Kit. HCV RNA corresponding to the 5′-non-translated region of HCV was measured by the TaqMan® quantitative real-time reverse transcription PCR (Applied Biosystems, Foster City, CA, USA) as described previously [33]. The level of HCV RNA was normalized for each sample using the comparative CT method with GAPDH as endogenous control in accordance with the manufacturer's specifications. The relative amount of HCV RNA in the absence of ATA was set at 100% and the presence of ATA was quantified relative to this control. The 50% effective concentration (EC50) value was determined from the ATA dose–response curve employing SigmaPlot 8.0 software. Each data point represents an average of at least two independent experiments.
Cell proliferation assays
BB7 and Huh-7.5 parental cells were plated in a 12-well plate at a density of 2×105 cells/well and treated with 0, 0.1, 1 and 10 μM ATA in triplicate. Fresh medium containing ATA was replenished every day. After 3 days, the cells were washed with PBS, harvested and counted. Cell numbers relative to those of cells without treatment were plotted against the concentration of ATA. Each data point represents an average of three replicates in cell culture. To monitor the effect of ATA on HCV NS5B expression, BB7 cells were seeded in a six-well plate and treated with increasing concentrations of ATA for a 72 h period with replenishment of fresh ATA every 24 h. In another set, the cells were treated with IFN-α at a concentration of 200 IU/ml. The cells were lysed in 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, and 1X Complete™ protease inhibitor cocktail. Cell lysates were clarified by centrifugation at 16,000 rpm for 15 min. Equal amounts of total protein from cell lysates were immunoblotted with anti-NS5B antibodies. GAPDH was used as a loading control. Two independent sets in duplicate were evaluated for this study.
Results
Identification of ATA as a potent inhibitor of the RdRp activity of HCV NS5B
In an ongoing quest for novel HCV NS5B inhibitors, we evaluated ATA (Figure 1A), a versatile compound with previously reported activities against polynucleotide-synthesizing enzymes. As a prerequisite, we purified to near homogeneity, functionally active recombinant HCV NS5B (genotype 1b) with an N-terminal His-tag and C-terminal 21-amino-acid deletion (NS5BCΔ21). N,N-disubstituted phenylalanine derivative number 14, a documented NS5B inhibitor [37], was included as an internal reference standard in this investigation and yielded an IC50 value of 0.3 μM under our experimental conditions, as previously reported [34]. The anti-NS5B activity of ATA was evaluated by the standard primer-dependent elongation reactions on poly rA/U12, as well as by the primer-independent de novo initiation mechanism on HCV 5′-UTR RNA template. Irrespective of the mechanism, ATA strongly inhibited the RdRp activity of NS5B in a dose-dependent manner on both these RNA TPs exhibiting IC50 values in the submicromolar range in the presence of Mn2+ as the divalent cation (Figure 1B). Furthermore, under these conditions, ATA exhibited a near twofold higher inhibitory potency on HCV 5′-UTR RNA template (IC50=149 nM) versus the homopolymeric poly rA/U12 TP (IC50=259 nM). However, it should be noted 15–20% NS5B RdRp activity was refractory to ATA treatment on both these RNA templates, even at the highest inhibitor concentration employed in this assay. This suggests that similar to benzothiadiazine and benzimidazole compounds [47], the activity of ATA might be affected by template RNA.
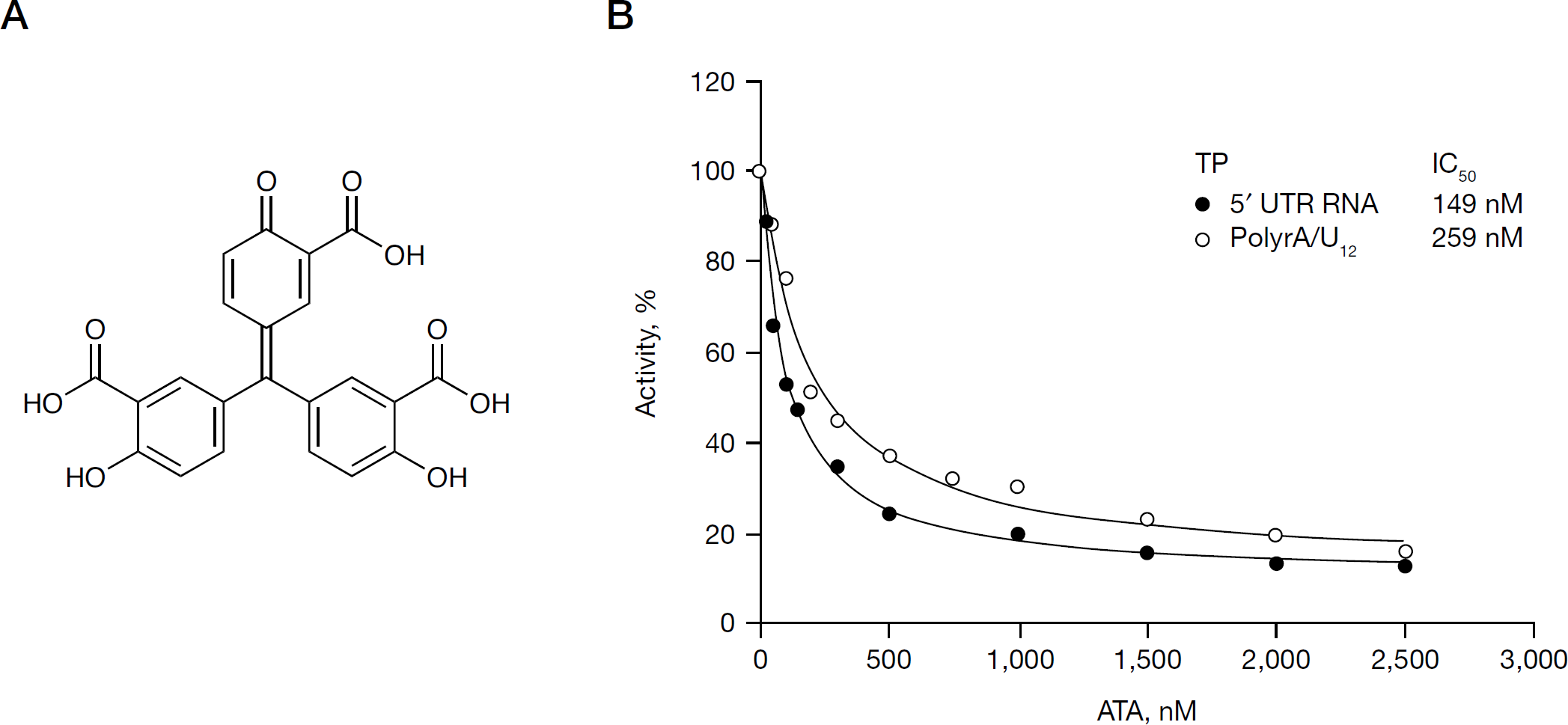
ATA inhibits hepatitis C virus NS5B RdRp
As the anionic ATA molecule harbours a keto carboxylic acid functional group, a pharmacophoric feature of the DKA series, we speculated that its inhibitory potency might be dependent on the nature of the divalent cation used in the assay, similar to that reported for some NS5B pyrophosphate mimetic inhibitors [14–17]. As expected, the IC50 value of ATA shifted to the low micromolar range with Mg2+ as the divalent cation, yielding values corresponding to 4.6 μM and 4.1 μM on 5′-UTR RNA and poly rA/U12 TP, respectively. More importantly, the 16–31-fold increase in the IC50 value in the presence of Mg2+ was consistent with the trend exhibited by other NS5B inhibitors belonging to the dihydroxypyrimidine and DKA series [14–17]; thus suggesting that ATA displays characteristics of NS5B pyrophosphate mimetic inhibitors.
We also analysed qualitatively, the size and pattern of the RNA products synthesized by NS5B in the presence of ATA on poly rA/U12 and HCV 5′-UTR RNA templates by gel-based analysis (Figure 2). Similar assays have been reported previously by us [35,36], as well as by several other groups on a variety of RNA TPs [38,39,48,49]. As seen in Figure 2, reactions carried in the absence of ATA (lane 1), yielded a predominant RNA product corresponding to the size of their respective templates (indicated by ‘P’), as discerned from the position of the labelled RNA template (not shown). In addition, on the HCV 5′-UTR RNA template, another prominent slower-migrating species (indicated by an asterisk) was also observed, its size corresponding to RNA twice as large as the template. A similar aberrant migration pattern of the HCV RNA product has been earlier reported by several other groups [39,48,49] and might possibly correspond to a RNA hybrid formed between the template RNA and its product initiated from the 3′-OH group of the template [39,48,49]. ATA inhibited full-length product formation on both RNA templates in a dose-dependent manner (lanes 2–10), with near absence of product synthesis seen at the highest concentration of ATA employed in the assay (lane 10). Furthermore, on HCV 5′-UTR RNA template, the intensity of the slower-migrating species also decreased concomitantly with increasing ATA concentration. Interestingly, reactions containing ATA did not appear to produce shorter products as a result of abortive initiation or premature termination.
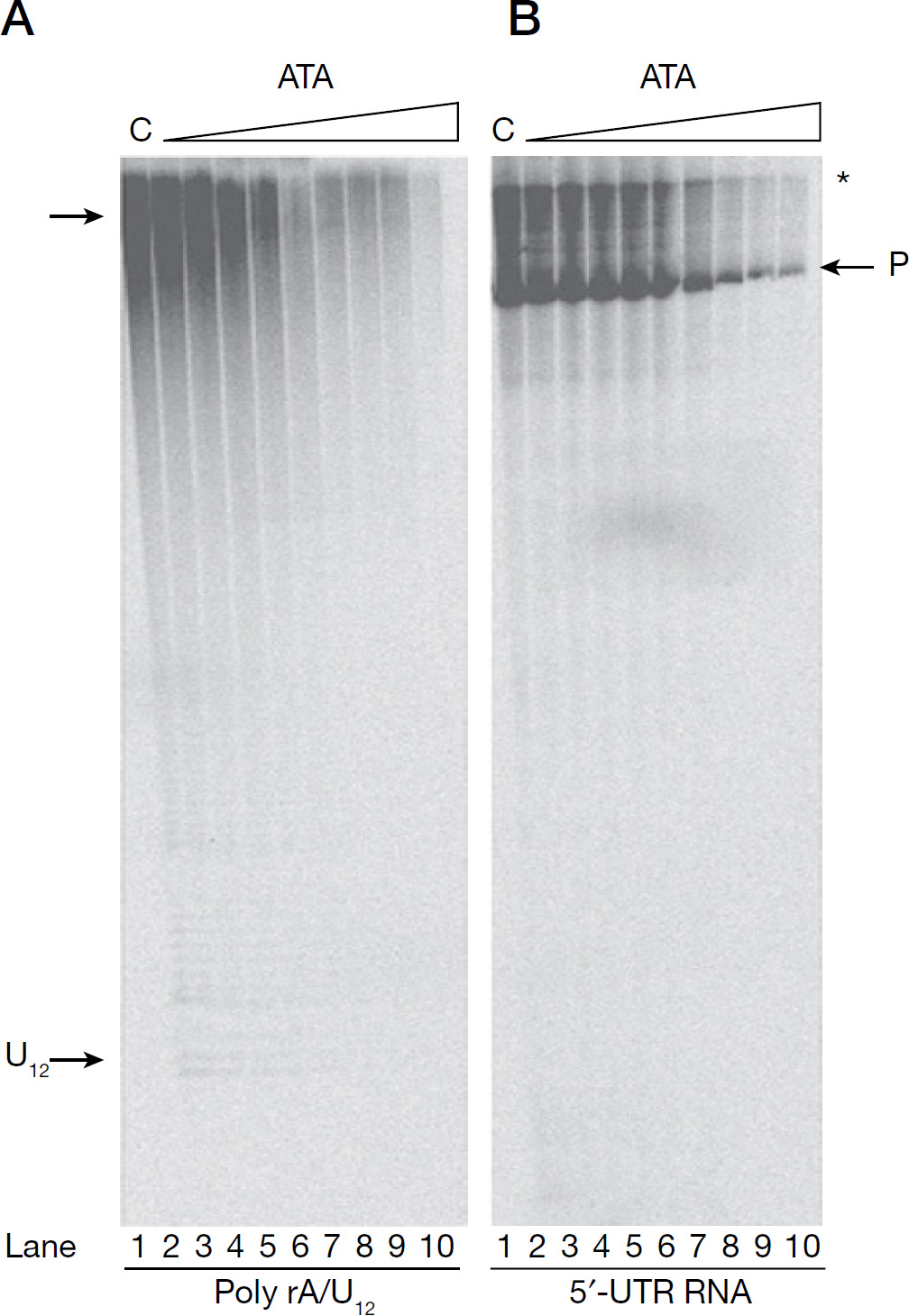
Product analysis of HCV NS5B RdRp reaction as a function of ATA concentration
Mechanism of inhibition
In order to investigate the mechanism by which ATA inhibits NS5B, two different series of kinetic analyses were performed and analysed by Cornish-Bowden and Dixon plots to evaluate the mode of inhibition and determine the Ki values. As seen from the reciprocal plots of reaction velocity, ATA exhibited a mixed mode of inhibition towards both the TP and UTP substrates, with a major competitive component and a minor uncompetitive component deduced from the location of the intercepts on the Dixon and Cornish-Bowden plots above and below the x-axis, respectively (Figure 3). The inhibition constants with respect to TP corresponded to Ki competitive =0.36 μM and Ki uncompetitive =3.20 μM, whereas these values with respect to UTP were Ki competitive =0.39 μM and Ki uncompetitive =3.00 μM. These data clearly indicate that although the mode of inhibition of ATA towards TP is similar to the HCV NS5B NNI class, such as coumestan, 4-thiazolidinone and benzimidazole derivatives [35,36,38], its mode of action in regards to UTP substrate differs markedly from all other HCV NS5B inhibitors investigated to date, irrespective of their class.
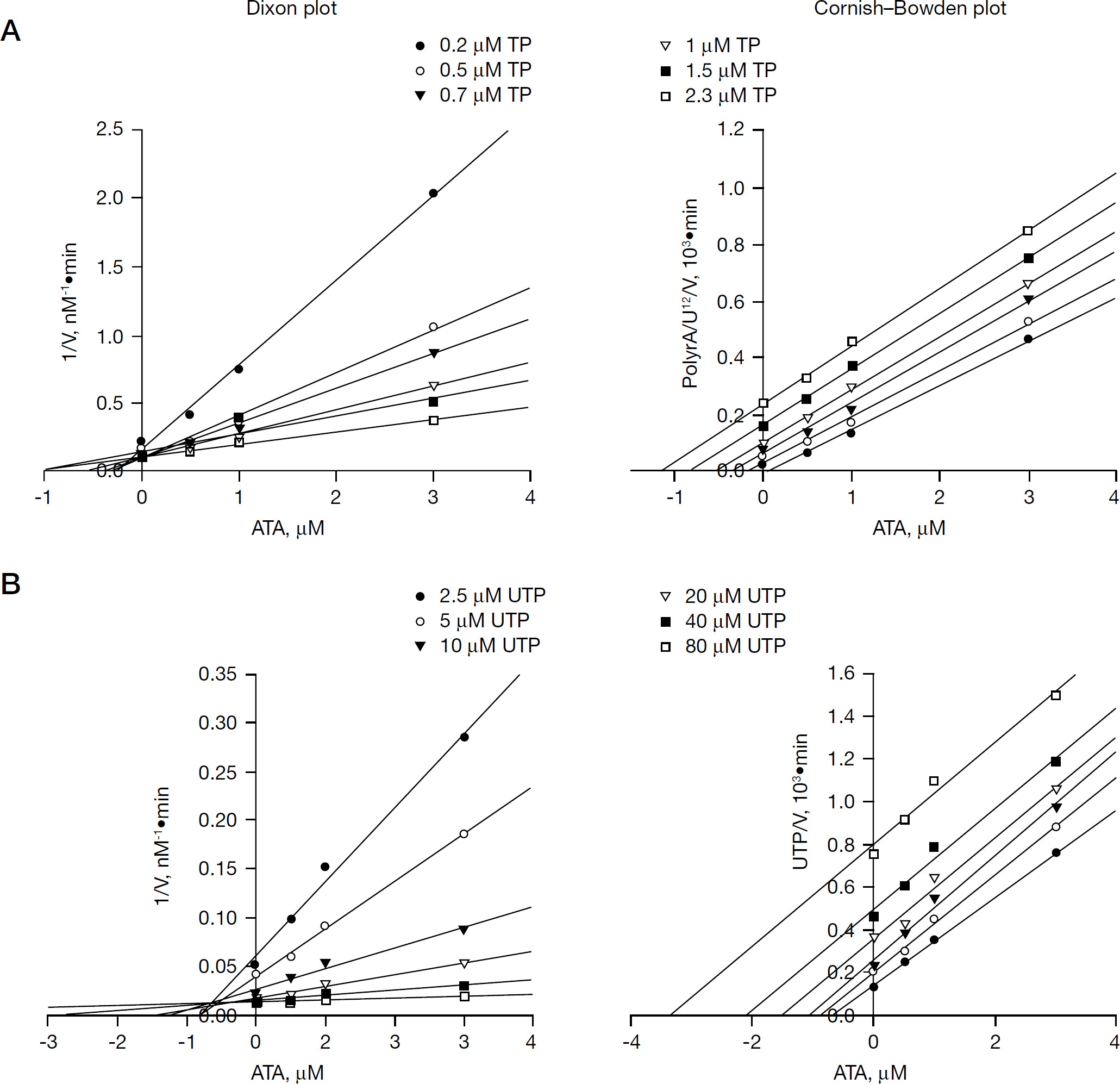
Evaluation of the mode of inhibition and kinetic parameters of ATA with regards to TP and UTP substrates
The above mechanistic studies revealed the ability of ATA to compete with both the TP and UTP substrates; however, it was unclear from this investigation whether ATA inhibits by preferentially binding to the NS5B apoenzyme prior to RNA binding or by forming a ternary complex with NS5B-bound RNA. To characterize these parameters, we carried out ATA inhibition assays under conditions of varying order of reagent addition (Figure 4). When NS5B was pre-incubated with ATA (E+I in Figure 4) before the addition of TP, its IC50 value dropped modestly from 259 nM and 149 nM to 160 nM and 127 nM, on poly rA/U12 and HCV 5′-UTR RNA T/P, respectively. In this scenario, a gradual near-complete inhibition of the NS5B activity was obtained at higher concentrations of ATA (data not shown), suggesting that binding of ATA to NS5B might occlude RNA binding thereby preventing RdRp reaction. Consistent with this observation, pre-incubation of NS5B with RNA (E+T in Figure 4) prior to the addition of ATA, exhibited a near 1.6- and 2.6-fold upward shift in its IC50 value on poly rA/U12 and HCV 5′-UTR RNA, respectively. Furthermore, under these pre-incubation conditions, a fraction of NS5B was refractile to ATA inhibition even at higher concentrations of the inhibitor, thus suggesting that pre-incubation with RNA might partially protect NS5B from ATA inhibition. To preclude the possibility that ATA–RNA interactions might affect inhibition characteristics, IC50 values were also determined under conditions of pre-incubation of ATA with RNA (I+T in Figure 4) prior to the addition of NS5B and substrates. Under these conditions, the IC50 value on poly rA/U12 was near similar to reactions carried out in the absence of pre-incubation (E+T+I in Figure 4), but exhibited a marginal 1.4-fold increase on HCV 5′-UTR RNA. Cumulatively, the pre-incubation data are consistent with the kinetic data and mode of inhibition, and suggest that ATA might exert its inhibitory effect by interfering with an early step in the RdRp reaction such as productive RNA binding to the enzyme.
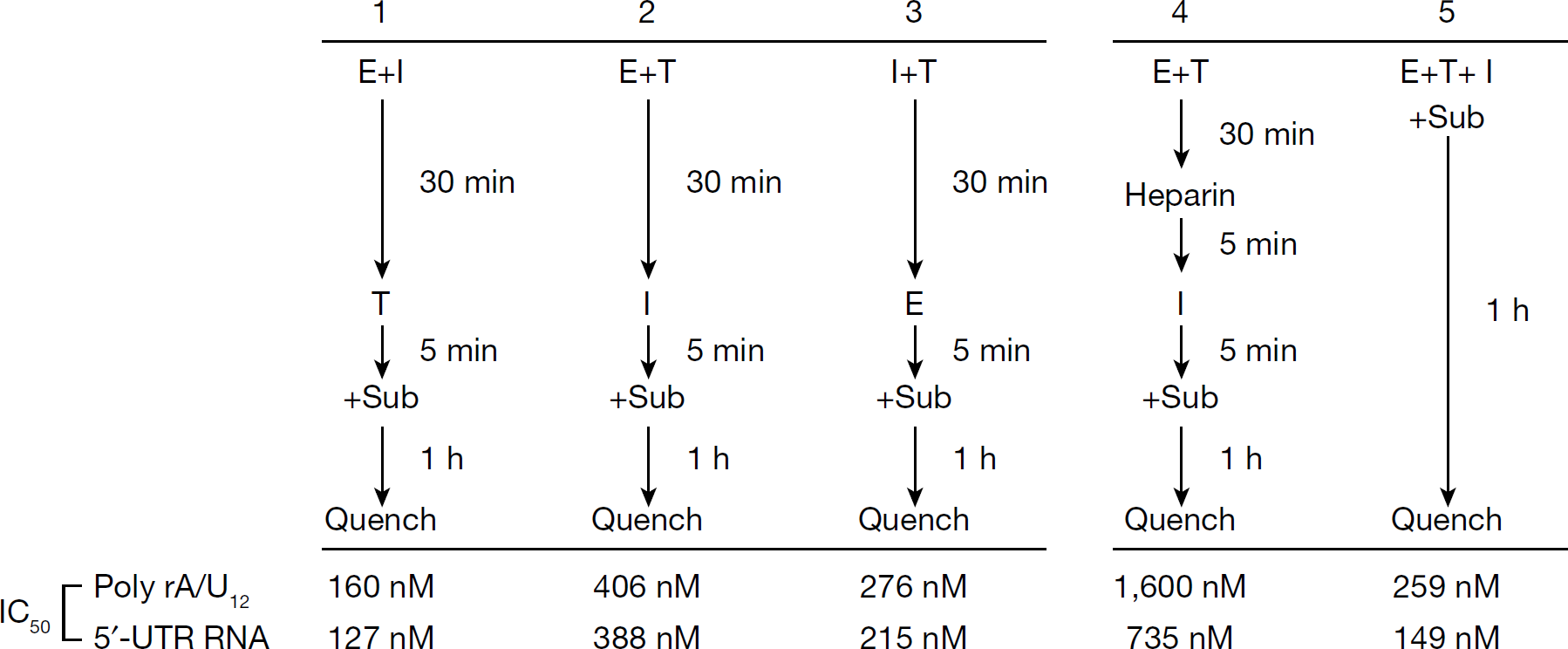
Effects of pre-incubation on ATA-mediated inhibition of NS5B activity
In order to further assess the potential of ATA to inhibit initiation or subsequent elongation from preformed HCV NS5B–RNA complexes under conditions that favour single-cycle RNA synthesis, the RdRp assays were modified to include incubation with heparin prior to the initiation of RNA synthesis in the presence of ATA. In this assay, heparin functions as a trap and prevent reinitiation events by quenching unbound NS5B and NS5B that might have dissociated from the RNA during elongation. The effectiveness of the heparin trap was evident from a near-complete absence RNA synthesis when NS5B was pre-bound to heparin, prior to the addition of RNA and initiation of RNA synthesis. This ensured evaluation of single-round of RNA synthesis occurring from already initiated complexes or complexes in the elongation phase, yielding IC50 values of 1.6 μM and 735 nM on poly rA/U12 and HCV 5′-UTR RNA, respectively (Figure 4, set 4). Although these values are ≥fivefold higher than the values obtained under continuous polymerization conditions (E+T+I in Figure 4), it is notable that ATA still exhibits potent inhibition of NS5B RdRp activity even under conditions involving single-cycle RNA synthesis. These data suggest that ATA inhibits initiation and elongation from pre-bound NS5B–RNA complexes.
ATA competes with RNA for NS5B binding
The mixed mechanistic mode towards the RNA template and the supporting NS5B–RNA pre-incubation data imply that ATA inhibits NS5B by potentially disrupting the binding of RNA to NS5B. To obtain more direct evidence in support of this occurrence, we resorted to UV-mediated cross-linking of NS5B–RNA binary complex as a function of ATA concentration. Similar assays have been previously described by us to study inhibitor interference at the step of NS5B–RNA binary complex formation [34,35]. As seen in Figure 5, cross-linking in the presence of ATA reduced the amount of NS5B–RNA binary complex species. Furthermore, this reduction directly correlated with ATA concentrations (Figure 5, lanes 2–8), with complete abolition of complex formation occurring at ATA concentration of ≥10 μM. These observations clearly indicate that ATA adversely affects the binding of RNA to NS5B. Interestingly, although NS5B exhibited <20% residual activity on both poly rA/U12 and HCV 5′-UTR RNA templates at ATA concentration of 2.5 μM (Figure 1B), a corresponding decrease in binary complex formation at equivalent ATA concentration was lacking (35% cross-linking, Figure 5 lane 4). To rule out the possibility that the lack of this correlation is not attributable to the shorter RNA TP (poly rA20/U12) employed in this experiment, we cross-linked poly rA/U12 with NS5B in the presence of ATA under identical conditions and obtained similar results (data not shown). The lack of concomitant decrease in binary complex formation with NS5B RdRp activity at equivalent ATA concentrations suggests that, although ATA inhibition might be mediated at the step of RNA binding, additional steps of the polymerase reaction, such as NTP-metal binding or elongation step, might possibly contribute to ATA-mediated inhibition. Some credence to this notion is supported by the mechanistically mixed mode of ATA inhibition towards UTP substrates, with a major competitive component.
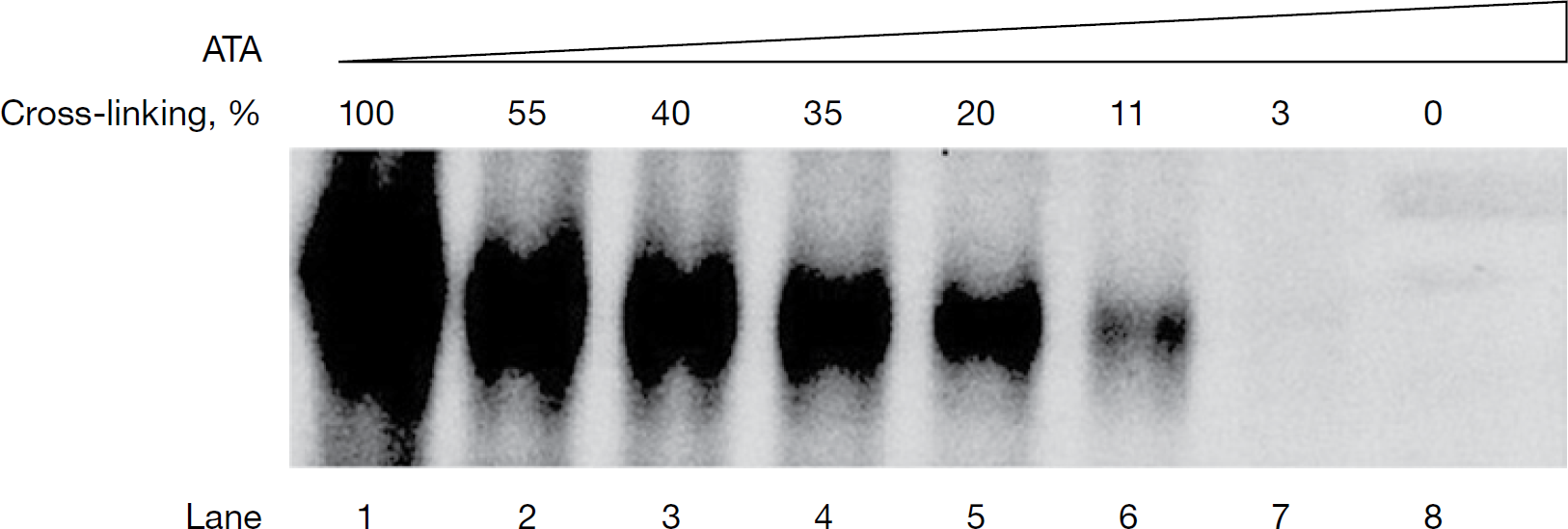
Effect of ATA on NS5B–RNA binary complex formation
Binding mode of ATA
To investigate the potential binding mode of ATA to HCV NS5B, we performed molecular modelling studies using Glide docking software. To rule out any bias, each of the three reported HCV NS5B NNI binding sites, represented by tetracyclic indole [42], N,N-disubstituted phenylalanine [37] and benzothiadiazine inhibitors [43], was examined for ATA binding. As a preliminary step, we validated the accuracy of our docking approach by determining how closely ‘the lowest energy pose (binding conformation)’ predicted by the object scoring function, Gscore in our case, resembles the experimental binding mode as determined by X-ray crystallography. As seen in Table 1, docking of ATA into NS5B NNI binding site for benzothiadiazine (allosteric pocket 3) yielded the most negative Gscore (−8.11 kcal/mol), followed by a Gscore of −7.41 kcal/mol for tetracyclic indole binding site (allosteric pocket 1). The N,N-disubstituted phenylalanine (allosteric pocket 2) seems to be the most unlikely site for ATA binding as it yielded a Gscore of −6.77 kcal/mol. To estimate the relative binding efficiency of ATA in the allosteric versus the active site pocket of NS5B, we compared our XP-Glide predicted allosteric binding energy data with the autodock predicted binding energy data recently reported by Yap and colleagues [50] for ATA in the active site pocket of NS5B. As seen in Table 1, the autodock score for ATA binding at the active site pocket was the least negative (−5.8), thus suggesting a much weaker binding strength for this pocket versus the allosteric sites of NS5B. This analysis suggests that ATA might bind in allosteric pocket 3 of NS5B.
Binding-energy data of ATA within each of the allosteric pockets of NS5B
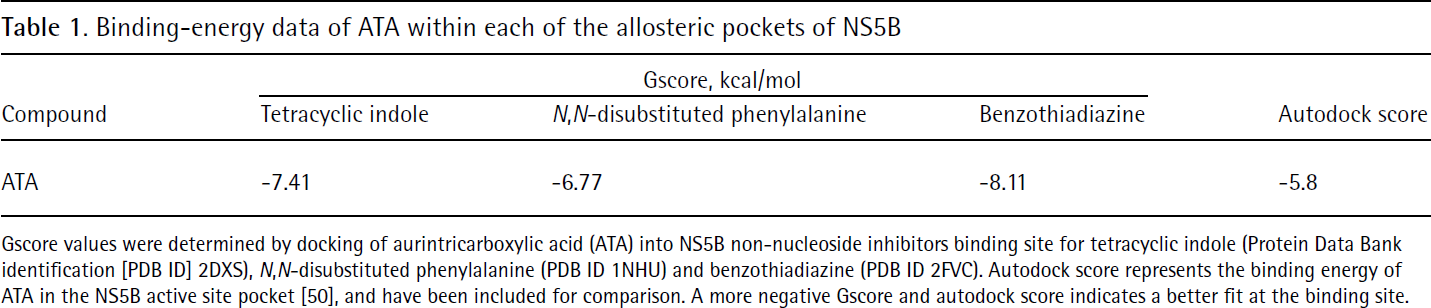
Gscore values were determined by docking of aurintricarboxylic acid (ATA) into NS5B non-nucleoside inhibitors binding site for tetracyclic indole (Protein Data Bank identification [PDB ID] 2DXS), N,N-disubstituted phenylalanine (PDB ID 1NHU) and benzothiadiazine (PDB ID 2FVC). Autodock score represents the binding energy of ATA in the NS5B active site pocket [50], and have been included for comparison. A more negative Gscore and autodock score indicates a better fit at the binding site.
To obtain evidence in support of our hypothesis that ATA might function as a NS5B pyrophosphate mimetic inhibitor, we used a competition displacement assay. This assay is based on the principle that if two compounds bind to a common site on the enzyme, they display mutually exclusive binding. The validity of this assay has been recently demonstrated by Pace et al. [17] in context of meconic acid derivative and DKA, two NS5B pyrophosphate mimetic inhibitors. In the present investigation, we examined the ability of ATA to compete with [14C]-foscarnet, a known pyrophosphate mimic and approved antiviral drug, for NS5B binding. As seen in Figure 6, binding of foscarnet to NS5B displayed an inverse correlation with ATA concentration. Maximum binding of foscarnet to NS5B was obtained in the absence of ATA, which decreased concomitantly with increasing ATA concentration, resulting in approximately 10% binding at 1 μM ATA concentration; thus suggesting that ATA competes with foscarnet for binding to a common site on NS5B. Furthermore, this analysis revealed that 50% of the [14C]-foscarnet was displaced at approximately 165 nM (50% displacement concentration) ATA concentration. The specificity of ATA–foscarnet binding to NS5B was evident from our observation that foscarnet did not bind BSA, employed as negative control, and coumestan, a NS5B non-nucleoside allosteric inhibitor, did not compete with foscarnet for NS5B binding (data not shown). Given the mutually exclusive binding between ATA and foscarnet to NS5B, this analysis reveals that ATA and foscarnet interact with NS5B at a common binding site.
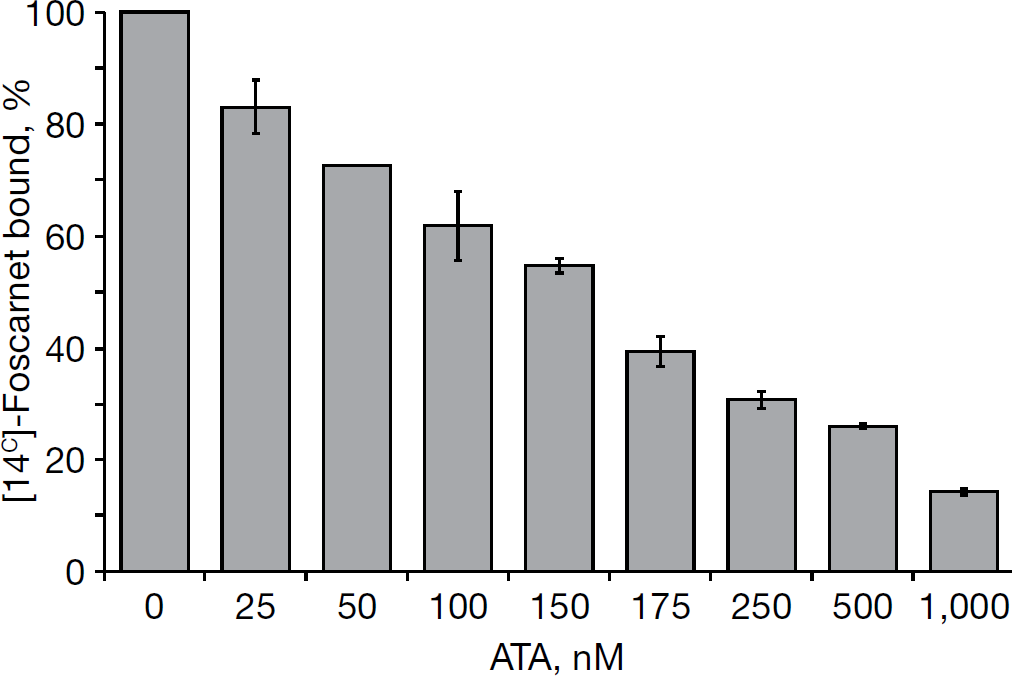
ATA competes with foscarnet for NS5B binding
ATA inhibits native HCV replicase in vitro
Given the potent inhibition of recombinant HCV NS5B RdRp by ATA, we were curious to examine its effect on the membrane-associated native HCV replicase complex. The replicase complex has been isolated in a functionally active form from cytoplasmic membrane fractions from Huh-7 cells harbouring a subgenomic NS3-5B replicon by a number of groups [45,46,51] and its activity has been demonstrated to be inhibited by HCV NS5B specific inhibitors in a cell-free assay system [45,46,51]. In the present study, we isolated the HCV CRC from two independent HCV replicon-bearing cells, NNeo/3-5B (RG) and MH14 cells, following previously published procedures. As controls, cytoplasmic membrane fractions were processed from Huh-7 and cured MH14 cells in a similar fashion. As the CRC-associated ribonucleoproteins of HCV are postulated to be composed of viral genomic RNA associated with NS5B, other HCV-encoded NS proteins and possibly host factors, we first confirmed the HCV protein composition of the isolated CRC by using western blot analysis. As an internal marker, we probed for the presence of GRP78, a chaperone endoplasmic reticulum-resident protein. As expected, we could detect the HCV NS protein components, such as NS5B, NS5A, NS3 and NS4B, in the CRC fraction from replicon-bearing cells (R in Figure 7A), but not in the corresponding membrane fractions isolated from the parent Huh-7 or cured MH14 cells (C in Figure 7A). The presence of GRP78 in both the CRC as well as the membrane fractions from Huh-7/cured MH14 cells (Figure 7A), confirmed the integrity of our isolation procedure. We also confirmed that the isolated CRC was functionally active and able to synthesize full-length copies of the endogenous replicon RNA template (data not shown).
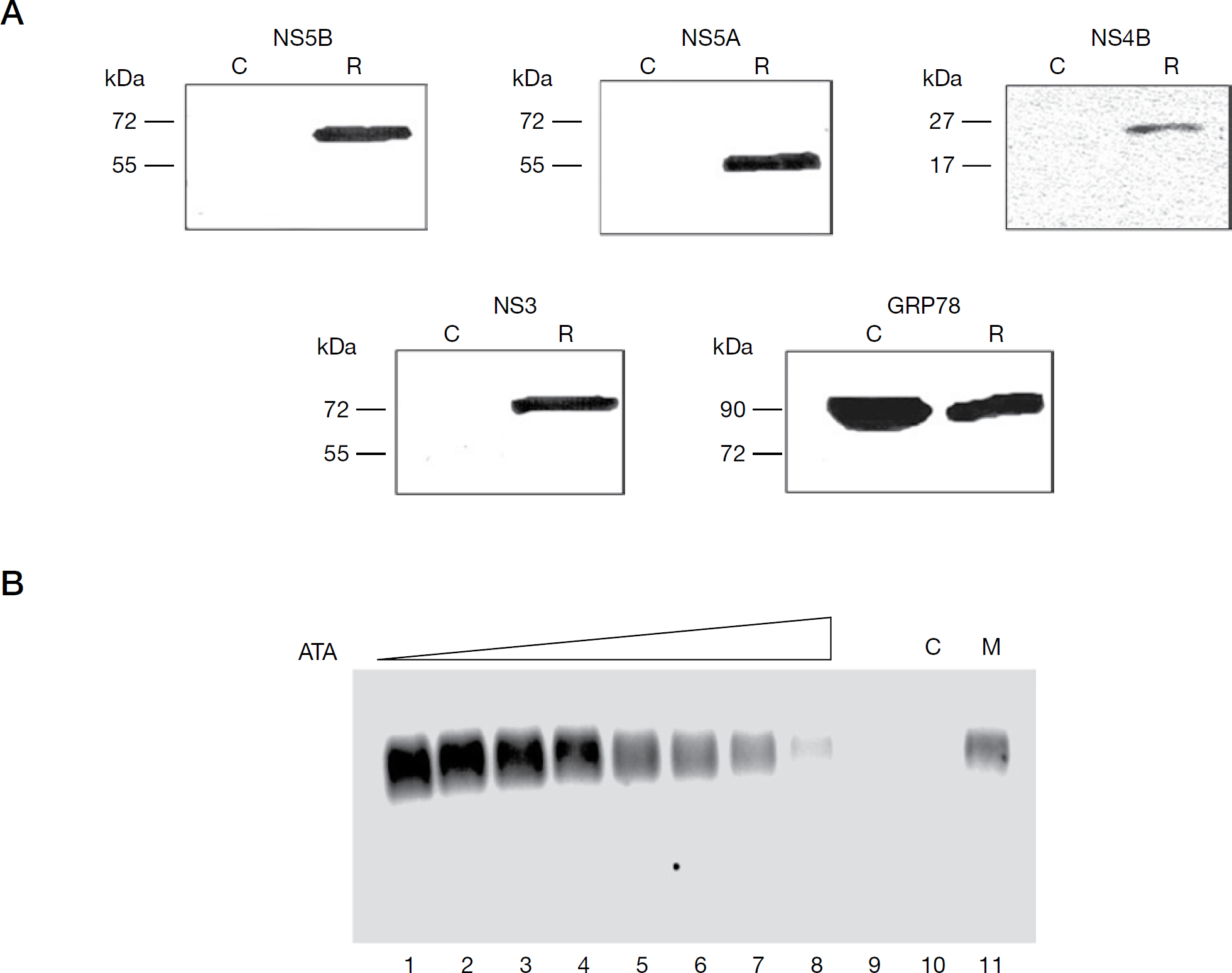
HCV cytoplasmic replicase complex is inhibited by ATA
We next assessed the effect of ATA on the RNA synthesis activity of the HCV replicase complex. To accomplish this, replication assays were set up with the isolated CRC in the absence and presence of increasing concentrations of ATA, and the resulting 32P-labelled, actinomycin D-resistant RNA products were resolved on a denaturing agarose gel and visualized by phosphorimaging. As seen in Figure 7B, in the absence of ATA and in reactions supplemented with exogenous NTPs (lane 1), the HCV CRC synthesized a major RNA product, corresponding to the expected size of the endogenous replicon RNA template. This was deduced from the migration of this band at equivalent position with an in vitro transcribed radiolabelled, single-strand subgenomic replicon RNA employed as marker (lane 11), thus confirming authentic HCV replication. The specificity of the HCV replicase reaction was further evident from the conspicuous absence of this RNA product when reactions were carried out with membrane fractions from cured cells (lane 10), or under conditions when only a single NTP was provided to the CRC (lane 9). The dose-dependent inhibitory effect of ATA on the RNA synthesis ability of the CRC was evident from the corresponding linear decrease of product formation at increasing ATA concentrations (lanes 2–8), yielding an IC50 value of 145 nM. An identical pattern of ATA-mediated inhibition of CRC was observed in the presence of heparin trap (data not shown), thus indicating that the HCV replicase complex was impervious to heparin as previously reported [45], and that ATA inhibited the elongation activity of the replicase complex. Furthermore, the near-similar inhibitory potency of ATA on the native replicase complex and the recombinant NS5B on HCV 5′-UTR RNA template (Figure 1B) suggests that the mode of ATA binding to NS5B might be similar in the replicase complex and the recombinant NS5B protein.
ATA suppresses replication of the HCV subgenomic replicon
Given the equipotent inhibitory activity of ATA in vitro against recombinant HCV NS5B as well as the native replicase complex, we were keen to investigate its antiviral activity against HCV. However, a concern existed whether the anionic ATA would be able to permeate the intact cell membrane and abrogate replication of HCV RNA in the HCV subgenomic replicon system, given the controversial reports in the literature regarding the membrane-penetrating ability of ATA. We therefore examined the effect of ATA treatment on HCV RNA replication and HCV NS5B expression in BB7 cells harbouring the HCV subgenomic replicon. BB7 cells were plated in 5% FBS medium at a confluency of 50% and treated daily with fresh serially diluted ATA to compensate for reduction in effective ATA concentrations either because of degradation or interaction with serum proteins. Direct quantification of HCV RNA levels was performed 72 h post-treatment by quantitative TaqMan® real-time PCR employing endogenous GAPDH mRNA as a control. This analysis revealed potent inhibition of the HCV RNA replication in BB7 cells, yielding EC50 value of 75 nM (Figure 8A). Interestingly, although a substantial reduction of the HCV RNA levels was observed at lower doses of ATA treatment (≤0.5 μM), this effect was surprisingly less pronounced at higher doses of ATA. This was reflected by approximately 67% reduction in HCV RNA levels at 1 μM ATA and 75% reduction at 10 μM ATA, thus amounting to an approximately 8% increase in inhibition though the dose of ATA was increased by 10-fold. It might be speculated that the diminished ability of ATA to suppress HCV RNA replication at higher concentrations could be because of its physicochemical properties to form aggregates, thereby impeding its ability to enter the cell.
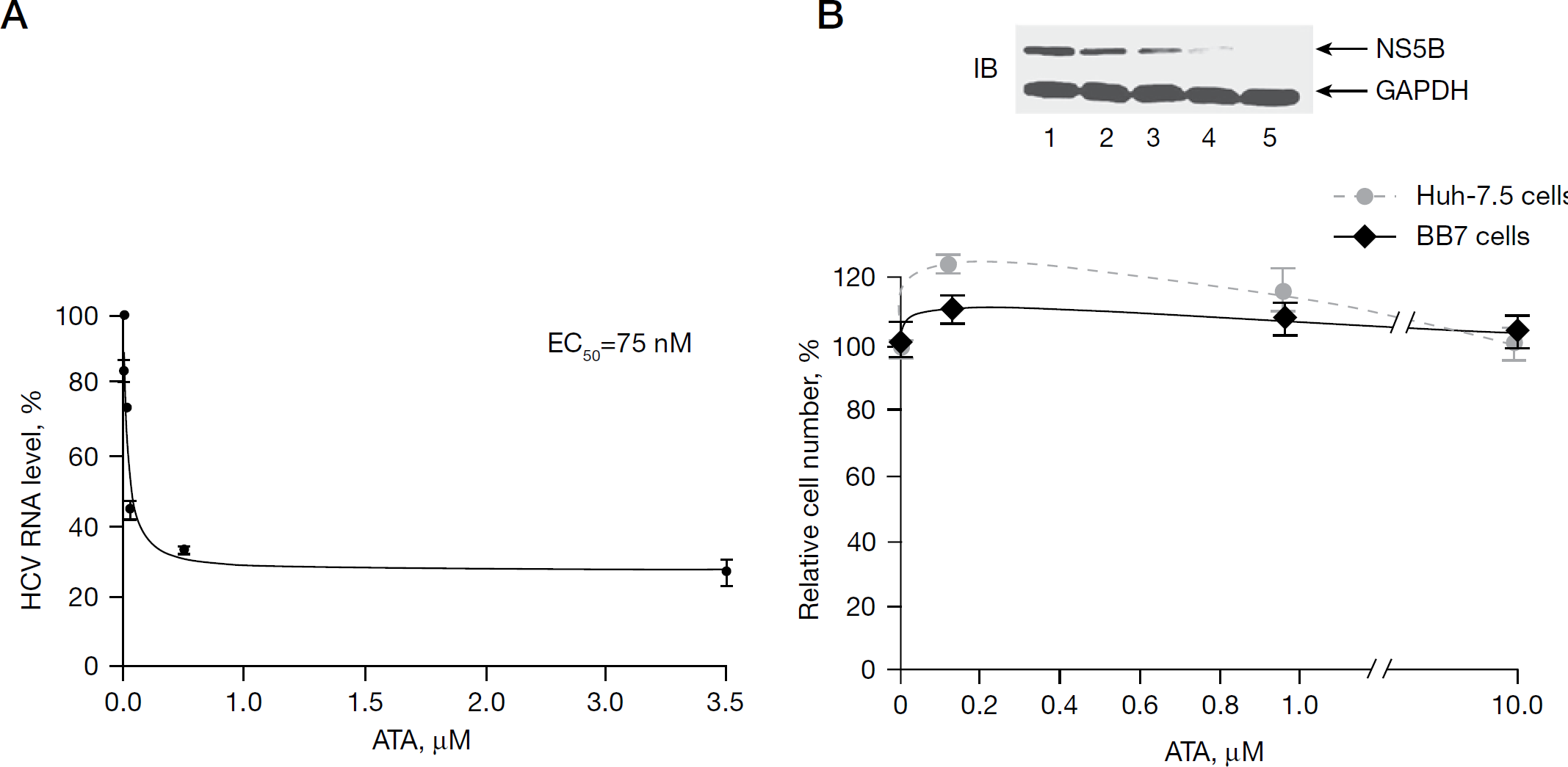
Antiviral activity of ATA
Consistent with the inhibition of HCV RNA replication as a function of ATA concentration, ATA treatment also decreased expression of HCV NS5B in a dose-specific manner, which was observed from immunoblots of total cell lysates of BB7 cells treated with increasing amounts of ATA (Figure 8B, inset). Thus, HCV NS5B became virtually undetectable at ATA concentration of ≥10 μM (lane 4), akin to abolition of its expression upon IFN-α treatment (lane 5). A similar pattern of NS5B reduction was also seen when NNeo/3-5B (RG) cells were treated with ATA (data not shown). The concomitant decrease in HCV NS5B with HCV RNA might be a direct consequence of decreased HCV NS5B production from reduced amounts of HCV RNA during subsequent rounds of replication and translation. These results provide strong evidence that ATA potently suppresses replication of the subgenomic 1b HCV replicon cells. This is also supported by our findings that ATA concentrations <10 μM did not affect the proliferation or cell viability of the BB7 replicon cells as well as the parental Huh-7.5 cells (Figure 8B), thus indicating that the anti-HCV activity of ATA in cell culture is not due to cytotoxic effect. Furthermore, ATA-treated cells (72 h) did not appear to be stressed even at 25 μM ATA concentration as deduced by a lack of any overt morphological changes in the cells. Consistent with this finding, ATA-treated cells displayed equal expression of GAPDH as untreated controls (Figure 8B, inset: lanes 1–4). Together these data indicates a direct effect of ATA on viral RNA replication.
Discussion
ATA, a unique compound with versatile biological activities, exhibits broad-spectrum antiviral potency against different virus families [21,28–30,52]. Here, we demonstrate for the first time that ATA is also a potent anti-HCV agent. Specifically, we demonstrate that ATA exhibits potent inhibitory activity against recombinant HCV NS5B and the HCV replicase complex, in addition to suppressing HCV RNA replication in the HCV replicon system.
ATA appears to inhibit recombinant NS5B by a bimodal mechanism because it displayed characteristics of the NNIs as well as the pyrophosphate isosteres. Data supporting the latter claim are clearly evident from the dependence of ATA inhibition on the divalent cations employed in the NS5B RdRp assay, a hallmark of the pyrophosphate mimics [14–17]. This translated to an overall 16–31-fold increase in ATA potency in the presence of Mn2+ versus Mg2+, a trend consistent with that exhibited by the dihydroxypyrimidine and DKA series of active site inhibitors, which have been proposed to interact with the metal ions present in the active site of NS5B [14–17]. Although there is no direct crystallographic evidence supporting the binding of ATA at the active site of NS5B, some clues in this regard come from ATA binding mode predictions performed by Yap and colleagues [50] on RdRps from pathogenic positive-strand RNA viruses, including SARS-CoV and HCV. These authors have proposed an evolutionarily conserved anti-parallel β-strand-turn-β-strand hairpin structure covering two of the three RdRp catalytic sites in the palm subdomain of these RdRps as a potential site of ATA binding. Furthermore, based on molecular docking and binding energies, they have also predicted the relative inhibition of these RdRps by ATA to be of the order: SARS-CoV RdRp≥HCV NS5B=HIV RT. Interestingly, our experimental findings for ATA-mediated inhibition of HCV NS5B (IC50=4.1 μM) and HIV RT (IC50=4.3 μM) with Mg2+ as the divalent cation is in agreement with their molecular model predictions. In addition, our competition displacement studies suggest that ATA and foscarnet share the same binding site on NS5B; thereby unequivocally substantiating ATA's role as a NS5B active site inhibitor capable of interacting directly with the metal ions.
Several laboratories, including ours, have kinetically characterized HCV NS5B inhibitors. These analyses have yielded two distinct modes of inhibition towards nucleotide substrates: a non-competitive or competitive mode. The former mode is displayed by several chemically distinct scaffolds of NNIs including coumestans and 4-thiazolidinones recently characterized by us [34,35]. The latter mode is displayed by the modified chain-terminating nucleoside analogues of NS5B [9,14,50]. More recently, the pyrophosphate mimics, like the derivatives of DKA, have been characterized with regards to their kinetic parameters and shown to bind competitively to the elongation NTP pocket in the active site of NS5B and inhibit both the initiation and elongation steps of polymerization [53]. Our investigation on the kinetic parameters of ATA-mediated NS5B inhibition has revealed a mixed mode towards nucleotide substrates, with a major competitive component. This mechanism displayed by ATA towards nucleotide substrates is distinct from all other HCV NS5B inhibitors investigated to date and to our knowledge represents the first demonstration of this kind for any HCV NS5B inhibitor. Interestingly, based on a review of published literature, it is apparent that ATA exhibits a varied mode of inhibition towards different polymerases. For instance, ATA has been shown to inhibit DNA polymerase α, DNA polymerase I and HIV-1 RT in a non-competitive fashion with respect to nucleotide substrates, whereas it inhibits RNA polymerase in a competitive fashion [18].
We envisage that the kinetic parameters of ATA-mediated inhibition of HCV NS5B might be highly complex. Under continuous polymerization conditions, a small fraction of NS5B RdRp activity was resistant to ATA inhibition suggesting that the activity of ATA might be affected by template RNA, which is observed with the benzothiadiazine and benzimidazole inhibitors [47]. In the modified order-of-addition experiments, ATA displayed diminished inhibition potency under conditions of NS5B–RNA pre-incubation, similar to the trend exhibited by coumestan, thiazolidinone and benzylidene derivatives [34,35,39]. This might be a consequence of decreased access of ATA to its binding site on NS5B, either because of occlusion by RNA or as a result of conformational change upon RNA binding. Additional support for the role of RNA in protecting NS5B from ATA inhibition is evident from the display of mixed mode towards TP with a significant competitive component similar to that reported for benzimidiazole, coumestan and thiazolidinone derivatives [34,35,38]. It is likely that ATA inhibits NS5B at an early step during initiation of RNA synthesis, possibly concurrent with NS5B–RNA complex formation. This speculation is consistent with our observation that no abortive or prematurely terminated products were observed in both the de novo or primer elongation reactions in the presence of ATA. Additionally, the decrease in NS5B–RNA cross-linking in the presence of ATA provides definite evidence of ATA inhibition occurring at the step of RNA binding. However, the lack of correlation between NS5B RdRp inhibition and binary complex formation at equivalent amounts of ATA, suggests that ATA might exert its effect at additional steps of the polymerase reaction following NS5B–RNA binding. This might include the substrate-binding step as inferred from the mixed mechanistic mode displaying a competitive component towards nucleotide substrates. Under single-cycle assay conditions, ATA inhibited both the initiation and elongation steps of the polymerization reaction, but its potency was substantially reduced compared with continuous polymerization conditions of a standard HCV RdRp assay. However, it might be noted that despite this decrease, ATA still exhibited potent inhibition of NS5B RdRp activity under single-cycle assay conditions (Figure 4, panel 4), which is evident from the nanomolar to low micromolar range of IC50 value. This shift in potency under single-cycle assay conditions is unique to ATA and is in marked contrast to other characterized NS5B inhibitors such as the DKA series of pyrophosphate mimics, which exhibit similar IC50 values under both single-cycle and continuous polymerization assay conditions [38,47], and benzimidazole series of HCV NNIs, which do not inhibit HCV NS5B RdRp activity from pre-bound RNA complexes [38].
Examination of the interaction of ATA at the three reported HCV NS5B NNI binding sites represented by tetracyclic indole (allosteric pocket 1), N,N-disubstituted phenylalanine (allosteric pocket 2) and benzothiadiazine (allosteric pocket 3) inhibitors (Table 1) have provided some clues regarding the potential binding mode of ATA to HCV NS5B. ATA clearly exhibited a better fit in allosteric pocket 3 versus allosteric pockets 1 and 2, which was implied by its relatively more negative Gscore in the former pocket versus the latter ones. Interestingly, the overall binding strength of ATA was also substantially higher at the three allosteric sites versus the active site of NS5B as is evident from their Gscore (Table 1). Although this analysis supports ATA's binding in allosteric pocket 3 located adjacent to the active site pocket of NS5B, the competition displacement assay (Figure 6) provides firm evidence in terms of ATA's ability to bind at the active site of NS5B. This apparent paradox regarding ATA binding can only be resolved by NS5B-ATA cocrystal structure. However, in the absence of this structure, it seems reasonable to speculate that ATA might exert its inhibitory effect by virtue of multiple modes of binding to NS5B, as suggested by its complex biochemical and mechanistic parameters of NS5B inhibition.
Previous studies have reported that the modified NNIs of HCV NS5B display equal sensitivities towards recombinant NS5B as well as NS5B associated with the native HCV replicase complex [45]. By contrast, the RNA synthesis activity of the native replicase complex is not amenable to inhibition by several structural classes of NNIs of HCV NS5B, although these compounds display potent inhibition towards the recombinant enzyme [45]. In case of the pyrophosphate mimics, to date there is no report in the literature documenting their effect towards the HCV replicase complex. We speculate that similar to the nucleoside analogue class of active site inhibitors, the pyrophosphate mimics are likely to inhibit recombinant NS5B and the native HCV replicase with relatively similar potencies. This is supported by the ability of ATA to inhibit native and recombinant NS5B with near equal potency (Figures 1 and 7). The sensitivity of the native replicase complex to ATA inhibition suggests that the presence of viral RNA and other NS viral proteins in the native replicase complex do not occlude the accessibility of ATA to its binding site on NS5B. In this mode, ATA might potentially exert its effect at the elongation step of the polymerase reaction from pre-bound RNA complexes.
The ability of ATA to translocate across the plasma membrane is documented by several studies. For instance, the survival-promoting action of ATA is attributed to its ability to permeate across the plasma membrane and prevent apoptosis [23]. In another study, ATA has been reported to exert its effect on vesicular stomatitis virus by interfering with its RNA transcription machinery [28]. Our findings that ATA suppressed the replication of the HCV subgenomic replicon in cell culture over a 72 h incubation period and also inhibited viral protein synthesis supports the notion that ATA exerts its effect upon permeating across the cell membrane. Similar inhibitory effects of ATA have been reported on SARS-CoV RNA replication and viral protein synthesis resulting in markedly decreased virus production [29]. Interestingly, ATA is also reported to exhibit antiviral activities against other viruses by diverse mechanisms. During HIV infection, ATA functions as an entry inhibitor and prevent virus binding by interfering with the gp120–CD4 interaction [52]. A similar mechanism of ATA action has also been proposed for human herpesvirus-7 infection [54]. In addition to its role as an entry inhibitor, ATA inhibits HIV-1 replication in vitro by inhibiting two key viral enzymes, HIV-1 RT and integrase [18,21,28].
The efficacy of an antiviral agent is governed by its ability to inhibit virus replication while minimally affecting host cell growth. In this regard, a growing body of evidence supports the low toxicity of ATA in cell culture and animal studies [30], and its potent antiviral activity against HIV, SARS-CoV and vaccinia virus in diverse cell types with relatively no cytotoxicity [21,29,30,52]. Our studies on the anti-HCV effects of ATA in HCV replicon-bearing BB7 cells are consistent with this trend. We speculate that the absence of ATA toxicity in cell culture at concentrations at which it exhibits its antiviral potency, despite its ability to inhibit purified cellular DNA and RNA polymerases in vitro, might be attributed to the differential accessibility of ATA to these cellular enzyme targets versus viral enzyme targets. The specificity of ATA for viral targets is evident from studies wherein aurin, an ATA analogue lacking the three carboxylic acid groups, and fuchsin acid, another structural analogue of ATA containing sulfonate instead of carboxylate, failed to inhibit HIV replication [52]. ATA has also been reported to potently and selectively inhibit vaccinia virus replication while displaying low toxicity [30]. The suitability of ATA as a drug candidate is evident from antiviral investigations against SARS-CoV by He and colleagues [29]. These authors demonstrated that the potency of ATA was 1 log unit higher than the two clinical antivirals, glycyrrhizin and nelfinavir. Furthermore, the therapeutic value of ATA for SARS patients was clearly evident from its 6–10-fold higher selectivity index over IFN-α and IFN-β in Vero cells [29]. In case of HCV, we found ATA to be non-cytotoxic to parental uninfected Huh7 cells and HCV replicon-bearing BB7 cells at concentrations 133-fold higher than that required to inhibit viral RNA replication by 50%. The 50% cytotoxicity of ATA in Huh7 cells is reported to be 1,250 μg/ml (approximately 3 mM) [30]. Considering this value, ATA might display a higher selectivity index against HCV in Huh7 cells than that reported for SARS-CoV in Vero cells, indicating its potential as a potent anti-HCV candidate with low toxicity.
In summary, we have demonstrated for the first time the potent activity of ATA against HCV NS5B RdRp in vitro in the HCV replicase complex as well as in the HCV replicon cells. In-depth biochemical, mechanistic, competition displacement and molecular modelling studies support the notion that ATA might bind to the benzothiadiazine allosteric pocket 3 of NS5B or at its catalytic centre and thereby inhibit its function. The potent anti-HCV activity of ATA in HCV replicon-bearing cells with low cytotoxicity suggests that the ATA scaffold might be further exploited and optimized on each of the aryl moieties to develop novel and potent anti-HCV NS5B compounds.
Footnotes
Acknowledgements
We thank Michael Lai and Jens Bukh for the plasmids pThNS5BCΔ21 and HCV pCVJ4L6S, respectively, and Craig Cameron for the anti-NS5A polyclonal antibody. We also acknowledge Stanley Lemon, Kunitada Shimotohno and Charles Rice for providing the subgenomic genotype 1b HCV replicon cell lines NNeo/3-5B (RG), MH14 and BB7, respectively. This work was supported by NIH grant DK066837 to NKB. Part of this work was presented at the 15th International Symposium on Hepatitis C Virus & Related Viruses 5–9 October 2008, San Antonio, Texas, USA.
The authors declare no competing interests.