
Abstract
Cell therapy is an evolving option for patients with end-stage heart failure. First-generation cell therapy trials have had marginal success. Our goal was to evaluate retrograde delivery of allogeneic umbilical cord subepithelial cells (UCSECs) in patients with heart failure. A prospective open-label dose escalation study of the safety and feasibility of UCSECs infused retrogradely into the coronary sinus was performed. Patients received a single dose of either 100 million (M), 200M, or 400M cells. The patients were followed for 2 years. Twenty-four patients were successfully enrolled in the study. The patients had UCSEC infusion without procedure-related complications. The ejection fraction in patients receiving UCSECs demonstrated improvement compared to baseline; from 25.4% (±5.5) at screening to 34.9% (±4.1) at 12 months. End-systolic diameter decreased significantly from 59.9 (±5.3) mm to 52.6 (±2.7) mm (p < 0.05). Retrograde UCSEC delivery was safe and feasible in all three dosage groups. Patients receiving 200M and 400M UCSECs showed signs of early improvement in left ventricular ejection fraction (LVEF) and remodeling. This study provides the basis for a larger clinical trial in heart failure (HF) patients using the middle or high dose of UCSECs.
Introduction
The overall patient mortality from coronary artery disease (CAD) has declined because of several factors including improvements in pharmacologic therapy and revascularization techniques 1 . However, cardiovascular disease still remains the leading cause of death in the world 2 . The life-saving advances in management of acute and chronic CAD have been associated with an increased prevalence of patients with heart failure (HF), with an estimated 250,000–350,000 patients who may be candidates for more advanced therapies 3 . Additionally, HF, like all cardiovascular diseases, is highly age dependent, and the prevalence will potentially double by 2030 with the advancing age of the US and world populations 4 .
Standard therapeutic options for patients who develop advanced HF are limited and include heart transplantation or the use of ventricular assist devices (VADs), both of which are limited by cost, availability, and complications 5 . This results in a large HF patient population with progressive symptoms and limited treatment options 6 . First-generation autologous cell therapies (derived from bone marrow, blood, or adipose tissue) for advanced HF have shown promise but have had limited and nondurable results due to lack of potency, low cell number, low retention, or limited biological function7–10. The goal of cell therapy, which utilizes the body's native repair mechanisms, is to reverse or restore the function of organs, tissues, and blood vessels. The newest generation of cell therapies will involve the use of autologous or allogeneic cultured and expanded cell products, ideally with defined potency, doses, low immunogenicity, and potential reproducibility. Umbilical cord tissue-derived cells, such as umbilical cord subepithelial cells (UCSECs), have some of these qualities, as they have been shown to have low to no immunogenicity in in vitro, preclinical, and early clinical studies11–19.
In addition to having better cell therapy options for HF patients, safer, more cost-effective, and reproducible delivery techniques are required. There are robust preclinical models of retrograde delivery of cells demonstrating safety and efficacy 20 . Our group has previously demonstrated the safety and reproducibility of retrograde delivery of first-generation cell types in both chronic angina and HF patients21,22. The goal of this study was to evaluate the safety and feasibility of retrograde delivery of a large dose range of xeno-free allogeneic UCSECs in patients with HF (RESCUE-HF).
Materials and Methods
Study Design
The institutional review board (IRB) at each participating institution including the data registry institutions approved the prospective open-label dose escalation study (clinicaltrials.gov NCT00130377), which was conducted to assess the safety and feasibility of retrograde infusion of allogeneic UCSECs via the coronary sinus in patients with HF. Ability to pay for treatment was not a requisite for inclusion in the study, and all patients gave written informed consent. All patients were deemed to have optimal medical treatment by an HF specialist for at least 1 month prior to enrollment. The primary endpoints of the study were the safety and feasibility of using UCSECs delivered retrogradely into the coronary sinus of patients with HF. Secondary endpoints were safety of retrograde delivery, changes in cardiovascular symptoms as measured by the New York Heart Association Class (NYHA) HF classification, B-type natriuretic peptide (BNP) levels, left ventricular ejection fraction (LVEF) by echocardiography, left ventricular end-diastolic diameter, and left ventricular end-systolic diameter. Additionally, the optimized standard of medical care was directed by an HF cardiologist throughout the trial and follow-up period. All clinical adverse events (AEs) were evaluated in real time by an independent reviewer, and a determination was made if the event was related or unrelated to the experimental treatment. Enrolled patients were stratified to receive one of three concentrations of the UCSEC product [100 million (M), 200M, or 400M cells in 60 cc of saline]. The reviewers were blinded to all imaging and follow-up information.
Patient Selection
Eligible patients were 18 years or older with congestive HF, an LVEF <40% by contrast echocardiography, NYHA class III or IV, stable on standard medical therapy for at least 1 month prior to screening, and with a life expectancy of 6 months or greater. Exclusion criteria included prior radiation treatment, hematological disorders, cirrhosis, bleeding/clotting disorders, acute myocardial infarction less than 7 days from treatment date, uncontrolled diabetes (HbA1c, >9), known active malignancy or risk of active malignancy (defined as abnormal PAP, CXR, mammogram, or positive fecal hemoccult), antibiotics within 7 days, high-dose steroids within 1 month before the procedure, dialysis, serum creatinine >3.0, or pregnancy.
Procedure
After written informed consent was verified, patients were taken to the catheterization laboratory where heart rate, blood pressure, pulse oximetry, and electrocardiogram (EKG) tracings were monitored. Cells were thawed and prepared into four 15-ml syringes and delivered onto the sterile field for infusion into the coronary sinus.
The coronary sinus was accessed via the right femoral vein using a 7F 8-mm × 40-mm balloon catheter (Cook Region Tech, Lafayette, IN, USA) under fluoroscopic guidance. Proper catheter position and balloon occlusion were confirmed with fluoroscopy. Each syringe of prepared UCSECs was infused continuously over a 2-min period per syringe, with a 15-s pause between syringes. The balloon remained inflated to occlude the coronary sinus for 10 min after the infusion under 2 atmospheres of pressure to overcome venous pressure in the coronary sinus, allowing for the migration of the cells into the cardiac tissue. Venography was performed at the 5-min interval to verify that no migration of the balloon occurred. After this dwell time, the balloon was deflated, and the catheter was removed 21 . Local hemostasis was obtained, and the patient was transferred to a telemetry monitoring unit for 24 h of observation. The average time to perform the cell delivery from venous access to catheter removal was 28 ± 11 min.
Safety Follow-Up
Patients returned for follow-up visits at 1, 4, and 12 months and a 24-month phone call was completed. Cardiac biomarkers, blood chemistry, and hematological lab tests were collected at their initial screening and subsequent 1-, 4-, and 12-month follow-up visits. Immune responses against the UCSECs were measured at 1, 4, and 12 months using anti-human leukocyte antigen (HLA) antibodies. Anti-HLA antibodies (Thermo Fisher Scientific, Pittsburgh, PA, USA) were measured by flow cytometry using recipient sera and panels with beads expressing HLA antigens and are defined as panel-reactive antibodies (PRA). A PRA >5% was considered a positive response. AEs were recorded at each visit. Echocardiography and EKGs were performed at the 1-, 4-, and 12-month follow-up.
Statistical Methods
Study data were collected using a dedicated electronic data capture system with real-time queries. Patient demographics, clinical characteristics, and safety data were summarized by dose (100M, 200M, or 400M). Descriptive statistics were provided in the analysis of all safety, cardiac function, laboratory, and health status endpoints in this study. Data analysis was performed as intent to treat. The descriptive statistics for continuous variables included mean, median, standard deviation, median, minimum, maximum, and sample size. Categorical variables were described with numbers and percentages. Incidence rates of AEs were tabulated by dose for the various types of AEs [e.g., all AEs, serious adverse events (SAEs), deaths, and nonfatal AEs possibly related to the procedure] and body systems. Severity, duration, and relationship of AEs to UCSECs were recorded on the electronic case report forms (eCRFs). The incidence of AEs by severity and relationship to UCSECs was compared by treatment group and ischemic stratification. Cardiac function and HF symptoms were tabulated and summarized by dose and time point with descriptive statistics. Statistical analysis was performed using SAS Version 9.3 (Cary, NC, USA). When presented, p values were two sided and were considered statistically significant at the two-sided 0.05 level of significance unless otherwise specified, as determined by paired t-tests and chi-square analyses. No imputation for missing data was performed, and no adjustment for multiple comparisons was made given the exploratory nature of this safety study.
Results
Disposition
Between March 2012 and December 2012, 42 patients gave consent and were screened, resulting in 24 patients being treated. There were eight patients in each dose group. There was a 1-week delay for the last treated patient in each group before advancing to the higher dose to assess acute and subacute safety (Fig. 1). Eighteen of 24 patients were male, and 16 of 24 total patients had ischemic HF. The mean age was 59.1 (±8.0) years. There were no significant differences in diabetes, alcohol, or tobacco use between the groups (Table 1). The baseline mean ejection fraction (EF) determined by echocardiography were 26.1 (±5.3), 25.5 (±6.4), and 24.7 (±4.7) for the three dose groups (100M, 200M, and 400M) by a blinded physician. All patients were NYHA class III or IV at the time of screening. The demographics of all patients by dose assignment are shown in Table 1.

Study patient flow over the 2-year period. All patients enrolled received the cells with no adverse events (AEs) related to the cell delivery. There were 24 treated patients, and 22 were available for the 2-year follow-up. *p < 0.05.
Patient Demographics

Values are mean [standard deviation (SD)] unless otherwise indicated.
All enrolled subjects were all alive and completed the 1-year follow-up except for one patient in the 100M cell dose group. One additional patient in the 100M cell dose group did not survive to the 24-month follow-up phone call. There were no technical difficulties with the cell delivery procedure, specifically no evidence of coronary sinus leakage or damage. Total occlusion of the distal coronary sinus was obtained in every case, with no AEs or hemodynamic changes related to the delivery of UCSECs.
Safety
All 24 patients experienced at least one AE. The incidence of nonserious AEs possibly related to the procedure among patients receiving UCSECs was 1/8 in the low dose (100M cell dose), 2/8 in the 200M dose, and 1/8 in the 400M cell dose. These nonserious events included elevated troponins in all patients, which resolved in 24 h (Table 2), catheterization site hematomas in 2 patients, and pain at the catheterization site in 5 patients.
Troponin I

The incidence of SAEs was 5 of 24 in all dosage groups combined. Five patients had a congestive HF (CHF) exacerbation requiring hospital admission, 3/8 in the 100M dose group, 2/8 in the 200M dose group, and 0/8 in the 400M dose group, which all resolved with medical treatment. All of the SAEs were classified by the PI as “unrelated” or “unlikely” to be related to the procedure. SAEs are shown in Table 3. The total number of deaths by dose at 2 years is shown in Figure 1. There were 2, 0, and 0 deaths at 2 years in the 100M, 200M, and 400M cell groups, respectively. The presence of donor-specific anti-HLA antibodies was less than 10% in all but one patient treated with UCSECs (Table 4).
Serious Adverse Events by Dose at 2 Years

Immune Response to UCSECs
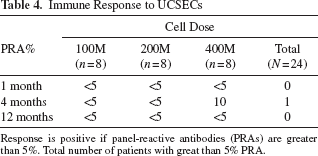
Response is positive if panel-reactive antibodies (PRAs) are greater than 5%. Total number of patients with great than 5% PRA.
Objective Measures
All patients enrolled had a transient elevated troponin periprocedure that decreased by discharge without any clinically significant AEs. The BNP levels for all dose groups were elevated at baseline with a mean of 574 (±184) ng/ml. However, by 4 months post-cell delivery, the 200M and 400M dose patients had a significant decrease in their BNP levels to 341 (±87) and 278 (±81) ng/ml, respectively (p < 0.05). At 1 year post-cell delivery, all groups had a significant decrease in their mean BNP level to 264 (±96) ng/ml (Fig. 2).
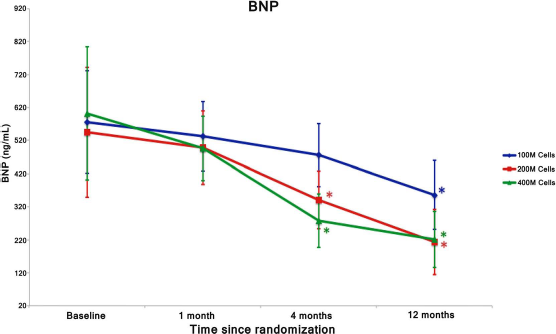
B-type natriuretic peptide (BNP) levels were observed from baseline to 12-month follow-up. Significant reduction in BNP was demonstrated in the 200M and 400M dose groups at 4 months post-cell delivery and in all groups at the12-month follow-up. *p < 0.05.
The EF of all patients at baseline was determined to be below 30% by echocardiography. The 100M cell dose group showed significant improvement only at the 12-month follow-up, changing from 26.1 (±5.3) to 30.2 (±5.1) (p < 0.05). The 200M cell dose group had significant improvement at the 4-month follow-up from 25.5 (±6.4) to 36.0 (±3.2), which continued to the 12-month follow-up at 36.8 (±2.6) (p < 0.05). Similarly, the 400M cell dose group had significant improvement at the 4-month follow-up from 24.7 (±4.7) to 35.7 (±1.9) (p < 0.05), which continued to the 12-month follow-up at 37.7 (±4.5) (p < 0.05) (Fig. 3).
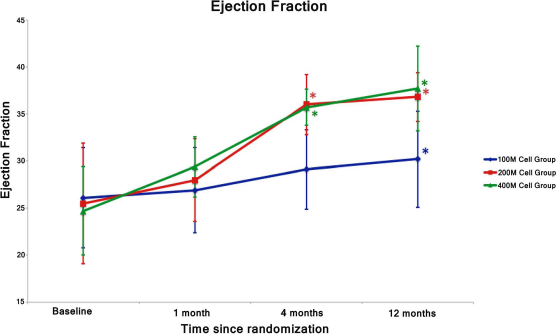
Ejection fraction (EF%) was followed from baseline to 12-month follow-up. Significant improvement in EF was demonstrated in the 200M and 400M groups at 4 months post-cell delivery and in all groups at the 12-month follow-up. *p < 0.05.
There was a significant decrease in end-systolic diameter as measured by echocardiography in all dose groups, which occurred by the 4-month follow-up period from a mean of 59.9 (±5.3) to 54.2 (±4.3) mm (p < 0.05) and continued to improve to 52.6 (±2.7) mm (p < 0.05) (Fig. 4). The same was not demonstrated in the end-diastolic diameter, which had no change in the 100M dose group, only significant change at 12 months in the 200M dose from 68.4 (±5.6) to 60.3 (±2.4) mm (p < 0.05), and significant change in the 400M dose from 68.4 (±5.6) to 63.9 (±4.9) mm (p < 0.05) and continued to 60.3 (±2.4) mm (p < 0.05) (Fig. 5).

Left ventricular end-systolic diameter (LVESD, mm) was followed from baseline to 12-month follow-up. Significant decrease in LVESD was demonstrated in all groups at the 4- and 12-month follow-up. *p < 0.05.

Left ventricular end-diastolic diameter (LVEDD, mm) was followed from baseline to 12-month follow-up. A significant decrease in LVEDD was demonstrated only in the 400M group at the 4-month follow up and in both the 200M and 400M groups at the 12-month follow-up. *p < 0.05.
Quality of Life
All patients were NYHA class III (20 patients) or IV (4 patients) at the time of screening. At the 12-month follow-up, 3, 13, 5, and 1 patients were NYHA class I, II, III, and IV, respectively. In the 100 M dosage group, 0/8 was class I, 2/8 class II, 3/8 class III, and 1/8 class IV at the 12-month follow-up. In the 200M dosage group, 1/8 was class I, 6/8 class II, and 1/8 class III at the 12-month follow-up. In the 400M dosage group, 2/8 were class I, 5/8 class II, and 1/8 class III at the 12-month follow up. NYHA class results are demonstrated in Table 5.
New York Heart Association (NYHA) Class
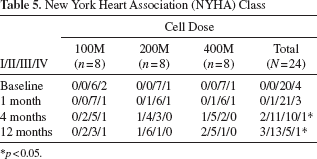
p < 0.05.
Discussion
This is the first prospective dose escalating trial using retrograde coronary sinus delivery of allogeneic cells in HF patients. The trial demonstrates both safety and feasibility in patients with NYHA class III/IV HF. Preclinical studies have demonstrated that total occlusion of the coronary sinus up to 60 min is safe because of the presence of Thebesian veins, which also drain cardiac venous blood. This allows biologics to be infused retrogradely under low pressure into the great and middle cardiac veins. Zakharova et al. also recently demonstrated preclinical feasibility of retrograde cell delivery with distribution throughout the left ventricle 20 . Retrograde delivery of cells was developed to minimize the risks associated with cardiac cell delivery. Most endocardial, epicardial, and intracoronary delivery techniques have limitations such as myocardial wall thickness, invasiveness, or significant coronary stenoses, which would cause perforation or poor cell distribution23–28. The number of cells delivered by these techniques is also potentially limited by microclumping leading to infarcts. Thus far, cell doses of no greater than 300M cells have been safely used because of these issues. Retrograde delivery enables a larger cell dose, which is translatable based on most preclinical models 21 . Optimal retention of cells still remains an ongoing investigation. Thus, retrograde coronary sinus delivery was chosen for the cell delivery route in this trial.
Our previous studies in both chronic angina and HF using retrograde delivery of bone marrow-derived mononuclear cells (BM-MNCs) were safe21,22. There have been a number of clinical trials using allogeneic umbilical cord cells for the treatment of cardiac disorders that have demonstrated early safety and interesting clinical outcomes that should be further investigated11,13,15,18,19.
There were clinical events in all arms of this current trial that would not be unexpected due to the severity of the patients. While there were AEs in all three dose groups, they were classified as unrelated to the procedure and should not raise safety concerns for this procedure. The rate of death in these patients should be considered a function of the poor health of this patient population that may improve with this procedure, rather than a function of the procedure or cell infusion itself, as it did not occur in the first 30 days post procedure. It is also interesting, and very relevant to the safety of the cells and dose, that only one patient in the entire study had a PRA level of 10% over the first year This is much less than that seen in the allogeneic bone marrow-derived mesenchymal stem cells (BM-MSCs) utilized by Perin et al. in a similar patient population using lower doses, which are currently being evaluated in a large-scale phase 3 clinical trial for HF 27 . The lower PRA may also be attributed to the fact that UCSECs are derived and cultured completely xeno free, which may minimize sensitization and immune response from the recipient of the cells, as they have minimal expression of major histocompatibility complex (MHC) class I and II antigens or costimulatory molecules on their surface 17 . The lack of any animal products from culture media or enzymes may also lead to younger cells, which could possibly contribute to better remodeling.
Although this study was not adequately powered to demonstrate a statistical significance in the use of UCSECs to treat patients with HF, there was no significant deterioration in EF or left ventricular chamber size in any patient group. The significant structural changes in the patients who received 200M and 400M cells are indicative that future studies are needed to further evaluate if a possible dose effect may exist without further toxicity.
There was a statistically significant decrease in BNP in patients receiving 200M and 400M cells. This decrease in BNP, coupled with the improved EF and reduction in left ventricular end-systolic volume (LVESV), potentially demonstrates an increased ability of the cardiac myocytes to tolerate stretch and volume overload. The combination of improving clinical measures and biomarkers is an interesting trend that would be better defined in a larger-dose study in similar patients. The exact mechanism for improvement with stem cell therapy remains controversial. Mirotsou et al. demonstrated that transplanted cells do not remain in the heart for more than a few days, and therefore, the benefit is likely related to the release of paracrine factors by stem cells into the injured myocardium that mediate neovascularization and remodeling 26 .
A number of different cell types have been used in HF. Perin et al. used allogeneic BM-MSCs delivered endocardially in HF patients, demonstrating a significant benefit in the 150M dose arm of their study 27 . However, Heldman et al. compared patients with ischemic cardiomyopathy receiving either intramyocardially delivery of mesenchymal stem cells or BM-MNCs to a placebo group, and demonstrated an improvement in the Minnesota Living with Heart Failure Questionnaire (MLWHFQ) score in both treatment groups, but not in the placebo group 24 . Henry et al. used a cultured, multicellular product delivered intramyocardially in patients with NYHA class III/IV, with an LVEF <30%. The patients with ischemic dilated cardiomyopathy appeared to benefit more than the nonischemic patients with fewer cardiac AEs, improved wall motion index scores, and improved MLHFQ scores 25 . A study by Vrtovec et al. using selected CD34+ bone marrow-derived cells in patients with nonischemic dilated cardiomyopathy showed significant improvement in ventricular function, without significant changes in left ventricular end-diastolic diameter LVEDD or left ventricular end-systolic diameter (LVESD) 28 .
Conclusions
This is the first prospective dose escalating clinical trial using allogeneic UCSECs infused into the coronary sinus of HF patients. This study met its primary endpoint demonstrating that 100M, 200M, or 400M cells can be safely delivered via retrograde coronary sinus delivery with no direct procedure-related AEs. Although this study was not powered to demonstrate statistical significance, results show early clinical signals that warrant further investigation. The present study was limited in its sample size and use of nonlabeled cells and was open labeled. However, the findings should lead to larger studies to further evaluate the potential benefits of UCSECs in patients with ischemic and nonischemic cardiomyopathy.
Footnotes
Acknowledgment
The study was funded by internal funds. J.T., A.C., J. Castillo, C.C., A.C., J.E., C.Y., and J. Cunza enrolled and treated the patients. J.T., and A.N.P. conceived the study. J.T., C.E.B., A.A.W., and A.N.P. all helped to draft the manuscript and were involved in the data analysis. All authors read and approved the final manuscript. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. A.N.P. has a patent filed on UCSEC. A.N.P. is a section editor of Cell Transplantation. Neither A.N.P. nor any member of the editorial board or editorial office affiliated with the authors' institutions were involved with the review process and/or decision making on this article. The authors declare no conflicts of interest.