
Abstract
Transferring exogenous mitochondria has therapeutic effects on damaged heart, liver, and lung tissues. Whether this protective effect requires the symbiosis of exogenous mitochondria in host cells remains unknown. Here xenogenic mitochondria derived from a hamster cell line were applied to ischemic rat brains and rat primary cortical neurons. Isolated hamster mitochondria, either through local intracerebral or systemic intra-arterial injection, significantly restored the motor performance of brain-ischemic rats. The brain infarct area and neuronal cell death were both attenuated by the exogenous mitochondria. Although internalized mitochondria could be observed in neurons and astrocytes, the low efficacy of mitochondrial internalization could not completely account for the high rate of rescue of the treated neural cells. We further illustrated that disrupting electron transport or ATPase synthase in mitochondria significantly attenuated the protective effect, suggesting that intact respiratory activity is essential for the mitochondrial potency on neural protection. These results emphasize that nonsymbiotic extracellular mitochondria can provide an effective cell defense against acute injurious ischemic stress in the central nervous system.
Introduction
The delivery of mitochondria from mesenchymal stem cells to injured cells may be one of the important aspects for the repairing potency of stem cells (12,23). Recent evidence also indicates that direct engraftment of autologous or allogeneic mitochondria into ischemic tissues such as heart (20,21), lung (12,24), and liver (16) successfully mitigated tissue injury. In addition, facilitating mitochondrial transport into the cells with pep-1, an artificial cell-penetrating peptide, effectively rescues the respiratory function of myoclonic epilepsy with ragged red fibers (MERRF) cell lines (5,6,17). These results illustrate the therapeutic potential of the transplantation of this organelle.
Although the underlying mechanism of the protective effects of mitochondrial transfer is largely obscure, previous reports indicated that exogenous mitochondria could be internalized by targeted cells (3,7,13), leading to the reestablishment of symbiosis. Engulfed mitochondria may also fuse with host mitochondria (14) and restore cellular respiration (22) and adenosine triphosphate (ATP) production (6,13,15).
However, the uptake efficacy of exogenous mitochondria was low or not determined in most studies (3,7,13,20,21). Whether the internalization of exogenous mitochondria and the reestablishment of symbiosis with host cells are essential for the therapeutic effect of mitochondria transplantation was also not investigated. In this study, we explore these questions in a rat brain stroke model induced by middle central artery occlusion (MCAO). In vitro rat primary cortical neurons were also subjected to this study under a nutrient-depleted environment. Xenogenic mitochondria, derived from hamster cells, were applied to test their therapeutic efficacy for mitigating ischemic stroke-related stress. The efficacy of neural protection, the histopathological features of the protective effects, the ratio of mitochondrial uptake in host cells and the underlying mechanism of mitochondrial therapy were investigated in this study.
Materials and Methods
Cell Culture
All the culture medium and supplements were purchased from Invitrogen-Gibco (Carlsbad, CA, USA), unless noted elsewhere. Baby hamster kidney fibroblast (BHK-21; male) cells (Food Industry Research and Development Institute, Shinchu, Taiwan) were cultured in high-glucose Dulbecco's modified Eagle medium (DMEM) supplemented with 5% fetal bovine serum (FBS), 1% penicillin and streptomycin (P/S), 1 mM L-glutamine, and 1 mM nonessential amino acids (NEAAs). For observing mitochondrial internalization and fusion in vitro, BHK-21 cells were transfected with a plasmid encoding mitochondrial signal peptide-fused green fluorescent protein (GFP) (Clontech Laboratories, Mountain View, CA, USA) before being extracted. BHK-21 cells were 70% confluent in a 10-mm culture dish (BD Biosciences, Franklin Lakes, NJ, USA) at transfection. Lipofectamine 2000 (8 μl; Invitrogen-Gibco) and 8 μg plasmid DNA in 392 μl Opti-MEM (Invitrogen-Gibco), respectively, were combined so that plasmid DNA was added into diluted Lipofectamine 2000 in a 1:1 ratio and incubated at room temperature for 5 min. The DNA–lipid complexes were added to the culture media of BHK-21 cells and incubated at 37°C for 24 h.
Primary cortical neurons were isolated from embryonic day (E) 18 Sprague–Dawley (SD) rat brains (n = 40; LASCO, I-Lan, Taiwan). Briefly, the cortex was carefully separated from the embryo in ice-cold Hank's balanced salt solution (HBSS), and the meninges were completely removed. Isolated cortical tissues were dissociated in HBSS with 2.7 mM CaCl2 (MDBio, Taipei, Taiwan), 0.5 mM ethylenediaminetetraacetic acid (EDTA) (AMRESCO, Solon, OH, USA), 1 mM NaH2PO4 (Sigma-Aldrich, St. Louis, MO, USA), 10 units/ml papain (Sigma-Aldrich), and 5 units/ml DNase I (Sigma-Aldrich). Dissociated cells were seeded into a 24-well tissue culture plate (BD Biosciences) at a density of 4 × 105 cells/well. The neural cells were maintained in high-glucose DMEM, supplemented with 10% FBS, 1% B27 supplement, 1% P/S, and 10 mM glutamate.
Mitochondria Extraction
In order to trace grafted mitochondria, BHK-21 cells were preincubated with 1 μM 5-bromo-2′-deoxyuridine (BrdU; Invitrogen-Gibco) for 24 h to tag mitochondrial DNA before mitochondria isolation. The reagents and methods for mitochondria extraction were described in a previous report (27). Briefly, dissociated 2 × 108 BHK-21 cells were washed with phosphate-buffered saline (PBS) and suspended with 2 ml SEH buffer [0.25 M sucrose, 0.5 mM ethylene glycol tetraacetic acid (EGTA), 3 mM N-(2-hydroxyethyl) piperazine-N′-ethanesulfonic acid (HEPES), pH 7.2; all from Sigma-Aldrich]. The cellular crude was homogenized by 10 ml Dounce grinders (Wheaton, Millville, NJ, USA). After homogenization, 10 μl of the cellular lysates was mixed with 30 μl of 0.4% trypan blue (Invitrogen-Gibco) and observed under a phase contrast microscope (CKX41; Olympus, Tokyo, Japan). Over 90% of the cells were disrupted by 25–30 strokes of the pestle in Dounce grinders. The crude lysates were centrifuged at 1,000 × g (MX-301; Tomy Digital Biology, Tokyo, Japan) for 15 min, and their supernatants were collected. Further centrifugation at 9,000 × g (Tomy Digital Biology) for 15 min concentrated the mitochondria from the supernatant in a pellet.
The mass of mitochondria was determined by Bradford method (Bio-Rad, Hercules, CA, USA), and the number of mitochondria was quantified by flow cytometry. To avoid the interference from the noise signal of cell debris, BHK-21 cells were stained with green MitoTracker before the mitochondria extraction. The total number of MitoTracker+ particles in 1 ml SEH buffer was determined by the flow analyzer (BD Influx; BD Biosciences). Examined by microscope examination and trypan blue exclusion assay, cellular nuclei and membrane-intact cells were barely observed. Referencing the obtained number of mitochondria particles in 10, 20, 50, 100, and 200 μg mitochondria, 75 μg mitochondria were interpolated to contain 1.2 ×1 06 mitochondria particles, according to flow cytometry analysis (data not shown).
Mitochondria Examination with Transmission Electronic Microscopy
Isolated mitochondria were fixed with 2.5% glutaraldehyde (Sigma-Aldrich) for 4 h at 4°C and then dehydrated with gradient concentrations of alcohol. After complete dehydration, mitochondria were embedded in 0.7 ml of LR white resin (Sigma-Aldrich) for 2 days at 4°C and then 12 h at 60°C. Embedded samples were sliced into 50- to 100-nm sections and then stained with 2% uranyl acetate (Sigma-Aldrich) and 0.2% lead citrate (Sigma-Aldrich). After staining, samples were visualized and examined under a transmission electron microscope (H-7000; Hitachi, Tokyo, Japan).
Respiratory Activity of Isolated Mitochondria
The respiratory activity of isolated mitochondria was revealed by the oxygen consumption rate, as determined by an XFe analyzer (Seahorse Bioscience, North Billerica, MA, USA) (24). The chemicals used in this section were all purchased from Sigma-Aldrich. In this study, 20 μg isolated mitochondria (about 3.2 × 105 mitochondria) were centrifuged at 2,000 × g (5500; Kubota, Osaka, Japan) to adhere them to the bottom of the assay plate. The procedure for analysis followed the manufacturer's suggested protocol. Mitochondrial respiratory chain modulators such as adenosine diphosphate (ADP, 2 mM), oligomycin (Omy, 2 μM), carbonyl cyanide-4-trifluoromethoxyphenylhydrazone (FCCP, 1 μM), and antimycin A (AntA, 4 μM) were prepared in mitochondrial assay buffer [70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, and 0.2% fatty acid-free bovine serum albumin (BSA), pH 7.2] and added into the reaction automatically.
The same concentrations of AntA and Omy in the respiratory activity assay were applied to the studies for cell viability assay under the oxygen glucose deprivation (OGD) condition.
Cerebral Stroke Model by Middle Cerebral Artery Occlusion (MCAO)
Adult male SD rats (n = 52; LASCO, I-Lan, Taiwan), weighing 250–300 g, were used in this study. We followed the MCAO method described in a previous study (18). Briefly, the right common carotid artery (CCA), external carotid artery (ECA), internal carotid artery (ICA), and the left CCA were exposed under a dissecting microscope (SM2445; Nikon, Tokyo, Japan). A 3-0 filament nylon suture (UNIK, New Taipei City, Taiwan) with a round tip was inserted into the right ECA to the original end of the MCA. The bilateral CCAs were ligated during MCAO to reduce blood support from the circle of Willis. After 1 h of occlusion, blood reperfusion was allowed by removing the suture as well as the ligations on the bilateral CCAs. The experimental protocols and procedures were approved by the Institutional Animal Care and Use Committee of National Chung Hsing University (IACUC 103-39).
Mitochondria Transplantation
After 24 h of MCAO, 75 μg of mitochondria (in 10 μl of SEH solution) was transplanted directly into the ischemic striatum [intercerebral injection (IC)]. Alternatively, 750 μg mitochondria (in 100 μl) was infused into the femoral artery [intra-arterial infusion (IA)] of the brain-ischemic rats. In the cerebral injection group, the injection site was as follows: AP: −0.4 mm; L: 3.4 mm; and DV: 5 mm. The injection rate was 1 μl/min. The syringe needle was held in place for an additional 5 min before withdrawal. In the arterial infusion group, mitochondria were injected into the lumen of the femoral artery by using an insulin syringe, and the infusion rate was 20 μl/min. The arterial infusion group received a boost of the same amount 14 days postoperation.
The total rat number was 52, grouped as Sham (n = 10), MCAO (n = 13), MCAO-IC (n = 15), and MCAO-IA (n = 14). Some of the animals were sacrificed 1 week postoperation for the measurement of brain infarct area and mitochondria tracing (MCAO, n = 4; IC, n = 5; IA, n = 4).
Animal Behavior Tests
The behavior tests of the MCAO rats were evaluated by using both grip (Muromachi, Tokyo, Japan) and rotarod equipment (Technical and Scientific Equipment, Bad Homburg, Germany). For the grip test, grip strength was measured by an electronic sensor linked with a pull bar. In the rotarod test, rats were pretrained to stay on an accelerating cylinder that was set to accelerate from 4 rpm to 40 rpm within 300 s. The recruited rats (Sham, n = 10; MCAO, n = 9; IC, n = 10; IA, n = 10) could grip 200 g with their forepaws and could stay on the rotarod cylinder for over 200 s.
Measurement of Brain Lesion
At 4 weeks postischemia, some of the rats (MCAO, n = 3; IC, n = 3; IA, n = 3) were euthanized with CO2, and their brains were quickly removed and sliced into six 2-mm-thick coronal sections. The brain slices were immersed in 2% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich) for 30 min at 37°C and then fixed with 4% paraformaldehyde (PFA; Sigma-Aldrich). TTC is reduced to red 1,3,5-triphenylformazan (TPF) by dehydrogenases in living tissue. The infarcted tissues exhibited a white color due to the nonreactiveness of dehydrogenases with TTC. Loss of brain areas was calculated by imaging analysis software, compared to the noninjured lateral cerebral hemisphere (Image-Pro Plus; Media Cybernetics, Sarasota, FL, USA).
Immunofluorescent Histological Staining
Some of the rat brains (MCAO, n = 5; IC, n = 6; IA, n = 6) were removed and fixed with 4% PFA for 4 h at 4°C. Brain samples were dehydrated in 12% sucrose solution for 24 h at 4°C. The samples were frozen at −80°C in Tissue-Tek OCT compound (Sakura Finetek, Tokyo, Japan) and sliced into 20-μm-thick sections. For antigen retrieval, sections were rinsed in PBS and then incubated in sodium citrate buffer (10 mM sodium citrate and 0.05% Tween-20, pH 6.0) at 95°C for 5 min. The tissues were washed in PBS and permeabilized with 0.1% Triton X-100 (USB Corporation, Cleveland, OH, USA) for 10 min. After incubation with 1% BSA in PBS for 1 h at room temperature, the samples were labeled with primary antibodies, including BrdU (1:500; BD Biosciences), glial fibrillary acidic protein (GFAP; 1:500; Millipore, Billerica, MA, USA), ionized calcium-binding adapter molecule 1 (Iba-1; 1:500; Novus Biologicals, Southpark Way, CO, USA), and microtubule-associated protein 2 (MAP-2; 1:500; Millipore) for 24 h at 4°C. The tissue slices were then washed with PBS and incubated with a rhodamine- or fluorescein isothiocyanate (FITC)-conjugated secondary antibody (1:300; Invitrogen-Gibco) for 1 h at room temperature.
A terminal deoxynucleotidyltransferase-mediated dUTP-fluorescein nick-end labeling (TUNEL) assay was performed with a commercial kit (In Situ Cell Death Detection Kit; Roche, Basel, Switzerland). Then 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) was used to stain cell nuclei. These fluorescently labeled samples were visualized and examined under a confocal fluorescent microscope (LSM 510; Carl Zeiss, Oberkochen, Germany) or a fluorescent microscope (Eclipse 80i; Nikon). The intensity of the fluorescent signal was measured by using imaging analysis software (Un-Scan-It 6.1; Silk Scientific, Orem, UT, USA).
In Vitro Ischemic Model
The method employed for the in vitro ischemic model for neurons was previously described (26). To establish ischemic conditions for the cells, primary neurons cultured from an E18 rat embryo were first amplified for 5 days and then cultured under serum-free and OGD conditions for 1, 2, 4, 6, or 12 h. The cells were cultured with DMEM 5030 (Sigma-Aldrich) supplemented with 1% NEAA and 1% P/S in an anoxic chamber (37°C, 5% CO2 and 95% N2; Modular Incubator Chamber; Billups-Rothenberg, Del Mar, CA, USA).
Cell Viability of Primary Neurons
At 1 h post-OGD treatment, mitochondria were added into the culture medium. Cell survival tests of the primary neurons were conducted at 24 h posttreatment of OGD. The cell survival rates were evaluated by Hoechst/propidium iodide (PI) double staining, 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay, AlamarBlue test, and lactate dehydrogenase (LDH) activity assay at the indicated times post-OGD. For Hoechst/PI double staining, nonfixed cells were incubated with 10 μg/ml Hoechst 33342 (Sigma-Aldrich) for 10 min at 37°C and then stained with 2 μg/ml PI (Invitrogen-Gibco) for 5 min. These fluorescently labeled samples were visualized and examined under a confocal fluorescent microscope (LSM 510). The intensity of the fluorescent signal was measured by using imaging analysis software (Un-Scan-It 6.1).
For the MTT assay, cells were incubated with 0.5 mg/ml MTT (USB Corporation) for 4 h at 37°C. After the reaction, the cells were washed twice with PBS and then harvested and dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich). The absorbance of the supernatant was measured at 570 nm (Paradigm Detection Platform; Beckman Coulter, Pasadena, CA, USA).
For the AlamarBlue assay, cells were incubated with 10% AlamarBlue (Invitrogen-Gibco) for 6 h at 37°C. After the reaction, the supernatant was loaded into a 96-well plate (BD Biosciences) for subsequent absorbance analysis at 570 nm and 600 nm (Paradigm Detection Platform).
The LDH activity assay was performed using a Cyto-Tox 96 nonradioactive cytotoxicity assay kit (Promega, Fitchburg, WI, USA). Total cell lysates (50 μl) were transferred into a 96-well plate and mixed with 50 μl of reconstituted substrate mix at room temperature for 30 min. The absorbance of the mixture was then measured at 490 nm (Paradigm Detection Platform).
Mitochondrial Internalization and Fusion
For observing mitochondrial internalization, primary neurons received GFP-tagged hamster mitochondria at 1 h post-OGD. After mitochondrial delivery, neurons were fixed with 4% PFA and incubated with primary antibody class III β-tubulin antibody (clone TuJ1, 1:500; Covance, Princeton, NJ, USA). The cellular samples were then washed with PBS and incubated with a rhodamine-conjugated secondary antibody (1:300; Invitrogen-Gibco) for 1 h at room temperature. Mitochondrial fusion was observed by first incubating primary neurons in media containing 250 nm MitoTracker Red CM-H2XRos (Thermo Fisher Scientific, Waltham, MA, USA) for 30 min at 37°C to tag the endogenous mitochondria, and then cocultured with GFP-tagged mitochondria for 24 h. The fluorescently labeled samples were visualized and examined under a confocal fluorescent microscope (LSM 510).
Statistical Analysis
One-way analysis of variance (ANOVA) and Tukey's post hoc analysis were used to determine the significance of differences between the experimental groups. Graphics creation and statistical analysis in this study were conducted by using GraphPad Prism 5 (GraphPad, La Jolla, CA, USA).
Results
Recovery of Motor Functions by Mitochondria Transplantation in Rats with Stroke
Before the evaluation of the efficacy of mitochondria transplantation in brain stroke, the quality of isolated mitochondria from BHK-21 cells was evaluated by assessing mitochondrial morphology and respiratory activity. The intact mitochondrial membrane and normal morphology of isolated mitochondria were validated under a transmission electronic microscope (TEM) (Fig. 1A, arrows). The purity of the mitochondria was 81.3 ± 4.1%. The isolated hamster mitochondria showed classical kinetic dynamics for the oxygen consumption rate after the treatment with electron transport modulators (24) (Fig. 1B). This evidence supports the integrity of the mitochondria and the normal functions of their respiratory complex proteins.

The isolated mitochondria displayed intact structures and classic respiratory activity. The morphology of the isolated mitochondria from BHK-21 cells was illustrated by examination under a transmission electronic microscope (A). Scale bar: 1 μm. The oxygen consuming rates (OCR) of the isolated mitochondria, representing the mitochondrial respiratory activity, were measured after treatment with respiratory protein complex modulators (B). The illustrated data represented the results from triplicate experiments.
We next examined the efficacy of xenogenic mitochondria transplantation on motor functional recovery in MCAO-treated rats. Brain-ischemic rats were severely impaired in motor performance during the first week postoperation (Fig. 2A, B). In contrast, the treated ischemic rats that received mitochondria transplantation at 24 h postoperation by cerebral injection (IC, 75 μg, 1.2 × 106 mitochondria) showed a significant recovery in rotarod performance at 7 days postoperation (Fig. 2A). Notably, providing the mitochondria via the blood vessel (arterial infusion, IA, 750 μg, 1.2 × 107 mitochondria) resulted in a similar level of motor activity recovery comparing to that of the IC injection group on 14 days postoperation (but not at 7 days) (Fig. 2A). In addition to rotarod analysis, the forelimb strength of MCAO-treated rats was quantified by the grip test. We found that the supplementation of exogenous mitochondria, either via local injection or systemic administration, successfully rescued the grip strength of MCAO-treated rats (p < 0.05) (Fig. 2B). Based on the motor activity analyses, we demonstrated that xenogenic mitochondria transplantation, either through brain injection or blood circulation, significantly improved the motor functions of brain-ischemic rats.

Mitochondria transplantation rescued the motor functions of middle cerebral artery occlusion (MCAO)-treated rats. (A) Motor coordination of the rats was evaluated by assessing the staying period on rotarod equipment within 1 month of treatment. (B) The motor activity of the forelimb was measured by a grip strength meter. Sham, nonoperated; MCAO, brain ischemia; MCAO-IC, brain ischemia with mitochondria transplantation via intracerebral injection; MCAO-IA, brain ischemia with mitochondria transplantation via intra-arterial injection. Pre-op, motor activities before MCAO operation. Statistical results are shown as means ± SE. *p < 0.05, compared with MCAO at the same time period. The number of rats: 8–10 in each group.
Reduced Brain Lesion by Mitochondria Transplantation
In addition to the behavioral examination, apoptosis in the acute phase of brain tissue injury was determined by a TUNEL assay. Examination of the peri-infarct area revealed that the rate of TUNEL+ nuclei was 20.3 ± 3.3% (n > 3000) in MCAO-treated neural cells 7 days postischemia. Providing mitochondria via brain injection (MCAO-IC) or arterial infusion (MCAO-IA) significantly decreased the rate of TUNEL+ cells to 6.8 ± 1.1% (n = 4,970) and 12.6 ± 2.8% (n = 4,845), respectively (p < 0.05) (Fig. 3A). In addition to counting the rate of TUNEL+ cells, the fluorescent intensity of TUNEL+ signals was examined in the slices of MCAO-treated brain (Fig. 3B). Compared with the untreated MCAO group, mitochondria-treated brains also exhibited significantly reduced apoptotic signals in their brain slices (p < 0.05) (Fig. 3B).
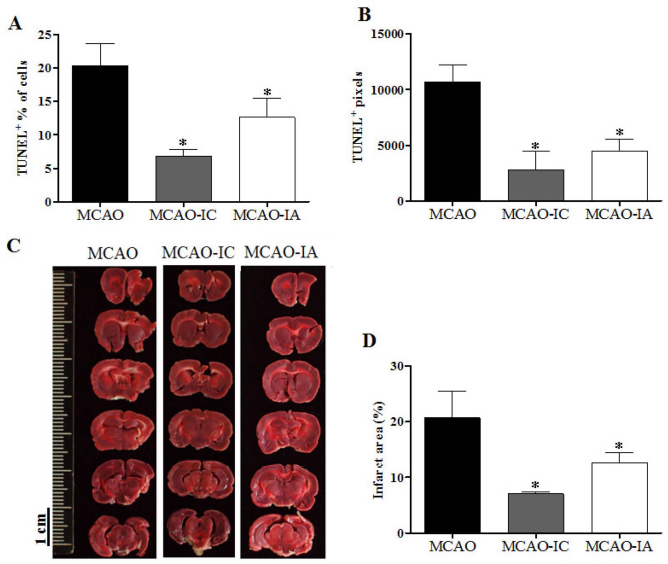
Mitochondria transplantation ameliorated cell death and infarct size in ischemic brains. Cell death in the peri-infarct area was assessed by terminal deoxynucleotidyltransferase-mediated dUTP-fluorescein nick-end labeling (TUNEL) assay at 1 week in post-MCAO brains. The ratios of TUNEL+ cells (A) and TUNEL+ pixels (B) are illustrated. Infarcted and missing areas, revealed by 2,3,5-triphenyltetrazolium chloride (TTC) stain (C), were calculated and compared with those of contralateral hemisphere (D) in 1 month post-MCAO brains. *p < 0.05, compared with MCAO. Three rat brains were examined in each group.
These early apoptotic cells contributed to the liquidation and loss of brain tissue at the late stage of ischemia. As determined by the liquidation and loss of brain tissue as revealed by TTC staining at 4 weeks postischemia, MCAO brains showed 20.7 ± 4.8% more tissue loss compared with the contralateral hemisphere (Fig. 3C). Providing hamster mitochondria significantly attenuated the size of the lesion to 7.1 ± 0.3% and 12.6 ± 1.8% in MCAO-IC and MCAO-IA rats, respectively (p < 0.05) (Fig. 3C, D). These results indicate that hamster mitochondria transplantation provides neural protection and motor functional recovery in brain-ischemic rats.
Distribution of Grafted Mitochondria in the Cerebrum
The grafted mitochondria, which were prelabeled with BrdU in BHK-21 cells for 24 h before extraction, were traced for their distribution and their internalization efficacy in the brain. In both MCAO-IC (Fig. 4A–C) and MCAO-IA (Fig. 4D–F), BrdU signals were detected in neurons (colocalization with MAP-2; 6.6 ± 1.9% and 3.5 ± 1.8%, respectively; n > 500) (Fig. 4A and D), astrocytes (colocalization with GFAP; 4.87 ± 1.54% and 1.93 ± 0.72%, respectively; n > 500) (Fig. 4B and E), and microglia (colocalization with Iba-1; 2.68 ± 2.08% and 2.87 ± 1.40%, respectively; n > 400) (Fig. 4C and F) in the peri-infarct area of the ischemic hemisphere at 4 weeks postischemia (summarized in Fig. 4G).
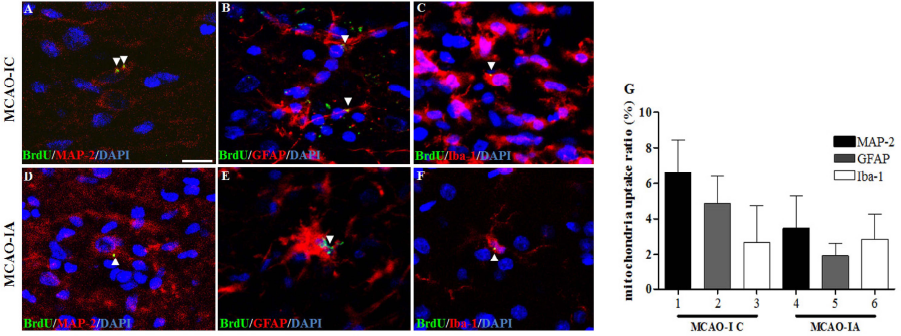
Cellular distribution of engrafted mitochondria in the ischemic brains. (A–F) The bromodeoxyuridine (BrdU)-labeled hamster mitochondria (arrowheads) were traced in neurons [microtubule-associated protein 2 (MAP-2+)] (A, D), astrocytes [glial fibrillary acidic protein (GFAP+)] (B, E), and microglial cells [ionized calcium-binding adapter molecule 1 (Iba-1+)] (C, F) of ischemic rat brains at 1 month post-MCAO. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). The ratio of BrdU in manually counted individual neural cells was statistically summarized (G).
In addition to the distribution of mitochondria in neural cells, the ratios of subpopulations of neural cells were also examined after MCAO. Compared with that in nontreated MCAO rats at 1 week postischemia (Fig. 5A), the proliferation of microglia was significantly enhanced in MCAO-IC rats (Fig. 5B). However, only a moderate elevation of microglial cell proliferation was observed in the MCAO-IA group (Fig. 5C). This pattern of cell proliferation was also observed for astrocytes in MCAO-treated brains (Fig. 5D–F). The altered cell population at 1 week post-MCAO was statistically analyzed (Fig. 5G–J, lines 1, 2, 3).
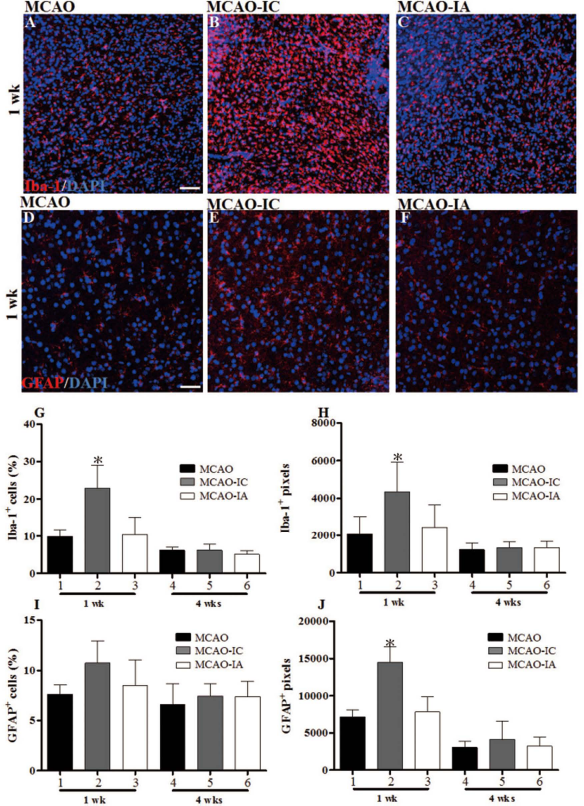
Transient proliferation of microglia and astrocytes after mitochondria transplantation into the ischemic brain. Brain slices at 1 week post-MCAO were stained for microglia (Iba-1) (A, B, C), astrocytes (GFAP) (D, E, F), and cell nuclei (DAPI). The brain samples include the slices from 1 week post-MCAO (A, D), from MCAO-IC (B, E) and MCAO-IA (C, F) rats. The ratios of microglial numbers (G) and their pixels (H) to the total number of brain cells and to histological photos were calculated, respectively. The same approach was also applied for astrocyte analysis (I, J). *p < 0.05, compared with MCAO.
However, at 4 weeks postischemia, no significant difference in astrocytic/microglial proliferation was detected between the experimental groups (Fig. 5G–J, lines 4, 5, 6). The roles of glial proliferation and activated microglial immune responses on the neural protection conferred by transplanted mitochondria require further investigation.
Hamster Mitochondria Protect Against the Stress of Serum-Free and Oxygen Glucose Deprivation Conditions
To further investigate the neural protective effect of grafted hamster mitochondria, primary neurons were cultured under serum-free OGD conditions to recapitulate the cell degeneration that occurs in the ischemic brain. The number of surviving cells was estimated by the intracellular LDH concentration in total cell lysates. Over 90% of the primary cells expressed the neuronal marker MAP-2, showing the high purity of cultured cortical neurons (Fig. 6A). After 4, 6, and 12 h of OGD treatment, intracellular LDH levels in the surviving cells at 24 h post-OGD dramatically reduced to 1.1 ± 0.9%, 0.7 ± 0.1%, and 0.6 ± 0.2%, respectively, normalized to the untreated control (Fig. 6B). Adding 20 μg of exogenous mitochondria into a well (24-well plate) at 1 h post-OGD significantly enhanced the survival rate up to 19.3 ± 3.8%, 19.1 ± 5.8%, and 16.0 ± 0.9% after 4, 6, and 12 h of OGD treatment, respectively (p < 0.05) (Fig. 6B). Moreover, AlamarBlue (Fig. 6C) illustrated similar results as the LDH assay and MTT assays showed enhanced cell viability at 4 h OGD (Fig. 6D). This evidence demonstrates that receiving exogenous hamster mitochondria protects primary rat neurons against OGD-triggered cell death.

Addition of hamster mitochondria mitigated oxygen-glucose deprivation (OGD)-induced stress in rat cortical neurons. (A) The high purity of E18 rat cortical neurons was revealed by expression of the neuronal marker MAP-2. Scale bar: 50 μm. (B) Providing hamster mitochondria enhanced cellular lactate dehydrogenase (LDH) activity in primary neurons after OGD. The mitochondrial activities of surviving cells, determined by AlamarBlue (C) and 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assays (D), are also illustrated. Statistical results are shown as the means ± SE. *p < 0.05 compared with OGD at the same time period. These results represent data from four experiments.
To trace the fate of internalized mitochondria in vitro, BHK-21 cells were transfected with a plasmid encoding mitochondrial signal peptide-fused green fluorescent protein (GFP). Primary cortical neurons (2 × 105 cells) were cocultured with a large amount of GFP-tagged hamster mitochondria (10 μg) for 24 h. Approximately 22% of primary neurons [β-III tubulin cells stained with antibody clone TuJ1 (TuJ1+), n > 300] harbored the applied mitochondria at 24 h coculture; this ratio was not dramatically altered between OGD-treated and nontreated neurons (Fig. 7). We also observed that the GFP+ neurons generally harbored less than five hamster mitochondria per cell (Fig. 7).

The uptake efficacy of hamster mitochondria was estimated in rat primary cortical neurons. Green fluorescent protein (GFP)-tagged mitochondria from BHK-21 cells were incubated with untreated (A) and OGD-treated (B–E) primary neurons (β-III tubulin+ cells stained with antibody clone TuJ1) for 24 h. Arrows indicate the internalized mitochondria in the neurons. The ratios of mitochondria internalization in primary neurons were estimated by manually counting under a fluorescent microscope (F). The result of (F) represents duplicate experimental data. Scale bar: 10 μm.
To examine the fusion between endogenous and exogenous mitochondria, the primary neurons were prelabeled with red fluorescent MitoTracker and then incubated with GFP-tagged mitochondria. Most hamster mitochondria appeared to scatter outside the neurons (Fig. 7); a few hamster mitochondria were internalized by primary neurons (Fig. 8A, B). In addition, few colocalized mitochondrial signals were detected within the primary neurons (yellow vs. green fluorescent mitochondria per cell, 2.7 ± 0.3%, n = 482) (Fig. 8C, D), suggesting that a low percentage of fusion occurred between the internalized mitochondria and the host mitochondria.

The colocalization of endogenous and exogenous mitochondria was observed in cortical neurons. Primary cortical neurons (2 × 105 cells) were first incubated with MitoTracker (250 nM) for 30 min to stain endogenous mitochondria and then cocultured with GFP-tagged hamster mitochondria (10 μg). Most hamster mitochondria scattered outside of the neurons (A) and few hamster mitochondria were internalized by the primary neurons (B). (C) Indicates that a rat neuron harbored the hamster mitochondria colocalized with rat mitochondria (arrowhead). A field of panel (C) is enlarged in (D) with 3D sections (thickness: 0.36 μm). Scale bar: 10 μm.
Functional Respiratory Activity Is Required for Mitochondria-Mediated Neural Protection
To determine whether providing intact mitochondria are essential for mitochondria-mediated neural protection, isolated mitochondria were pretreated with AntA and Omy to block electron transport and ATP synthesis, respectively (1). Flow cytometry analysis (Fig. 9) revealed that after 4 h of OGD treatment, the ratio of membrane-disrupted cells, which are permeable to PI (Fig. 10), was robustly elevated (80% of total cells) at 24 h post-OGD (Fig. 9A–C and Fig. 10A, B). The delivery of hamster mitochondria significantly reduced the ratio of PI+ cells in a time-dependent manner (Figs. 9D–F, 10C and F). Interestingly, AntA- and Omy-treated mitochondria failed to effectively protect primary neurons against OGD stress (PI+ cells, 60% and 70%, respectively) (Figs. 9G, H, 10D–F).

Intact mitochondrial respiratory activity is essential for coping with OGD-induced stress. Live primary neurons were treated with propidium iodide (PI) and analyzed with fluorescent-activated cell sorting (FACS) to estimate the number of dead cells after OGD. The PI-permeable cells were quantified after OGD for 1 h (A), 2 h (B), and 4 h (C). The alteration of PI+ cells by hamster mitochondria treatment was shown by FACS (D–F), including the groups containing antimycin A (AntA) (G) or oligomycin (Omy) (H)-treated mitochondria after 2 h of OGD-induced stress.

The respiratory activity of mitochondria is a determining factor for coping with OGD-induced stress. (A–E) Live primary neurons were treated with PI/Hoechst 33342 to estimate the ratio of dead cells after OGD. The PI-permeable cells were observed after treatment with 2 h OGD (B–E). The ratio of PI+ cells was manually counted under a fluorescent microscope, including the groups containing AntA- or Omy-treated mitochondria (F). Scale bar: 50 μm. Intracellular LDH activity of the surviving cells was also measured after 2 h of OGD treatment (G). *p < 0.05 compared with OGD. #p < 0.05 compared with OGD/MT/AntA. &p < 0.05 compared with OGD/MT/Omy. The results of (F) and (G) represent data from four experiments.
We further conducted an LDH assay to assess the protective effects of mitochondria against OGD. Hamster mitochondrial treatment significantly retained the intracellular LDH concentration after 1, 2, 4 and 6 h of OGD (p < 0.05) (Fig. 10G). Treating mitochondria with AntA or Omy showed a moderate benefit for cell survival at 1 h of OGD but exhibited little effects against longer periods of OGD (Fig. 10G). These results suggest that dysfunctional mitochondria are unable to cope with OGD-induced stress.
Discussion
Our findings demonstrated that xenografting hamster mitochondria via brain injection or systemic administration mitigated brain lesions in MCAO-treated rats. The recipients of mitochondria exhibited significant functional recovery of their motor activities. We also applied OGD in primary neuronal culture to mimic focal ischemia in vitro and illustrated that providing xenogenic mitochondria effectively improved neuron survival after the induction of ischemia in vitro.
We argue that the isolated mitochondria in the crude cell extracts are mainly responsible for the long-term protection against brain ischemia. The normal morphology and functional respiratory complexes of the isolated mitochondria support that the transplanted mitochondria have integrity and are intact. A trypan blue exclusion assay also indicated that cellular nuclei and membrane-intact cells were barely observed in the mitochondrial fraction under microscope examination. Although recent evidence indicates that some cellular organelles such as exosomes contain genetic material and may contribute to neural protection (8,28), the low centrifugal force (9,000 × g for 15 min) used in this study should exclude these small vesicles from the isolated mitochondria pellets, leaving most exosomes in the discarded supernatants (10).
Previous studies in ischemic hearts (20,21) and livers (16) have shown that mitochondria transplantation provides effective prevention against perfusion-triggered mitochondrial disruption. However, mitochondria transplantation for the treatment of ischemic lesions after perfusion has not been reported. Here we demonstrate that mitochondrial engraftment through IC injection significantly protected rats against ischemia-induced damage after perfusion. Transferring mitochondria effectively rescued forelimb and hindlimb motor functions at 7 days postoperation and sustained this recovery for 1 month.
In contrast to the direct IC injection, providing the mitochondria via the blood (IA) did not rapidly rescue the motor activities in ischemic rats on day 7 postperfusion. Nevertheless, significantly improved motor function in the recipients of mitochondria via IA was observed at 2 weeks postoperation. Further investigations are required to optimize the dosage of mitochondria, number of boosts, and different routes of administration via the blood to achieve better improvements with mitochondrial therapy against brain ischemia.
A recent study has revealed that mitochondrial transfer from engrafted mesenchymal stem cells to alveolar cells is essential for therapeutic effects in lipopolysaccharide-induced lung injury (12). The internalized mitochondria can restore cell growth and metabolism, possibly through supplying ATP, reducing host mitochondrial damage or increasing numbers of mitochondria (12). To examine the necessity of mitochondrial symbiosis for functional restoration, xenogenic mitochondria transplantation was used in this study. Our results illustrate that the xenogenic mitochondria show strong potency for providing neural protection in ischemic stroke. However, only a low internalization ratio of the engrafted mitochondria was detected in neural cells both in vivo and in vitro. These results suggest that fusion between host and engrafted mitochondria may not be necessary for xenogenic mitochondrial-mediated neural protection.
A recent report indicated that DsRed2-labeled human mitochondria could be internalized into a rat cardiomyoblast cell and a human endometrial mesenchymal cell (15). Interestingly, although less than 10% DsRed-positive cells were detected when 5 μg of mitochondria was cocultured, the mitochondria significantly improved the cell survival and dramatically enhanced the cellular bioenergetics (15). An in vitro study also indicated that incubation with xenogenic mouse mitochondria can restore the respiratory function of human mitochondria-defective (Rho0; ρo) cells (13). Species compatibility between the nucleus and the mitochondria is crucial for respiratory protein complex functions and mitochondrial renewal (9); therefore, internalized hamster mitochondria could not be sustained long term in the rat cells (29). Although the detailed mechanisms underlying neural protection by xenogenic mitochondria remain obscure, exogenous mitochondria might serve as an ATP source or reactive oxygen species (ROS) scavenger to protect the cells from damage caused by free radicals and to sustain the integrity of endogenous mitochondria during ischemic stroke.
Although we did not investigate the immune response in the rat recipients after the hamster mitochondria transfer, the xenogenic mitochondria may activate innate immune cells, such as macrophage and microglia cells, and consequently enhance the cross-species transfer of mitochondria into phagocytic cells. It is also possible that rat antibodies against hamster mitochondria can be induced and then enhance the phagocytosis of exogenous mitochondria. Nevertheless, in this study, we mainly focus on the Fc receptor-free neurons under ischemic stress in vivo and in vitro. Antibody-dependent enhancement of mitochondria engulfment may not contribute to this cross-species transfer in neurons. Interestingly, our recent research indicates that systemic transfer of allogenic mitochondria significantly reduces the immune cell infiltration and inflammation in rats with acute respiratory distress syndrome (ARDS) (24). This downregulated inflammation may cause the direct protection on damaged lung tissue, rather than inhibitory modulation of the immune system.
Taken together, the results of this study first demonstrate the feasibility of mitochondria transplantation for treating ischemic stroke. Engrafted hamster mitochondria successfully mitigated the brain lesion and rescued the motor activity of MCAO-treated rats. In contrast to neural cell transplantation, mitochondrial transfer causes less concern of tumor formation (2,4), immunorejection (25), tissue misintegration, and abnormal neural circuit creation (11,19). Facilitating mitochondrial transfer into the cells and investigating the potential mechanisms of protection should accelerate the application of mitochondria for the treatment of numerous clinical diseases caused by dysfunctional mitochondria.
Footnotes
Acknowledgments
We thank the contribution from the Professor Chi-Me Hsueh at the Department of Life Sciences National Chung Hsing University on the OGD culture. This work was supported by grants from the National Science Council of Taiwan (NSC 101-2811-B-005-015-MY3 and 102-2628-B-005-007-MY3) and the Taichung Veterans General Hospital/National Chung Hsing University Joint Research Program (TCVGH-NCHU 1057606 and 1027607). This research was also funded in part by the Ministry of Education, Taiwan, Republic of China, under the ATU plan. The authors declare no conflicts of interest.