
Abstract
Efficient maintenance of the undifferentiated status of embryonic stem cells (ESCs) may be important for preparation of high-quality cell sources that can be successfully used for stem cell research and therapy. Here we tried to identify a compound that can enhance the quality of pluripotent stem cells. Treatment of ESCs and induced pluripotent stem cells (iPSCs) with 3,2′-dihydroxyflavone (3,2′-DHF) led to increases in cell growth, colony formation, and cell proliferation. Treatment with 3,2′-DHF resulted in high expression of pluripotency markers (OCT4, SOX2, and NANOG) and significant activation (STAT3 and AKT) or suppression (GSK3β and ERK) of self-renewal-related kinases. 3,2′-DHF-treated high-quality pluripotent stem cells also showed enhanced differentiation potential. In particular, treatment of iPSCs with 3,2′-DHF led to elevated expression of ectodermal differentiation markers and improved differentiation into fully matured neurons. Next, we investigated the in vivo effect of 3,2′-DHF-pretreated iPSCs (3,2′-DHF iPSCs) in a peripheral nerve injury model and found that transplantation of 3,2′-DHF iPSCs resulted in more efficient axonal regeneration and functional recovery than in controls. Upon histopathological and gene expression analyses, we found that transplantation of 3,2′-DHF iPSCs stimulated expression of cytokines, such as TNF-α, in the early phase of injury and successfully reduced convalescence time of the injured peripheral nerve, showing an effective neuroprotective property. Taken together, our data suggest that 3,2′-DHF can be used for more efficient maintenance of pluripotent stem cells as well as for further applications in stem cell research and therapy.
Keywords
Introduction
Embryonic stem cells (ESCs) are capable of self-renewal and can differentiate into numerous cell types (60,61). These abilities are affected by many factors, such as growth factors and feeder cells in culture, and by various signaling pathways, such as survival-, apoptosis-, self-renewal-, and differentiation-regulating pathways (50). Maintenance of the undifferentiated status of ESCs requires the preservation of self-renewal by inhibiting differentiation and regulating senescence and apoptosis (57). Although ESCs are thought to serve as an unlimited cell source for regenerative medicine, techniques for maintaining undifferentiated ESCs are still inefficient, which results in inhomogeneous populations of cells.
In a recent series of studies, mouse and human fibroblasts were reprogrammed into pluripotent stem cell-like cells [induced pluripotent stem cells (iPSCs)] through the transfection of a diverse set of stem cell-related transcription factors, such as mouse octamer-binding protein 4 (mOct4), mouse sex-determining region Y box 2 (mSox2), mouse Kruppel-like factor 4 (mKlf4), and mc-Myc (47,75). The iPSCs derived from mouse and human fibroblasts proved to be highly similar to ESCs in genetic, epigenetic, and developmental criteria (23). The application of iPSCs has several advantages, including easy attainment of cells, existence of various sources, freedom from ethical issues, and possible therapeutic potential. Numerous studies have been performed to test the ability of various supplements, such as human serum albumin, vitamins, antioxidants, trace minerals, lipids, and cloned growth factors to support growth and maintenance of ESCs and iPSCs (60). Moreover, a cell-based screen of chemical libraries was carried out to identify small molecules that control self-renewal of pluripotent stem cells (11).
Flavonoids are naturally arising. These are widely distributed plant pigments that are present in a wide range of foods (46,65,66,74). The main dietary sources of flavonoids are fruits and vegetables, as well as popular beverages such as wine, tea, and coffee. Certain flavonoids have been observed to exhibit several physiological functions, such as antihypertensive, antiviral, and anti-inflammatory properties (12). In particular, their antioxidant activity has been receiving attention as a possible dietary source of protection against cardiovascular and neurodegenerative diseases (13). Recent studies have shown that flavonoids and their metabolites exert several intracellular effects, including the ability to directly modulate cell-signaling pathways (50). Previously, we reported that various flavonoids had antiapoptotic, antioxidant, and cell differentiation-regulating activities (29,30,34-39), and the hydroxylation patterns of the flavonoid B ring were known to play a critical role in their function. Although many other studies have also shown that substituted functional groups cause diverse effects on flavonoid function (14,31), the effect of differential hydroxylation of the flavonoid B ring has not been well characterized in pluripotent stem cells.
Here we used flavonoids containing differential hydroxylation in the B ring of the diphenylpropane (C6C3C6) skeleton and attempted to identify a flavonoid that enhances cell growth and pluripotency marker expression in pluripotent stem cells. As a result, we have determined that 3,2′-dihydroxyflavone (3,2′-DHF) significantly increases the cell growth, pluripotency marker expression, and neural differentiation of ESCs and iPSCs. We also investigated the in vivo effect of 3,2′-DHF-pretreated iPSCs (3,2-DHF iPSCs) in a sciatic nerve injury model and found that transplantation of the 3,2′-DHF iPSCs could promote functional recovery and axonal regeneration involving a mechanism thought to be associated with its neuroprotective property.
Materials and Methods
Cell Culture
D3 mouse embryonic stem cell line from male Sv129 mice (CRL-1934; American Type Culture Collection, Rockville, MD, USA) and iPSCs were seeded on 30-mm culture dishes (BD Biosciences, San Jose, CA, USA) at 1.2 χ 104 cells on feeder layers of mitomycin C (Sigma-Aldrich, St. Louis, MO, USA)-treated C57/BL6 (Biomedical Mouse Resource Center in Korea Research Institute of Bioscience and Biotechnology, Cheongwon, Chungbuk, Korea) male/female mixed mouse embryonic fibroblast (MEF) cells. D3 cells and iPSCs were maintained in ESC culture media, which is Dulbecco's modified Eagle's media (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 20% fetal bovine serum (Hyclone, Logan, UT, USA), 2 mM l-glutamine (Sigma-Aldrich), 0.1 mM β-mercaptoethanol (Sigma-Aldrich), 1% nonessential amino acids (Sigma-Aldrich), 50 U/ml penicillin, 50 μg/ml streptomycin (Invitrogen), and leukemia inhibitory factor (LIF; 1,000 units/ml of ESGRO; Chemicon, Temecula, CA, USA) at 37°C in 5% CO2.
Lentiviral Infection and iPSC Generation
MEFs were infected with self-inactivating lentiviral GFP-expressing vectors (System Biosciences, Mountain View, CA, USA) encoding Oct4, Sox2, Klf4, and c-Myc under the control of the elongation factor-1 α promoter. Approximately 14 days after infection, ESC-like colonies were observed and individually transferred to separate dishes containing mitomycin C-treated MEF feeder cells. These cells were incubated with ESC culture media that was changed daily.
Flavonoid Treatment and MTT Assay
The flavonoids 3-hydroxyflavone (3-HF), 3,2′-DHF, 3,3′-dihydroxyflavone (3,3 -DHF), and 3,4 -dihydroxyflavone (3,4′-DHF) were purchased from the INDOFINE Chemical Company (Hillsborough, NJ, USA). ESCs and iPSCs were trypsinized (Gibco, Grand Island, NY, USA), and 3 χ 103 cells were plated in 96-well plates (BD Biosciences) containing ESC culture media with or without the individual flavonoids indicated above. Cell viability was evaluated using an MTT assay. In brief, after incubation, the cells were treated with MTT solution (Sigma-Aldrich) at a final concentration of 0.25 mg/ml for 3 h at 37°C. The MTT assay for live cells resulted in the formation of dark blue formazan crystals, which were dissolved in dimethyl sulfoxide (Sigma-Aldrich) upon completion of the incubation period. The absorbance of wells was determined using a Bio-Rad (Hercules, CA, USA) plate reader (Model 680) at 540 nm.
Embryonic Body (EB) Formation and Spontaneous Differentiation
To assess EB formation, iPSCs were harvested and suspended in low attachment dishes (BD Biosciences) for 4 days. Aggregated EBs were plated on 0.1% gelatin (Sigma-Aldrich)-coated tissue culture plates (BD Biosciences) to initiate spontaneous differentiation. The cells were then cultured for an additional 7 days in ESC culture media without LIF before fixation. Culture media were changed every other day.
Mature Neuronal Differentiation
To induce differentiation into mature neurons, equal numbers (3 χ 104 cells/35-mm culture dish) of cells were cultured on poly-L-ornithine (Sigma-Aldrich)/laminin (Sigma-Aldrich)-coated plates (BD Biosciences) with neural induction media containing DMEM/F12 (Gibco), B27/N2 supplement (Sigma-Aldrich), and 10 ng/ml basic fibroblast growth factor (R&D Systems, Minneapolis, MN, USA) for 3 days (ND1). At day 5, 500 ng/ml sonic hedgehog (R&D Systems) and 1 μM retinoic acid (Sigma-Aldrich) were added to the ND1 media (ND2), and the cells were cultured for an additional 5 days to induce differentiation into neuronal progenitors. The neuronal progenitors were cultured in neurobasal media consisting of N2 supplement, 1 μM cyclic adenosine monophosphate (Sigma-Aldrich), 10 ng/ml brain-derived neurotrophic factor (BDNF; R&D Systems), 10 ng/ml glial cell line-derived neurotrophic factor (GDNF; R&D Systems), and 10 ng/ml neurotrophin-3 (NT-3; R&D Systems) (ND3). The cells were then cultured for 1 to 2 more weeks in ND3 media.
Alkaline Phosphatase (AP) Assay
AP activity was quantitated using the Sigma Diagnostics Alkaline Phosphatase Kit (Sigma-Aldrich) in accordance with the manufacturer's protocol. Briefly, ESCs were fixed with a citric acid-acetone formalin (Sigma-Aldrich) fixative and then stained with Naphthol/FRV-Alkaline solution (Sigma-Aldrich). The activity of AP was evaluated microscopically (Olympus USA, Melville, NY).
RT-PCR and Real-Time PCR Analyses
Total RNA was isolated using the TRI reagent (easy-blue; Intron, Seoul, Korea), according to the manufacturer's protocol. cDNA synthesis was performed with 1 μg of total RNA using a cDNA synthesis kit (Promega, Madison, WI, USA). cDNA was subjected to PCR amplification with specific primers. Primer sequences are listed in Tables 1-3. PCR was carried out using Accuprime Taq polymerase (Invitrogen), and quantitative real-time PCR was performed with Fast SYBR Green Master Mix (Applied Biosystems, Stockholm, Sweden). Quantitative gene expression data were normalized to the expression level of housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (Gapdh).
Western Blotting
To prepare whole-cell extracts, cells were washed three times in ice-cold phosphate-buffered saline (PBS; Hyclone), scraped from the dishes, and suspended in lysis buffer (1% Triton X-100; Amresco, Solon, OH, USA), 100 mM Tris-HCl (Sigma-Aldrich), pH 7.5, 10 mM NaCl (Sigma-Aldrich), 10% glycerol (Junsei Chemical, Tokyo, Japan), 1 mM sodium orthovanadate (Sigma-Aldrich), 50 mM sodium fluoride (Sigma-Aldrich), 1 mM p-nitrophenyl phosphate (Sigma-Aldrich), and 1 mM phenylmethylsul-fonyl fluoride (Sigma-Aldrich). After incubation on ice for 30 min, lysates were centrifuged, and protein in the supernatants was quantified using the Bradford Protein Assay Reagent (Bio-Rad). An equal amount of protein was then separated on a 10% SDS-PAGE gel (30% polyacrylamide (Sigma-Aldrich), 1.5 M Tris (Sigma-Aldrich), 10% SDS (Sigma-Aldrich), 10% ammonium persulfate (Sigma-Aldrich), tetramethylethylenediamine (Sigma-Aldrich) followed by electrophoretic transfer to nitrocellulose membranes (Whatman, Maidstone, UK). Membranes were blocked in 5% nonfat dry milk (Amresco) and 0.1% Tween-20 (Amresco) in Tris-buffered saline (Sigma-Aldrich). Primary antibodies used for Western blotting were anti-OCT4, anti-SOX2, anti-NANOG, anti-β-ACTIN, anti-phosphorylated glycogen synthase kinase 3 β (p-GSK3β), anti-GSK3β, anti-phosphorylated extracellular signal-regulated kinase (p-ERK), and anti-ERK (1:1,000; all Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-phosphorylated signal transducer and activator of transcription 3 (p-STAT3), anti-STAT3, anti-phosphorylated v-akt murine thymoma viral oncogene homolog (p-AKT), and anti-AKT (1:1,000; Cell Signaling, Danvers, MA, USA); and secondary antibodies were anti-mouse immunoglobulin G horseradish peroxidase (IgG-HRP) or anti-rabbit IgG-HRP (1:2,000; Santa Cruz Biotechnology). Immunoblotting was followed by detection using an enhanced chemiluminescence kit (Amersham Biosciences, Piscataway, NJ, USA).
Primer Sequences for Markers of ESC Pluripotency and Germ Layer Differentiation

Oct4, octamer-binding protein 4; Sox2, sex-determining region Y box 2; Klf4, Kruppel-like factor 4; Fgf4, fibroblast growth factor 4; Gapdh, glyceraldehyde 3-phosphate dehydrogenase; Afp, a fetoprotein; Cdx2, caudal type homeobox 2; Bmp4, bone morphogenetic protein 4.
Primer Sequences for Markers of Neuronal Differentiation

Otxl, orthodenticle homeobox 1; Enl, engrailed-1; Wntl, wingless-type mouse mammary tumor virus (MMTV) integration site family member 1; Lmxlb, LIM homeobox transcription factor 1; Th, tyrosine hydroxylase; Dcx, double cortin; Chat, choline acetyl transferase; Ddc, dopa decarboxylase.
Primer Sequences for Markers of Neuroprotection and Nerve Regeneration

Tnf, tumor necrosis factor; Vegf, vascular endothelial growth factor; Gap43, growth-associated protein 43; Bdnf, brain-derived neurotrophic factor; Gdnf, glial cell line-derived neurotrophic factor; Nt-3, neurotrophin 3; Ncam, neural cell adhesion molecule; Ne-fl, neurofilament light polypeptide.
RNA Sequencing (RNAseq)
For RNAseq analysis, the total RNA samples extracted from control ES or 3,2′-DHF-treated ESCs were submitted to the sequencing company, BML (Seoul, South Korea). As previously described (53), RNA libraries for RNAseq were prepared from 100 ng of total RNA from each sample according to the manufacturer's instructions. Briefly, first-strand cDNA was generated from total RNA using DNA/RNA chimeric primers and reverse transcriptase, creating a cDNA/RNA hybrid. The second-strand cDNA was synthesized from the DNA/RNA duplex. The resulting double-stranded cDNA molecule was amplified, and the amplified products were modified by random priming and extension to create double-stranded products for generating libraries for sequencing. The double-stranded products underwent blunt-end repair, and adapter molecules were ligated to the 5 and 3 ends of each fragment to generate the final library. Three distinct indexed libraries were loaded per flow cell and sequenced on an Illumina HiSeq 2000 (Illumina, Inc., San Diego, CA, USA) using TruSeq SBS sequencing software (version 3) (Illumina, Inc.) and SCS data collection software (version 1.4.8; Illumina, Inc.). Base calling was performed using Illumina RTA (version 1.12.4.2). An average of 130 million reads per sample was achieved, resulting in 97% mapped reads. Then raw reads were aligned to the reference sequence by TopHat software (version 2.0.11) (Baltimore, MD, USA).
Immunocytochemistry
For immunocytochemistry, cells were fixed in 4% paraformaldehyde (Sigma-Aldrich) in 0.1 M PBS and permeabilized with 0.2% Triton X-100 for 10 min. After washing with PBS, the cells were incubated in 10% normal goat serum (Vector Labs, Burlingame, CA, USA) for 1 h to block nonspecific binding. Cells were then incubated with the primary antibody at 4°C overnight. After washing with PBS, cells were then incubated with a secondary antibody for 1 h. Nuclei were stained using 10 μg/ml 4′,6-diamidino-2-phenylindole (DAPI; 1:1,000; Invitrogen). Primary antibodies used were mouse monoclonal anti-OCT4, anti-stage-specific embryonic antigen 1 (SSEA-1; Santa Cruz Biotechnology, 1:500), anti-α-fetoprotein (AFP; 1:500; R&D Systems), anti-β-tubulin III (1:500; Sigma-Aldrich), anti-nestin (1:500; Abcam, Cambridge, UK), rabbit polyclonal anti-Brachyury (1:500; Santa Cruz Biotechnology), anti-homeobox 9 (HB9; 1:500), anti-choline acetyltransferase (CHAT; 1:200), and anti-microtubule-associated protein 2 (MAP2; 1:500; all from Abcam, Cambridge, UK). Secondary antibodies used were Dylight 549-conjugated goat anti-mouse and Dylight 649-conjugated goat anti-rabbit (1:1,000; Jackson ImmunoResearch Labs) antibodies.
Sciatic Nerve Injury Animal Model and Transplantation
Thirty 6-week-old male Sprague-Dawley rats weighing an average of 200 g were purchased from Orient Bio (Seongnam, Kyungkido, Korea). Surgical procedures were performed under general anesthesia induced by intraperitoneal injection of tiletamine zolazepam (Virbac Korea, Seoul, Korea) and xylazine HCl (Bayer Korea, Kyungkido, Korea). Bilateral sciatic nerves were exposed by lateral incision of the hindlimb parallel to the femur and crushed for 10 min using Halsted mosquito forceps, straight (Roboz Surgical Instrument, Gaithersburg, MD, USA). After the crushing injury, 5 χ 106 iPSCs with and without 3,2′-DHF pretreatment were injected to the center of the lesion using a 50-μl Hamilton syringe (Hamilton Company, Reno, NV, USA). The cell-grafted sites were coated with PBS-diluted Matrigel (1:1; BD Biosciences) from the proximal to distal regions of the lesion. Rats were divided into five groups: handling only (no injury, sham), injury (injury control), Matrigel (injury with matrigel coating, matrigel control), iPSCs (iPSCs + Matrigel coating), and 3,2′-DHF iPSCs (3,2′-DHF-pretreated iPSCs + Matrigel coating). All experimental protocols were approved by the Institutional Animal Care and Use Committee of Konkuk University (KU13006). Rats were sacrificed at 10 and 28 days after surgery, and gross lesion morphology of the bilateral sciatic nerves was examined macroscopically and photographed. The bilateral sciatic nerves were collected and evaluated by histopathological and gene expression analyses.
Accelerating Rota-Rod Examination for Evaluation of Motor Nerve Function
After surgery, motor nerve function was examined using a Rota-Rod (Single station Rota-Rod for Rat; Med Associates Inc., Burlington, VT, USA) at 1-week intervals (over 1 week to 4 weeks), and necropsy was performed at day 10 or day 28. The range of the Rota-Rod speed was set to the accelerating mode (4.0-10 rpm over 5 min), and three trials were performed with 15-min intertrial intervals. The latency times at which each rat fell from the rod were recorded, and the average time of the three trials was evaluated.
Histopathological and Immunohistochemical Analysis
The almost total sciatic nerves were excised, fixed in 10% neutralized buffered formalin (BBC Biochemical, Mt. Vernon, WA, USA), processed using the standard method, and embedded in paraffin. Sections (4 μm in thickness) were stained with hematoxylin (Sigma-Aldrich) and eosin (BBC Biochemical) (H&E). The longitudinal sections of the samples were examined to define the correct injured sites. For single immunohistochemistry, the sciatic nerve sections were deparaffinized and incubated in 3% hydrogen peroxide (Daejung, Kyungkido, Korea) in methanol (Duksan, Kyungkido, Korea) for 30 min. After washing with PBS, the sections underwent a combination of citric acid (Sigma-Aldrich) and trisodium citrate (Sigma-Aldrich) for heat-induced antigen retrieval for immunohistochemical reactions to liberate the cross-linked epitopes. The sections were then incubated in blocking solution (10% normal goat serum; Vector Labs) for 30 min. The primary antibodies used were mouse monoclonal anti-green fluorescent protein (GFP) antibody (1:200; Santa Cruz Biotechnology), anti-cluster of differentiation 45 (CD45; 1:100; Santa Cruz Biotechnology), anti-nestin (1:500; Abcam), mouse monoclonal anti-CD3 (Abcam; 1:200), and rabbit polyclonal anti-CD31 (1:100; Santa Cruz Biotechnology). The antibody-labeled sections were then incubated with an avidin-biotin-peroxidase complex (ABC) solution using an ABC kit (Invitrogen). 3,3′-Diaminobenzidine (Zymed Laboratories, San Francisco, CA, USA), Vector NovaRed (Vector Labs), and Vector SG (Vector Labs) were used for visualization. The sections were counterstained with Mayer's hematoxylin (Sigma-Aldrich) and nuclear fast solutions.
Statistical Analysis
Each experiment was repeated a minimum of three times, and data are presented as mean ± standard deviation. Statistical differences were determined by unpaired Student's t-test using Origin software (OriginLab Corporation, Northampton, MA, USA) or InStat 3 (GraphPad Software, La Jolla, CA, USA).
Results
Effect of Flavonoid Treatment on Cell Viability and Colony Formation of ESCs
Previously, we reported that various cellular functions can be modulated by treatment with the flavonoids, 3-HF, 3,2′-DHF, 3,3′-DHF, and 3,4′-DHF (Fig. 1A), which have differential hydroxylation in the B ring of the diphenylpropane (C6C3C6) skeleton (9,55). Here we analyzed the effect of these flavonoids on the proliferation and differentiation of pluripotent stem cells. There was significantly improved cell viability when mouse ESCs were treated with 5 μM of 3,2′-DHF (Fig. 1B). Treatment with 3,2′-DHF led to an approximately 30% increase in cell growth (Fig. 1C) and proliferation rate at low cell densities (100 cells/cm2) (Fig. 1D). Furthermore, 3,2′-DHF-treated ESCs exhibited a higher colony formation efficiency than control ESCs (Fig. 1E).

Chemical structures and effects of flavonoids on the viability of embryonic stem (ES) cells (ESCs). (A) Chemical structures of 3-hydroxyflavone (3-HF), 3,2′-dihydroxyflavone (3,2′-DHF), 3,3′-DHF, and 3,4′-DHF. (B) ESCs were exposed to different concentrations (1-10 μM) of several flavonoids for 48 h. Cell viability was determined using the MTT assay. (C) Growth curves of ESCs under different culture conditions with or without 3,2′-DHF. ESCs maintained under the leukemia inhibitory factor (LIF) conditions were used as a positive control. (D) The rate of cell proliferation at a low cell density (100 cells/cm2). (E) Quantitative analysis of the colony formation efficiency of ESCs. Cells were collected every 2 days, and cell numbers were counted using a hemocytometer. Error bars represent ±SD from at least three independent experiments (*p <0.01).
Increased Expression of ESC Pluripotency Markers with 3,2′-DHF Treatment
We performed further investigation on the effect of 3,2′-DHF treatment on ESC culture. Compared with control cells, in the presence of LIF, 3,2′-DHF-treated ESCs exhibited more compact and rounder colony morphology, as well as larger and more homogeneous colony sizes, and a higher cell proliferation rate, although we could observe the beneficial effect of 3,2′-DHF treatment even in the absence of LIF (Fig. 2A, B). Next, we accessed the expression of pluripotency markers (Oct4, Sox2, Klf4, c-Myc, Cripto, and Fgf4) and differentiation markers [Afp, bone morphogenetic protein 4 (Bmp4), and β-tubulin III] (Fig. 2C, D) in the absence or presence of LIF and 3,2′-DHF. We could confirm that LIF and 3,2′-DHF treatment led to synergistic increases in the expression of pluripotency markers and a decrease in the expression of differentiation markers (Fig. 2C, D).
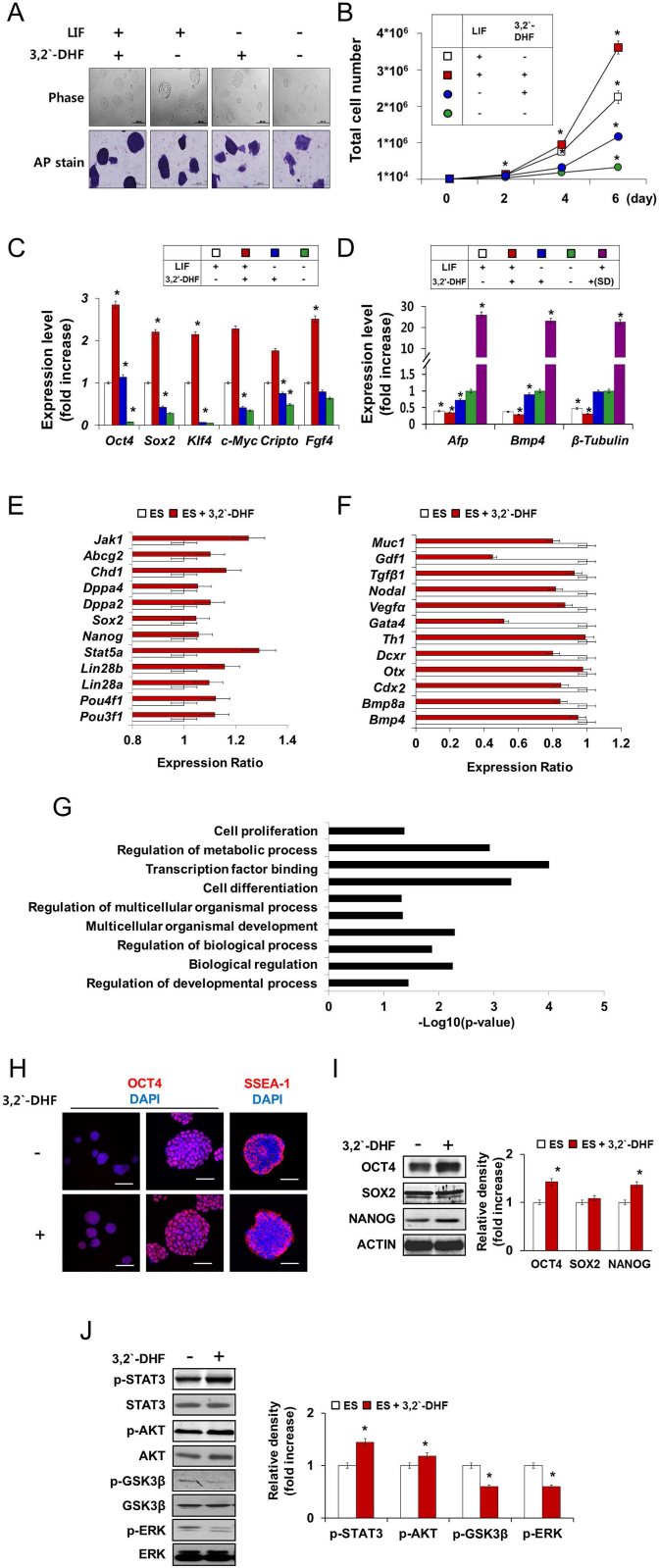
Enhancement of ESC pluripotency markers expression by 3,2′-DHF treatment. (A) ESC colonies are shown in phase contrast and stained with alkaline phosphatase (AP). Scale bars: 200 μm in Phase and 500 μm in AP stain. (B) Total cell number of ESCs under different culture conditions with or without 3,2′-DHF in the absence or presence of LIF. ESCs maintained under the LIF conditions were used as a positive control. (C, D) Quantitative real-time PCR of pluripotency marker gene expression [octamer-binding protein 4 (Oct4), sex-determining region Y box 2 (Sox2), Kruppel-like factor 4 (Klf4), c-Myc, Cripto, and fibroblast growth factor 4 (Fgf4)] and differentiation marker gene expression [α fetoprotein (Afp), endoderm; bone morphogenetic protein 4 (Bmp4), mesoderm; and β-tubulin III, ectoderm] in ESCs. For comparison, the expression level of differentiation markers during spontaneous differentiation (SD) was presented. Individual PCR reactions were normalized against internal control (Gapdh). (E, F) Based on the RNAseq analysis, transcriptional profile of the selected pluripotency (E) and differentiation (F) marker genes was presented. Expression level in 3,2′-DHF-treated ESCs relative to that in control ESCs was provided. Error bars represent ±SD of each gene. (G) Through GO analysis using Network Ontology Analysis program (http://app.aporc.org/NOA/), GO categories for genes that were differentially expressed upon 3,2′-DHF treatment were presented (lower than −1 log2 value). (H) Immunocytochemical analysis of mouse pluripotency markers OCT4 and SSEA-1. Nuclei were stained with DAPI. Scale bars for high-magnification images (middle and right panels): 500 μm, and low-magnification images (left panel): 50 for the immunocytochemical images. (I, J) Western blot analysis of the expression of pluripotency markers (OCT4, SOX2, and NANOG) and the phosphorylation of self-renewal-related kinases [signal transducer and activator of transcription 3 (p-STAT3), v-akt murine thymoma viral oncogene homolog (AKT), glycogen synthase kinase 3 β (GSK3β), and extracellular-signal regulated kinase (ERK)]. The bar graph shows quantitative results of densitometric analysis of Western blots. *p <0.01 compared with control values. Jak1, Janus kinase 1; Abcg2, ATP-binding cassette subfamily G member 2; Chd1, chromodomain-helicase-DNA-binding protein 1; Dppa2, developmental pluripotency-associated protein 2; Pou4f1, POU class 4 homeobox 1; Muc1, Mucin 1, cell surface associated; Gdf1, growth differentiation factor 1; Tgfβ, transforming growth factor β; Vegfα, vascular endothelial growth factor A; Gata4, guanine-adenine-thymine-adenine-binding protein 4; Th1, tyrosine hydroxylase 1; Dcxr, dicarbonyl/L-xylulose reductase; Otx, orthodenticle homeobox; Cdx2, caudal type homeobox 2.
Furthermore, we compared global gene expression patterns between control ESCs or 3,2′-DHF-treated ESCs by RNAseq analysis. The genome-wide expression profiles demonstrated that ESCs and 3,2′-DHF-treated ESCs have a different gene expression pattern. In 3,2′-DHF-treated ESCs, we could observe upregulated expression of the multiple pluripotency-regulating genes, including POU class 3 homeobox 1 (pou3f1; OCT6), Lin28a, Stat5a, developmental pluripotency-associated protein 2 (Dppa2), chromodomain-helicase-DNA-binding protein 1 (Chd1), ATP-binding cassette subfamily G member 2 (Abcg2), and Janus kinase 1 (Jak1) (Fig. 2E). Moreover, 3,2′-DHF-treated ES cells showed downregulated expression of differentiation-associated genes, including Bmp4, caudal type homeobox (Cdx), orthodenticle homeobox (Otx), tyrosine hydroxylase 1 (Th1), guanine-adenine-thymine-adenine-binding protein 4 (Gata4), vascular endothelial growth factor A (Vegfa), growth differentiation factor (Gdf), transforming growth factor β (Tgfβ), Nodal, and Mucin 1, cell surface associated (Muc1) (Fig. 2F). Functional annotation with gene ontology (GO) analysis showed that the differentially expressed genes upon 3,2′-DHF treatment were related to cell proliferation, transcription factor binding, cell differentiation, or developmental process (Fig. 2G).
Immunocytochemistry and Western blotting for pluripotency marker expression also indicated that 3,2′-DHF treatment led to an apparent improvement in the ESCs morphology and pluripotency marker expression (Fig. 2H, I). Treatment of 3,2′-DHF also resulted in activation (STAT3 and AKT) or suppression (GSK3β and ERK) of self-renewal-related kinases (Fig. 2J), supporting the beneficial effect of 3,2′-DHF treatment on ESC culture.
Enhancement of Proliferation and Pluripotency Marker Expression of iPSCs by 3,2′-DHF Treatment
As iPSCs are known to have characteristics that are very similar to ESCs and offer several advantages over use of ESCs, including being free from ethical issues and potential therapeutic applicability, we also investigated the effects of 3,2′-DHF in iPSCs. Initially, we established iPSC lines from MEFs using a lentiviral Oct4, Sox2, Klf4, and c-Myc expression system (data not shown). Next, we investigated whether 3,2′-DHF affects the maintenance of iPSCs. Treatment with 3,2′-DHF led to enhanced cell proliferation and colony formation (Fig. 3A, B) and also resulted in higher expression of pluripotency markers, such as Oct4, Sox2, Nanog, Fgf4, and Cripto, compared to control cells (Fig. 3C, D). Moreover, 3,2′-DHF-treated iPSCs showed upregulated phosphorylation of STAT3 and AKT and downregulated phosphorylation of GSK3β and ERK (Fig. 3E), confirming that 3,2′-DHF can be used for the maintenance of high-quality pluripotent ESCs and iPSCs.

Effect of 3,2′-DHF treatment on cell viability and pluripotency markers expression of induced pluripotent stem (iPS) cells (iPSCs). (A) iPSCs were treated with the indicated amounts of flavonoids for 48 h in three independent experiments. Cell proliferation was measured using the MTT assay. Error bars represent ±SD from at least three independent experiments (*p <0.01). (B) Quantitative analysis of colony formation in iPSCs. (C) Expression levels of pluripotency markers (Oct4, Nanog, Sox2, Fgf4, and Cripto) in iPSCs were analyzed using quantitative real-time PCR. Individual PCR reactions were normalized against internal controls (Gapdh). *p <0.01 compared to the control values. (D, E) Western blot analysis of the expression of pluripotency markers (OCT4, SOX2, and NANOG) and the phosphorylation of STAT3, AKT, GSK3β, and ERK in iPSCs.
3,2′ -DHF-Pretreated iPSCs Exhibit Efficient Ectoderm Lineage Differentiation
We expected that high-quality 3,2′-DHF iPSCs may also show more efficient differentiation potential. We maintained the culture of iPSCs in 3,2′ -DHF-containing media and removed the 3,2′-DHF from the cells by washing with PBS. After trypsinization, centrifugation, and washing with media, the 3,2′-DHF iPSCs were prepared for spontaneous differentiation, which was conducted in 3,2′-DHF-free media (Fig. 4A). We detected enhanced expression of markers for all three germ layers, endodermal (Afp and Cdx2), mesodermal (Brachyury and Bmp4), and ectodermal (nestin and β-tubulin III), in the 3,2′-DHF iPSCs compared to control iPSCs (Fig. 4A, B). In particular, we found a significant improvement in ectodermal differentiation (Fig. 4B). We also examined the time-dependent expression of Bmp4, β-tubulin III, and Afp during spontaneous differentiation and detected an increase in ectodermal marker expression (Fig. 4C). The 3,2′-DHF iPSCs also showed enhanced expression of other neuroectodermal markers [orthodenticle homeobox 1 (Otx1), Sox1, engrailed-1 (En1), wingless-type mouse mammary tumor virus (MMTV) integration site family member 1 (Wnt1), LIM homeobox transcription factor 1 (Lmx1b)] upon spontaneous differentiation (Fig. 4D).

Characterization of the ectodermal lineage of differentiated 3,2′-DHF iPSCs. (A) Scheme for spontaneous differentiation. iPSCs were treated with 3,2′-DHF during maintenance and washed before differentiation. Spontaneous differentiation of the pretreated iPSCs was performed in the absence of 3,2′-DHF. Quantitative real-time PCR analysis was conducted to access the expression levels of germ layer differentiation markers [endoderm, Afp, Cdx2; mesoderm, Brachyury (Bra), Bmp4; ectoderm, Nestin, β-tubulin III]. Total RNA was extracted from the spontaneously differentiated 3,2′-DHF iPSCs. (B) Immunocytochemical analysis of the expression of the three germ layer markers (endoderm, AFP; mesoderm, BRACHYURY; ectoderm, NESTIN) in 3,2′-DHF iPSCs. Nuclei were stained with DAPI. Scale bars: 100 μm in AFP and BRACHYURY and 50 μm in NESTIN. (C) Quantitative real-time PCR analysis of germ layer differentiation markers (endoderm, Afp, Cdx2; mesoderm, Brachyury, Bmp4; ectoderm, Nestin, β-tubulin III) at time points during differentiation. (D) Expression level of neuroectodermal markers [orthodenticle homeobox 1 (Otx1), Sox1, engrailed-1 (En1), wingless-type mouse mammary tumor virus (MMTV) integration site family member 1 (Wnt1), LIM homeobox transcription factor 1 (Lmx1b)] in treated and untreated iPSCs during spontaneous differentiation. (E, F) Expression levels of pluripotency marker (Oct4) and differentiation marker (β-tubulin III) were monitored before and during spontaneous differentiation using quantitative real-time PCR. (G) Scheme for mature neuronal differentiation. iPSCs were pretreated with 3,2′-DHF during maintenance and washed before differentiation. Mature neuronal differentiation of the pretreated iPSCs was performed in the absence of 3,2′-DHF. Expression levels of mature neuronal markers [β-tubulin III, tyrosine hydroxylase (Th), double cortin (Dcx), choline acetyltransferase (Chat), and dopa decarboxylase (Ddc)] were analyzed. All individual real-time PCR reactions were normalized against internal controls (Gapdh). *p <0.01 compared with the control values. (H) In vitro mature neuronal differentiation potential of 3,2′-DHF iPSCs was analyzed using neuron specific markers [β-TUBULIN III, microtubule-associated protein 2 (MAP2), and CHAT]. Nuclei were stained with DAPI. Scale bars: 100 μm in MAP2 and 50 μm in CHAT.
As we monitored the expression level of Oct4 and β-tubulin III before and during spontaneous differentiation of iPSCs, although 3,2′-DHF-pretreated iPSCs maintained higher Oct4 expression and lower β-tubulin III expression in the presence of LIF, upon differentiation condition in the absence of LIF and 3,2′-DHF, the 3,2′-DHF iPSCs showed a rapid decrease in Oct4 expression and enhanced increase in β-tubulin III expression compared with control iPSCs (Fig. 4E, F), confirming that the high-quality 3,2′-DHF iPSCs have more efficient differentiation potential.
Next, we performed mature neuron differentiation of the 3,2′-DHF iPSCs in 3,2′-DHF-free neural induction media (Fig. 4G). As expected, we detected significantly enhanced expression of doublecortin (Dcx, a migrating neuronal marker), β-tubulin III (a neuronal extension marker), Th (a neurotransmitter-related marker), Chat (a neurotransmitter-related marker), dopa decarboxylase (aromatic l-amino acid decarboxylase) (Ddc, a neurotransmitter-related marker), and MAP2 (a mature neuronal marker) in the 3,2′-DHF iPSCs (Fig. 4G, H), suggesting that high-quality 3,2′-DHF iPSCs also have a highly enhanced differentiation potential, especially for the neuroectodermal lineage.
Transplantation of the 3,2′-DHF iPSCs Enhances Motor Function in a Sciatic Nerve Injury Model
High-quality 3,2′-DHF iPSCs are expected to have improved in vivo therapeutic effects in animal models of sciatic nerve injury. Following injury by sciatic nerve crushing (Fig. 5A), we injected 3,2′ -DHF iPSCs into the injury lesion and performed Rota-Rod testing for the clinical assessment of motor function. At days 7 and 10, all injury groups exhibited shorter latency times compared with the sham group, with no discernible difference between the 3,2′-DHF iPSC and control iPSC groups. The latency time of the 3,2′-DHF iPSC group was significantly longer than that of the control groups at day 14 and recovered gradually during all time points of the experiment (Fig. 5B). In terms of clinical convalescence, the 3,2′-DHF iPSC group showed a more significant improvement in recovery (p <0.05) at day 28 compared with that in the other groups (Fig. 5B).

Sciatic nerve injury model and examination of motor function. (A) Diagram of the surgical procedures for the sciatic nerve injury model. (a) Bilateral sciatic nerves were exposed at the lateral surface of the femur and crushed using hemostatic forceps for 10 min. (b) After sciatic nerve crush injury, the lesions appeared as thin films at the gross level (white box). (c) A controlled number of cells were injected through the center of the lesions via a Hamilton syringe. The sciatic nerves were then coated with diluted Matrigel. (B) All sciatic nerve injury groups were tested for motor function using a Rota-Rod (treadmill) test. (C) Necropsy of the sciatic nerve after 10 and 28 days.
We collected and examined excised nerve tissues at day 10 to evaluate the early to intermediate phase of peripheral nerve regeneration, as well as on day 28 for evaluation of the intermediate to late phase. In contrast to the sham group, macroscopic examination of the crush lesion in all the injured groups revealed hypertrophy with pigmentation at day 10 (Fig. 5C). The magnitude of the hypertrophic lesion was much more significant in the iPSC and 3,2′-DHF iPSC groups than in the control injury groups. At day 28, the diameter of the crush lesion became normal in all groups, and no significant pathological indicators were detected.
Transplantation of 3,2′-DHF iPSCs Slightly Improved Axonal Regeneration via BMSC Recruitment in the Early Phase and Improved Angiogenesis in the Late Phase
In all groups, the central region of the nerve exhibited similar histopathological features, such as edema and hydropic changes resulting in neuronal hypertrophy at day 10 (Fig. 6A). Marked degrees of hypertrophic and edematous changes were observed in the Matrigel, iPSC, and 3,2′ -DHF iPSC groups compared with the injury-alone group. Wallerian degeneration, a salient feature of peripheral nerve injury, was detected in the distal region of the injury sites with similar degrees of injury observed among all the groups. Up to day 28, hydropic changes remained in the central region of the injured nerve in all groups, while severity was much reduced in the iPSC and 3,2′-DHF iPSC groups compared to that in the injury-alone group. Histopathological indicators, such as the infiltration of inflammatory cells, were much more obvious in the perineurium compared to the control group. At day 10, neovascularization with severe infiltration of basophilic cells was observed in the perineural region, especially in the iPSC and 3,2′-DHF iPSC groups. Moreover, in the Matrigel-coated groups (Matrigel, iPSC, and 3,2′-DHF iPSCs), basophilic cells infiltrated near the remnants of the Matrigel. All groups exhibited hypertrophy in perineurium at day 28, with marked decreases in basophilic cell numbers. In addition, various sizes of neovasculature were found in the iPSC and 3,2′-DHF iPSC groups. We also found that cells infiltrating the perineurium were positive for CD45 (bone marrow cell marker) and GFP (transplanted cells) in the iPSC and 3,2-DHF iPSC groups (Fig. 6B). The number of immune-positive cells was far greater in the iPSC group than in the 3,2′-DHF iPSC group, indicating the localization of cellular aggregation at the perineurium. Instead of a small number of CD45-positive cells located in the perineurium, the majority of the infiltrating cells were CD3-positive. These results indicate that the implantation of iPSC and 3,2′-DHF iPSCs into the area of nerve injury affects the recruitment of CD3-positive T-lymphocytes in the early to intermediate phase of peripheral nerve regeneration.

Histological and immunohistochemistry analyses after transplantation. (A) H&E staining of sciatic nerves (nerve central and perineurium region) at 10 and 28 days after injury and transplantation. (B-D) Immunohistochemical analyses of transplanted and infiltrating cells. (B) To identify infiltrating cells, immunohistochemistry was performed using CD45 (bone marrow cell marker) and CD3 (T-cell marker). The relationship between recruitment of bone marrow cell (CD45) and transplanted cells (GFP) were analyzed by double immunostaining in perineurium region at day 28. (C) The expression of the neural marker NESTIN in the nerve center and perineurium regions. (D) Relationship between neovessel formation (CD31) and transplanted cell (GFP) was assessed by double immunohistochemical analysis. The expression of CD31 and Nestin expression in the perineurium region at day 28. For visualization, Nova red (red color) was used for CD45 and CD3, and diaminobenzidine (DAB; brown color) for Nestin. Nova red and Vector S/G (bluish gray color) was used for double immunohistochemistry; CD45/GFP, CD31/GFP, and CD31/Nestin. Scale bars: 100 μm.
The central regions of the injured nerves were immunoreactive for nestin in all groups. However, there were no significant differences among experimental groups at days 10 and 28 (Fig. 6C). The number of nestin-positive neovessels was greater in the perineurim of the iPSCs and 3,2′-DHF iPSC groups at day 10 compared to the injury-alone and Matrigel groups. The 3,2′-DHF iPSC group showed a remarkable increase in nestin expression compared with that in the iPSC group. In contrast to the injured, Matrigel, and 3,2′-DHF iPSC groups, the iPSC group exhibited nestin-positive round cells located in the surrounding adipose tissues near the perineurium at day 28. In the 3,2′ -DHF iPSC group, nestin-positive round cells were not observed, but nestin-positive neovasculature was observed. We then analyzed the effect of implanted iPSCs on angiogenesis using double immunohistochemistry for CD31 (angiogenesis marker), nestin, and/or GFP (transplanted cells) at day 28. Only a small number of the CD31-positive neovessels were found in the injured and Matrigel groups; however, the 3,2′-DHF iPSC group showed elevated numbers of various sized neovessels in the perineurium (Fig. 6D). In the iPSC group, GFP-positive cells aggregated near CD31-positive neovessels in the perineurium. Furthermore, nestin-positive cells were colocalized with GFP-positive cells near CD31-positive neovessels. These results indicate that, unlike the 3,2′-DHF iPSC group, the iPSC group still exhibits nestin- and GFP-positive cells in the perineurium region 28 days after nerve injury, especially near the surrounding tissue with CD31-positive neovessels.
3,2′-DHF iPSCs Enhance Neuroprotection
According to previous studies, tumor necrosis factor (TNF-α) can effect neuroprotection by reducing the levels of nitric oxide and free radicals (19), as well as by enhancing the release of neurotrophic factors. Moreover, the angiogenic protein VEGF (10) has also been reported to have a neuroprotective effect and promotes neurogenesis and angiogenesis in a variety of tissues (58). Our results showed that Tnf-α expression was increased in all injury groups at day 10, while its expression was significantly reduced at day 28 (Fig. 7A). Interestingly, although the amount of reduction in Tnf-α expression was minimal in the Matrigel and other control groups, a prominent decrease was observed in the 3,2′-DHF iPSC group. The expression levels of Vegf in the iPSC and 3,2′-DHF iPSC groups were similar to those observed in the control groups (no injury, no treatment) and were different from those of the injury groups. Moreover, expression levels of the Schwann cell marker S100b (63) were significantly different among all groups; however, weak expression was observed at day 10 and was slightly increased at day 28, indicating that nerve regeneration was in progress (Fig. 7B). Furthermore, the expression level of the immature axonal regeneration marker growth-associated protein-43 (Gap43) was significantly decreased in the 3,2′-DHF iPSC group compared with the injury-alone group. In terms of the intermediate to late phase of nerve regeneration, all groups showed lower expression levels of Gap43 at day 28 than at day 10 (the early phase). The expression levels of the neurotrophic factors brain-derived neurotrophic factor (Bdnf) and glial cell line-derived neurotrophic factor (Gdnf) (73), which are related to early to intermediate phase of nerve regeneration, were upregulated at day 10 in the injured and 3,2′-DHF iPSC groups (Fig. 7C). The iPSC group showed significantly decreased expression of Bdnf at day 10. In contrast, neurotrophpin 3 (nt-3) (71) expression was similar to the control and sham groups in the injury group at day 10, while the Matrigel, iPSC, and 3,2′-DHF iPSC groups showed significantly decreased expression compared to the injury group at day 10. Meanwhile, the degree of neuronal regeneration was analyzed by assessing the expression of the immature and mature neuronal markers neural cell adhesion molecule 1 (ncam) (62) and neurofilament light polypeptide (ne-fl), respectively. The expression of Ncam was not significantly altered in any of the groups. However, Ne-fl expression was increased at day 10 and decreased at day 28 in all groups except the 3,2′-DHF iPSC group (Fig. 7D). The expression of Ne-fl in the 3,2′-DHF iPSC group was consistently elevated at days 10 and 28.

Expression analyses of neuroprotection-, inflammation-, or neuronal regeneration-related genes after transplantation. Quantitative real-time PCR analysis was performed to access the expression levels of the mouse- or rat-originated genes using the primers listed in Table 3. We designed rat-specific or mouse-specific primers separately for tumor necrosis factor-α (Tnf-α), growth-associated protein 43 (Gap43), brain-derived neurotrophic factor (Bdnf), and neurofilament light polypeptide (ne-fl), which showed lower than 95% gene homology between mouse and rat ones. However, in the case of Vegf, S100b, glial-derived neurotrophic factor (Gdnf), neurotrophin 3 (nt-3), and neural cell adhesion molecule (ncam), the homology between two species was too high to design primers separately, so we used the primers matching with both mouse and rat ones. Expression levels of the neuroprotection- or neuroinflammation-related markers, Tnf-α and Vegf (A), the Schwann cell and axonal regeneration markers, S100b and Gap43 (B), the neuronal regeneration-associated neurotrophic factor markers, Bdnf, Gdnf, and Nt-3 (C), and the immature/fully mature neuronal markers, Ncam and Ne-fl (D). Error bars represent ±SD from at least three independent experiments. +, compared with Control; #, compared with Sham group; *, compared with Injury group. *p <0.05, **p <0.01.
Discussion
Our previous report showed that ESCs cultured on a nanopatterned polydimethylsiloxane (PDMS) substrate showed enhanced differentiation into three germ layers (27) during culture maintenance, suggesting that maintenance of a high-quality undifferentiated state of ESCs may be important for efficient differentiation. Other groups have also reported that nanoscale substrates can produce an environment that enables efficient control of ESC self-renewal and fate (6). Moreover, small molecules such as trimipramine and ethopropazine have been shown to promote the maintenance of the undifferentiated state of ESCs and iPSCs (33) and to induce specific lineage differentiation (7). Here we have observed that the flavonoid 3,2′-DHF can be used for the maintenance of high-quality pluripotent ESCs and iPSCs by promoting the expression of pluripotency markers, such as Oct4, Sox2, Nanog, Fgf4, and Cripto, and through the significant activation (STAT3 and AKT) or suppression (GSK3β and ERK) of self-renewal-related kinase (Figs. 1-3).
Flavonoids are natural products that are presently garnering a great deal of attention, as they can be used in the treatment of many important common diseases (24). They are safe and nonimmunogenic natural substances that exhibit a variety of biological effects, including anti-inflamniatory and antiviral activities (28,32,34,59). Several studies have reported that certain flavonoids can support embryonic development and exert antioxidant, antiapoptotic, or antitumor activity (24,40). Moreover, several flavonoid compounds have been observed to have an effect on stem cell differentiation. For example, the flavonoid wogonin induced the differentiation of neural progenitor cells both in vitro and in vivo (44), whereas neuronal differentiation was induced by baicalin in human cord blood mesenchymal stem cells (22). Recently, quercetin was reported to be used in the selective removal of undifferentiated human pluripotent stem cells after transplantation (41).
We expected that high-quality ESCs or iPSCs, well maintained in the undifferentiated state, might also exhibit efficient differentiation potential. We originally confirmed that high-quality 3,2′-DHF iPSCs exhibit a highly enhanced differentiation potential, particularly for the neuroectodermal lineage (Fig. 4). Moreover, we found that transplantation of 3,2′-DHF iPSCs could promote functional recovery and axonal regeneration in sciatic nerve injury, which is likely to be mediated through their neuroprotective property (Figs. 5-7).
Peripheral nerve injury is not only a serious clinical problem for individuals but also a great socioeconomical burden for society (8,26,42,54). Damage to the peripheral nervous system is primarily caused by traumatic injuries such as accidents with glass, traffic accidents, and iatrogenic surgery (5,11), as well as neurodegenerative diseases such as multiple sclerosis (56). Nerve injury results in patients suffering from pain, loss of sensory feedback from peripheral tissues, and the inability to control distal muscles and joints.
The peripheral nervous system is capable of undergoing unique processes of degeneration and regeneration, including Wallerian degeneration, which takes place in regions distal to injury sites, and chromatolysis, which occurs retrograde to injury sites and affects cell bodies in the dorsal root ganglia (18,42). Despite the spontaneous regeneration occurring in the peripheral nervous system, functional recovery does not progress to preinjury states and occurs over a very long time frame, often requiring years of recovery following injury (20).
Numerous studies were performed to enhance the speed of axonal regeneration and to reduce neural degeneration (52). Most studies can be largely classified into two categories: those focusing on the replacement or repair of the neuronal components (neurorepair/neuroregeneration) and those attempting to induce microenvironments favorable for regeneration (neuroprotection) (1,2,16). For neurorepair and neuroregeneration, many studies were performed that utilized techniques such as microsurgery, nerve grafts, tubulization techniques, and stem cell therapy (15,45,64,77). Several stem cell sources, including bone marrow stem cells (43), neural stem cells (3), and ESCs (15), have been reported to show beneficial effects on peripheral nerve regeneration. With stem cell therapy, although a variety of stem cells have been used, their usage is often limited by factors such as ethical issues associated with ESCs, limited amounts of cells due to their source (umbilical cord stem cells), and the need for invasive and painful surgery to obtain bone marrow stem cells (17,69,72). From this viewpoint, the application of iPSCs has the advantages of cells being easily obtained in plentiful amounts, freedom from ethical issues, and possible therapeutic potential.
In advancing neurorepair/neuroregeneration strategies, there are many natural materials that could be used to induce favorable microenvironments in peripheral nerve injury, such as platelet-rich plasma, ascorbic acid (vitamin C), α-tocopherol (vitamin E), (-)-epigallo-catechin gallate (green tea polyphenol), and mushroom extracts (51,67,68,76). For example, Yu et al. suggested that platelet-rich plasma could promote nerve regeneration via growth factors and proteins derived from a granules (76). Indeed, it is obvious that a single therapeutic strategy for peripheral nerve injury will be limited in that it cannot address the entire nerve regeneration processes.
In our study, we evaluated the differences between 3,2′-DHF iPSCs and nontreated iPSCs in the rat sciatic nerve crush injury model. We found that high-quality 3,2′-DHF iPSCs showed significantly improved in vivo therapeutic effects in the animal model of sciatic nerve injury. Although transplantation of 3,2′-DHF iPSCs resulted in an improved axonal regeneration rate, the effect was not thought to be due to neural replacement, but rather to neuroprotection resulting from angiogenesis and the recruitment of myeloid cells, as shown in a previous report (48).
The enhanced expression of CD45 in the 3,2′-DHF iPSC group indicated that the transplantation of 3,2′-DHF iPSCs attracts CD3 T-lymphocytes to the injury site to facilitate Wallerian degeneration. According to previous studies, lymphocyte recruitment, especially of T-lymphocytes, affects the clinical course of axonal regeneration, with the recruitment largely originating from hematopoietic lineages of bone marrow stem cells (4,21).
Previously, nestin, formerly known as a neural stem/progenitor cell marker, was suggested to be a possible vascular marker for the early to intermediate phases of angiogenesis (25,49,70). In our results, expression of nestin was apparent in the 3,2′-DHF iPSC group, indicating that 3,2′-DHF iPSCs could modulate angiogenesis during nerve regeneration.
TNF-α is thought to have a dual role in nerve degeneration (neuroinflammation) and regeneration (neuroprotection). In our study, Tnf-α expression levels in the 3,2′-DHF iPSC group were higher in the early to intermediate phases of regeneration, while decreasing significantly in the late phases, which strongly implies that transplantation of 3,2′-DHF iPSCs may induce neuroinflammation at the injury site until the intermediate phase of nerve regeneration and facilitates Wallerian degeneration via the attraction of myeloid cells.
Taken together, the novel research presented herein suggests that 3,2′-DHF can be used to enhance the efficiency of pluripotent stem cell maintenance as well as for further application in stem cell research and therapy.
Footnotes
Acknowledgment
This work was supported by grants from the National Research Foundation (NRF) funded by the Korean government (MEST) (No. 2010-0020348 and No. 2013M3A9D3045880 to S.-G. Cho) and by the Bio-Industry Technology Development Program, Ministry of Agriculture, Food and Rural Affairs (No. 312062-05 to S.-G. Cho and S. H. Do) and by the Public Welfare and Safety research program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2012-0008610 to S. H. Do). The authors declare no conflicts of interest.