
Abstract
The objective of this study was to assess the effect of high-dose atorvastatin treatment on endothelial progenitor cell (EPC) recruitment and angiographic and clinical outcome in coronary artery disease (CAD) patients treated with the Genous™ EPC-capturing stent. The HEALING IIB study was a multicenter, open-label, prospective trial that enrolled 100 patients. Patients were started on 80 mg atorvastatin qd, at least 2 weeks before the index procedure and continued for at least 4 weeks after the index procedure. Eighty-seven patients were included in this analysis. EPC levels significantly increased as early as 2 weeks after the start of atorvastatin. Remarkably, among this group, 31 patients proved to be nonresponders to atorvastatin treatment (i.e., no increase in EPC levels), while 56 patients were responders (i.e., rise in EPC count between week −2 and 0). Compared to responders, nonresponders had a significantly higher baseline EPC count (76 ± 10 vs. 41 ± 5, p < 0.01) with a lower late luminal loss (LLL) at 6- and 18-month follow-up (FU) (0.61 ± 0.07 vs. 0.88 ± 0.08, p < 0.05, and 0.50 ± 0.08 vs. 0.82 ± 0.08, p < 0.01 respectively). Furthermore, baseline EPC count was inversely correlated with LLL at 6-month FU (R = −0.42, p < 0.001). Patients with a higher EPC count at baseline showed no increase in EPC recruitment in response to statin treatment but had favorable LLL at 6- and 18-month FU, whereas patients with lower EPC count were responsive to statin therapy, but EPCs might be less functional as they had higher LLL at 6- and 18-month FU. These data imply that although statin treatment can enhance EPC titer in patients with low baseline levels, there is no indication for a possible beneficial clinical effect with EPC capture stents.
Introduction
In recent years, there has been a tremendous development in the treatment of coronary artery disease (CAD), from balloon angioplasty to bare metal stent placement to stents with active coatings. The introduction of bare metal improved the outcome of percutaneous coronary intervention (PCI), but in the long term, restenosis of the stent can occur due to vascular smooth muscle cell (VSMC) proliferation. Presently, more advanced stents bearing cytostatic compounds have been developed. These are known as drug-eluting stents (DESs) and have largely overcome the problem of restenosis, as they inhibit the proliferation of VSMCs. However, DESs have been associated with stent thrombosis, as they also inhibit the regrowth of the endothelium. The endothelium plays a pivotal role in maintaining vascular integrity and function, while injury or dysfunction can trigger the onset of cardiovascular disease. It is therefore important to maintain vascular integrity by rapid restoration of any damage to the endothelium. Vascular repair can be ensured by proliferation and migration of adjacent mature endothelial cells or incorporation of circulating endothelial progenitor cells (EPCs). EPCs were first described by Asahara et al. in 1997 and were defined as cluster of differentiation 34-positive (CD34+) or kinase insert domain receptorpositive (KDR+) cells with the ability to differentiate into endothelial cells in vitro (2). Furthermore, they showed in vivo that those cells contributed to neoangiogenesis in a murine and a rabbit hindlimb ischemia model. Since this initial study, many researchers have reported a beneficial effect of EPCs on neovascularization, reendothelialization, and reduction of neointima formation in animal studies (12–14,22). Observational clinical studies showed that circulating EPC levels were correlated with cardiovascular risk factors and outcome (5,20,23).
EPCs can enhance the arterial repair, so an anti-human antibody-coated stent (Genous” Bioengineered R stent, OrbusNeich, Fort Lauderdale, FL, USA) was designed to bind circulating CD34+ hematopoietic cells and facilitate accelerated vascular repair and decrease neointima formation after stent placement (1,16). The Healthy Endothelial Accelerated Lining Inhibits Neointimal Growth (HEALING) II study showed that use of the Genous stent for the treatment of de novo coronary artery disease is safe and feasible with an in-stent late luminal loss (LLL) after 6 months of 0.78 ± 0.39 mm (6,7). Remarkably, patients with low circulating EPC levels responded relatively poorly to the Genous stent with worse clinical and angiographic outcome. We therefore hypothesized that an increase in the number of circulating EPCs would improve the response to the Genous stent and subsequently improve clinical and angiographic outcome. Since statins can increase circulating EPCs (17,21,23), we designed the HEALING IIB study in which we evaluated the efficacy of the Genous stent in conjunction with a high-dose statin (atorvastatin 80 mg qd) therapy. We previously reported the clinical and angiographic outcome of the HEALING IIB study (4). Here we present the in-depth analysis of the effect of atorvastatin treatment on circulating EPC levels and correlate these findings with patient clinical outcome of the HEALING IIB trial.
Materials and Methods
Study Population and Protocol
The design and clinical results of the HEALING IIB trial were recently published (4). Briefly, 100 patients, from 13 sites in seven European countries, were enrolled with a diagnosis of de novo stable or unstable angina or silent ischemia with a maximum of two de novo lesions in two independent native coronary arteries eligible for coronary stenting. Patients were started on 80 mg atorvastatin (Pfizer, Capelle aan de Ijssel, the Netherlands) qd at 2 weeks before index procedure and continued for at least 4 weeks after index procedure. Patients already on statin therapy were switched to 80 mg atorvastatin qd aspirin (Meda Pharma BV, Amstelveen, the Netherlands) treatment, which was initiated 12 h before the procedure (75 mg qd), whereas a loading dose of 300 mg of clopidogrel (Sanofi Aventis, Gouda, The Netherlands) was administered prior to the procedure, proceeded by 75 mg qd for the period of 4 weeks.
Anginal status (according to the Canadian Cardiovascular Society Classification of Angina and the Braunwald Classification for Unstable Angina) and the documentation of Major Adverse Cardiac Events (MACE) were assessed at 1-, 6-, 12-, 18-, and 24-month follow-up (FU). Circulating CD34+ EPC titers were quantified by flow cytometric analysis at screening (baseline), 2 weeks after initiation of high-dose atorvastatin, and at 30 days FU of the index procedure.
Quantitative angiographic analysis was performed at 6- and 18-month FU. Coronary angiograms were obtained in perpendicular views following an intracoronary injection of nitrates. Offline quantitative analyses of preprocedural, postprocedural, 6- and 18-month FU angiographic data were performed at an independent imaging core laboratory (Cardialysis, Rotterdam, the Netherlands).
The trial was reviewed and approved by the local Medical Ethics Review Committees, and written informed consent was obtained from all patients.
Genous™ R Stent: Endothelial Progenitor Cell Capture Technology
The Genous™ Bioengineered R stent is based on a 316-L stainless steel stent platform with study devices available in diameters of 2.5, 3.0, and 3.5 mm and lengths of 9, 18, 23, and 33 mm (OrbusNeich Medical, Fort Lauderdale, FL, USA) with a polysaccharide matrix coating covalently coupled with murine anti-human CD34 antibodies, which specifically target the CD34+ circulating endothelial cell progenitor population.
Blood Sample Preparation
Venous blood was drawn under sterile conditions and collected in ethylenediaminetetraacetic acid (EDTA) collection tubes (BD Biosciences, Breda, The Netherlands). Whole blood was used to measure circulating EPC levels. Low-density lipoprotein (LDL), high-density lipoprotein (HDL), cholesterol, and triglyceride levels were measured by the clinical chemistry laboratorium of the hospital.
Quantification of Circulating Endothelial Progenitor Cells
EPCs were measured using the Stem Cell Kit (Beckman Coulter Comp, Villepente, Roissy CDG, France) and a modified International Society of Hematotherapy and Graft Engineering (ISHAGE) protocol. Whole blood was incubated with complete antibody mix, that is, allophycocyanin (APC)-labeled anti-hCD45, fluorescein isothiocyanate (FITC)-labeled anti-hCD34 (BD Pharmingen; Breda, the Netherlands), and phycoerythrin (PE)-labeled anti-hKDR (R&D systems, Abingdon, UK). As a negative control, we used PE labeled antihuman immunoglobulin G (hIgG; BD Pharmingen). Blood was incubated for 20 min at room temperature. Dead cells were excluded using 7-aminoactinomycin D (7-AAD; BD Biosciences) viability dye. Samples were lysed for 10 min at room temperature using 2 ml of lysis buffer (BD Biosciences). Hundred microliter counting beads (from Stem Cell Kit) were added to correlate cell amounts to blood volume. Duplicate samples and one isotypic control were analyzed on an automated flow cytometer (FACSCanto®, Becton & Dickinson, Breda, the Netherlands). Final data analysis was done with Flowjo software (Tree Star, Inc., Ashland, OR, USA) (see Fig. 1). We first excluded dead cells by gating on a 7-AAD versus side scatter (SSC) dot plot. The remaining viable cells were displayed on a CD45 versus SSC dot plot. We gated the CD45+ and dim population to select lymphocytes and excluded red blood cells and debris. On the third plot, showing the CD34 versus SSC dot plot, we gated the bright CD34 cell population and further excluded irrelevant cells by a lymphogate. EPCs were characterized as 7-AAD-, CD45dim/+, CD34+, FSC/SSClow, and KDR+ [vascular endothelial growth factor receptor 2 (VEGFR2)]. The EPC gate is set according to the IgG1-PE isotypic control.

Measurement of EPCs in whole blood using a modified International Society of Hematotherapy and Graft Engineering (ISHAGE) protocol. We used a novel method to measure endothelial progenitor cells (EPCs) in whole blood using a modified ISHAGE protocol. Dead cells are excluded with a 7-aminoactinomycin D (7-AAD) viability dye. Lymphocytes are selected as cluster of differentiation 45 (CD45) dim and positive cells. The bright CD34 cell population is selected, and a lymphogate excludes irrelevant cells. EPCs are characterized as 7-AAD-, CD45dim/+, CD34+, forward scatter/side scatterlow (FSC/SSClow), and kinase insert domain receptor positive (KDR+). The gate is always set according to the immunoglobulin G1-phycoerythrin (IgG1-PE) isotypic control. APC, allophycocyanin; FITC, fluorescein isothiocyanate.
Statistical Analysis
Continuous data are presented as mean ± standard deviation (SD), and categorical variables are expressed as numbers and percentages. The Kolmogorov–Smirnov test was used to analyze whether continuous data were normally distributed. Differences in baseline characteristics between responders and nonresponders were analyzed by Student's t tests, the Mann–Whitney U test, chi-square tests, and Fisher's exact test as appropriate. EPC counts at different time points were compared using repeated measurements ANOVA for normally distributed variables with Newman–Keuls as post hoc test and Friedman test for nonnormally distributed variables with Dunn's multiple comparison test as post hoc test. The Spearman correlation coefficient was used to correlate circulating EPC counts with clinical outcome.
We applied a univariable logistic regression (LR) analysis to detect clinical characteristics that influenced the risk of being a nonresponder to atorvastatin treatment. To determine the independent risk factor of being a nonresponder, we applied a multivariable LR analysis. Nonresponder was considered the dependent variable, and the following independent variables: baseline EPC count, age, sex, hypertension, diabetes mellitus, hypercholesterolemia, previous statin use, BMI, and smoking history were considered potential determinants. The relatively small sample size meant that we performed multivariable LR with backward deletion, and all variables with a value of p < 0.15 were maintained in the model.
We report crude and adjusted odds ratios (aORs) along with their 95% confidence intervals (CIs). For all tests, a two-sided value of p < 0.05 was considered significant. All statistical analyses were performed using SPSS 17.0 for Windows (SPSS, Inc., Chicago, IL, USA) and GraphPad Prism 4 (San Diego, CA, USA).
Results
Baseline Characteristics
We included 100 patients in the HEALING IIB (HIIB) study. Four patients were excluded from the postprocedural analysis; two patients did not meet the inclusion and exclusion criteria, one patient was not on atorvastatin during the 2 weeks preprocedure as required per protocol, and one patient had a postprocedural diameter stenosis of 32% despite postdilation. Another nine patients were excluded from this analysis because of missing values in their EPC count on baseline or 2 weeks FU.
Among the final 87 patients, 31 patients were nonresponders to atorvastatin treatment (i.e., no rise in EPC count between week −2 and week 0), and 56 patients were responders (i.e., rise in EPC count between week −2 and 0). Baseline characteristics for the nonresponder and responder groups are summarized in Table 1. Baseline characteristics between nonresponders and responders were relatively similar except for baseline EPC count, sex, and smoking status. There was no difference in medication use between the two groups (data not shown). Seventy-one percent of the patients in the responder group and 74% of the patients in the nonresponder group were already on statin therapy before the start of the study (p = 0.78) and were switched to 80 mg of atorvastatin treatment. At the index procedure, 100% of the patients were initiated on high-dose atorvastatin therapy (80 mg qd) for at least 2 weeks according to the protocol outline. At 1 month FU, 91% of the patients were still being treated with atorvastatin 80 mg qd.
Patient Demographics and Clinical Characteristics

Numbers are % (counts/available field sample size) or mean ± 1 SD. MI, myocardial infarction; CABG, coronary artery bypass graft; PTCA, percutaneous transluminal coronary angioplasty.
Effect of Atorvastatin Treatment on Serum Lipid Profile in Responders and Nonresponders
As shown in Table 2, atorvastatin treatment resulted in a significant decrease in total cholesterol, LDL cholesterol, and triglyceride levels in the responder group, while there was no effect on HDL cholesterol. Likewise, the nonresponder showed a significant decrease in total cholesterol and LDL cholesterol and a trend toward a decrease in triglyceride level. HDL cholesterol was not affected by atorvastin in the nonresponders.
Serum Lipid Levels Before and After Start of 80 mg Atorvastatin Treatment

LDL, low-density lipoproteins; HDL, high-density lipoproteins.
Effect of Atorvastatin on Circulating Endothelial Progenitor Cell Levels
First, we assessed the effect of atorvastatin treatment on circulating EPC levels for the complete HIIB cohort. As shown in Figure 2A, baseline EPC levels were 53.7 ± 5.4 cells/100 μl. Two weeks of atorvastatin treatment resulted in a significant increase in EPC levels to 74.9 ± 6.3 cells/100 μl (p < 0.05 by repeated measurements ANOVA), and this was maintained until 6 weeks after the start of treatment (85.2 cells ± 8.5 cells/100 μl, p < 0.05 by repeated measurements ANOVA and compared to baseline). When we divided the patients into responders to atorvastatin treatment and nonresponders, the responders had a significant increase in EPC levels from 41.1 ± 5.2 cells/100 μl to 89.4 ± 7.9 cells/100 μl at 2 weeks (p < 0.001) and 96.41530±153011.8 cells/100 μl at 6 weeks (p < 0.001) (see Fig. 2B), while the nonresponders showed a significant decrease upon atorvastatin treatment from 76.4 ± 10.9 cells/100 μl to 48.6 ± 8.3 cells/100 μl at 2 weeks (p < 0.001) and 65.7 ± 9.9 cells/100 μl at 6 weeks (p < 0.05) (see Fig. 2C). At baseline, there were significantly higher numbers of circulating EPCs in the nonresponder group compared to the responder group (76.4 ± 10.9 cells/100 μl vs. 41.1 ± 5.2 cells/100 μl, p < 0.01). Two weeks of atorvastatin treatment reversed this, and the nonresponders had a significantly lower number of circulating EPCs (48.6 ± 8.3 cells/100 μl vs. 89.4 ± 7.9 cells/100, for nonresponders and responders, respectively, p < 0.001) (see Fig. 2D).

Circulating EPC levels following statin treatment. (A) Statin increased circulating EPC levels for the complete Healthy Endothelial Accelerated Lining Inhibits Neointimal Growth (HEALING) IIB cohort as early as 2 weeks and was maintained until 6 weeks. (B) Responders to statin therapy had a significant increase in circulating EPC levels, 2 and 6 weeks after start of treatment. (C) Nonresponders showed even a significant decrease in circulating EPC levels upon statin treatment. (D) Nonresponders had a significantly higher baseline EPC count compared to responders. Two weeks after the start of treatment, this was reversed, and responders had a significantly higher EPC count. The key in (D) is for (D) only.
Determinants of Response to Atorvastatin Treatment
Baseline EPC count and the number of patients that never smoked were significantly higher in nonresponders, whereas there were fewer men. When we applied our LR model, only baseline EPC count (aOR: 1.02 and 95% CI: 1.007–1.031) and having never smoked (aOR 0.27 and 95% CI: 0.082–0.913) were independent risk factors for being a nonresponder (see Table 3).
Multivariate Regression Model
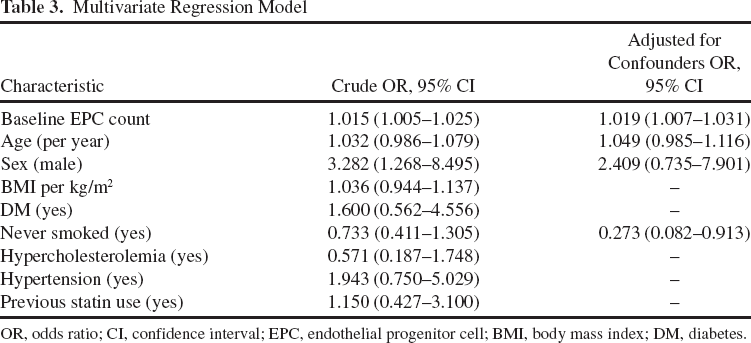
OR, odds ratio; CI, confidence interval; EPC, endothelial progenitor cell; BMI, body mass index; DM, diabetes.
Circulating Endothelial Progenitor Cell Levels and Clinical and Angiographic Outcome
Complete H2B Cohort. Baseline EPC count inversely correlated with late luminal loss (LLL) at 6 months (Spearman r = −0.41, p < 0.001) (see Fig. 3A). EPC levels at baseline did not correlate with major adverse cardiac events (MACE) or target lesion revascularization (TLR) at 6 months. Although statin significantly increased circulating EPC levels for the complete HIIB cohort, this was not accompanied by an improvement in LLL, as LLL was comparable with the HII study.

Correlation between EPC counts and late luminal loss (LLL) at 6 months follow-up (FU). (A) Baseline EPC count significantly correlates with LLL in the complete HEALING IIB cohort. (B) Baseline EPC also significantly correlates with LLL in the nonresponder group but not in the responder group (C).
Nonresponders Versus Responders
Baseline EPC count of nonresponders inversely correlated with LLL (Spearman r = −038, p < 0.05) (Fig. 3B). Responders did not show such a correlation (Spearman r = −0.26, p > 0.05) (Fig. 3C). LLL at 6 and 18 months were significantly lower in nonresponders compared to responders (0.61 ± 0.07 vs. 0.88 ± 0.08, p < 0.05 and 0.50 ± 0.08 vs. 0.82 ± 0.08, p < 0.01) (see Fig. 4). There were no TLRs in the nonresponder group compared with six in the responder group (p = 0.09).
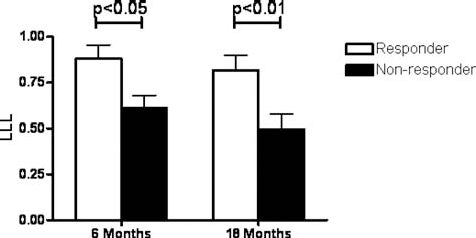
LLL values at 6 and 18 months FU. Nonresponder group has a significantly lower LLL at 6 and 18 months FU.
Discussion
In this subanalysis of the HEALING IIB study, we evaluated the effect of high-dose atorvastatin treatment on circulating EPC levels and correlation with angiographic and clinical follow-up. Prati and coworkers already showed that high-dose atorvastatin treatment could reduce in-stent restenosis and LLL (19). However, in contrast to the RESTenosis AfteR sTenting (RESTART) study, we specifically looked at the effect of high-dose atorvastatin treatment on EPC levels and its effect on LLL. Our study supplies data on the possible role of EPCs in LLL and demonstrates that statin treatment in patients might contribute to vascular healing via EPC recruitment.
In line with earlier studies (17,21,23), statin treatment significantly increased circulating EPC levels for the complete HEALING IIB cohort, as early as 2 weeks after the start of statin treatment. When we evaluated EPC levels for each individual patient at every time point, we could divide the cohort into patients responding to statin treatment with a rise in EPC levels (responder group) and patients who did not respond to statin treatment with an increase in EPC levels (nonresponder group). Interestingly, the nonresponder group had a significantly higher baseline EPC count in comparison to the responder group, suggesting that the response to statin treatment on EPC level depends on baseline EPC count and that patients with a high baseline EPC count are unable to recruit or produce more EPCs despite high-dose statin treatment. An alternative and maybe more logical explanation would be that nonresponders react to vascular disease with a more efficient vascular repair response. That would explain the high basal level of EPC count in the blood of nonresponders versus responders (76.4 ± 10.9 cells/100 μl vs. 41.1 ± 5.2 cells/100 μl, p < 0.01), which indicates that the nonresponders are more capable of recruiting EPCs from the bone marrow into the bloodstream. Likewise, the blood circulatory pool of EPCs of nonresponders may be more efficient in homing to the affected vascular area, an intrinsic capacity that was further amplified by statin treatment (3,15). For the responders, the data might imply that although recruitment of EPCs from bone marrow by statin treatment is successful, the recruited EPCs in the bloodstream remain inefficient in homing capacity to the target vessel wall. Further research is required to assess this hypothesis on the complex effect of statins on EPC recruitment and homing.
EPC Levels and Cardiovascular Risk Factors
Earlier publications showed that several cardiovascular risk factors (e.g., hypertension, diabetes, age, smoking status, or increased body mass index) could affect EPC titer. Vasa and coworkers reported that patients with coronary artery disease had lower EPC levels compared to healthy volunteers (21). Moreover, the number of risk factors was significantly correlated with lower EPC levels, and multivariate analysis showed that smoking status was an independent predictor of reduced EPC levels. Later, Hill and coworkers showed, in a relatively small group of healthy volunteers, that the number of EPC colonyforming units was significantly reduced in patients with diabetes, hypercholesterolemia, or hypertension (11). When they adjusted for age, only hypercholesterolemia remained significant. Multivariate analysis revealed that only flow-mediated brachial reactivity, a composite measure of endothelial integrity, was an independent predictor of reduced EPC colonies. Here we assessed whether cardiovascular risk factors could affect being a nonresponder or responder. We found a significant difference between responders and nonresponders in baseline EPC count, smoking status, and sex. However, when we applied our LR model, only baseline EPC count and smoking status remained significant predictors for being a nonresponder, that is, a higher baseline EPC count.
EPC Levels and Cardiovascular Outcomes
Baseline EPC count inversely correlated with LLL for the complete HEALING IIB cohort. Furthermore, patients from the nonresponder group had a lower LLL at 6 months FU compared to patients from the responder group. This difference in LLL was even more pronounced at 18 months FU. As Ellis and coworkers showed that LLL can serve as a surrogate clinical endpoint (8), we evaluated the occurrence of target lesion revascularization (TLR). Indeed, we found a trend toward more TLRs in the responder group. These results suggest that baseline EPC count can predict the response to the Genous stent in terms of LLL and perhaps clinical benefit. There is much debate about the significance of baseline EPC count in the pathophysiology of cardiovascular disease. It has already been shown that low circulating EPC levels are associated with less favorable cardiovascular outcomes (5,9,20). Circulating EPCs play an important role in maintaining the integrity of the endothelial layer by accelerating reendothelialization in injured arteries, so low levels of EPCs could result in delayed reendothelialization and increased cardiovascular risk. Our study confirmed these results but furthermore showed that baseline EPC count is correlated with angiographic outcome, not only at 6 months FU but also at 18 months FU. Pelliccia and coworkers were the first to prospectively assess circulating EPC levels and the occurrence of in-stent restenosis or progression of coronary artery disease in patients treated with a bare metal stent (18). In contrast to our findings, they showed that high circulating EPC count correlated with the occurrence of in-stent restenosis and high late LLL, possibly due to engraftment of EPCs in the neointima and, subsequently, differentiation into vascular smooth muscle cells. There was no correlation between EPC count and progression of atherosclerosis, stable coronary artery disease, or healthy controls. There are some differences between our work and the work of Pelliccia et al. (18). First of all, as with many papers on EPCs, there is a difference in characterization of EPCs, as there is no standardized way to measure EPCs. Originally, EPCs were described as CD34+ or KDR+ cells, and now, different combinations of cell surface markers have been advocated to define EPCs, most commonly CD133+/CD34+/KDR+, CD34+KDR+, CD34+/KDR+/CD45-, or CD14+/CD34low. We measured EPCs using a modified ISHAGE protocol using whole blood in which the EPC population was defined as CD45+/CD34+/KDR+, whereas Pelliccia and coworkers used mononuclear cells, defining EPCs as CD45-/CD34+/KDR+. Second, Pelliccia included only stable angina patients, whereas we also included patients with unstable angina and silent ischemia. Third, our patients were treated with the Genous stent, and Pelliccia used the bare metal stent. However, it seems unlikely that this influenced baseline EPC count, as blood was drawn at least 1 day before PCI. More recently, in contrast to our work and the work of Pelliccia, Haine et al. reported that they found no correlation between circulating EPC levels and angiographic and clinical restenosis (10), making it even more complex to understand the role of EPCs in cardiovascular disease and outcome. We believe that, at least for the clinical outcome after Genous stent placement, baseline EPC count is a valid biomarker for, and reflects the regenerative potency of, the vascular bed.
Interestingly, although statin treatment significantly increased circulating EPC count in the responder group, this increase was not reflected in a favorable LLL. One explanation could be that the responder group not only had lower baseline EPC count but also less functional EPCs and that statin therapy can increase EPC count but cannot improve the angiogenic capacities of these cells. However, we can only speculate on this as we only performed a quantitative analysis of the EPCs but no functional analysis. Future studies to elucidate the angiogenic potential of EPCs of this specific subset of nonresponder patients will help to understand the complex biology behind these clinical findings.
Conclusion
To our best knowledge, this is the first prospective study to show that there is a significant difference in baseline EPC count between responders and nonresponders to atorvastatin 80 mg treatment for EPC recruitment in coronary artery disease patients treated with the Genous stent. Furthermore, higher baseline EPC count correlated with favorable angiographic outcome with less LLL at 6 and 18 months FU compared to lower baseline EPC count. Although statin treatment significantly increased circulating EPC levels in the responder group, this was not reflected in improved angiographic outcome, suggesting that low EPC count reflects less functional EPCs. EPCs might be used as predictor for angiographic outcome for patients treated with the Genous stent.
Footnotes
Acknowledgments
Study supported by OrbusNeich Medical Technologies, Fort Lauderdale, FL, USA. Independent Imaging Core lab (QCA/QCU) and data monitoring by Cardialysis (Rotterdam, The Netherlands). The authors declare no conflicts of interest.