
Abstract
We show for the first time that signaling through the TLR4/TRIF pathway plays a critical role in allogeneic bone marrow cell (BMC) rejection. This appears to be unique to BMCs as organ allografts are rejected mainly via MyD88 signaling. Using T- or T-/B-cell-deficient mice, we found that BMC allorejection occurred early before T-cell activation and was T- and B-cell independent, suggesting an effector role for innate immune cells in BMC rejection. We further demonstrated the innate immune signaling in BMC allorejection by showing superior engraftment in mice deficient in TRIF or TLR4 but not in MyD88 or TLR3. The restored cytotoxicity in TRIF-deficient recipients transferred with wild-type F4/80+ or NK1.1+ cells suggests TRIF signaling dependence on macrophages or NK cells in early BMC rejection. Production of the proinflammatory cytokine IL-6 and TRIF relevant chemokine MCP-1 was significantly increased early after bone marrow transplantation. In vivo specific depletion of macrophages or NK innate immune cells in combination with anti-CD154/rapamycin resulted in additive-enhanced allogeneic engraftment. The requirement for irradiation was completely eliminated when both macrophages and NK cells were depleted in combination with anti-CD154/rapamycin to target T- and B-cells, supporting the hypothesis that two barriers involving innate and adaptive immunity exist in mediating the rejection of allogeneic BMCs. In summary, our results clearly demonstrate a previously unappreciated role for innate immunity in BMC allorejection via signaling through a unique MyD88-independent TLR4/TRIF mechanism. These findings may have direct clinical impact on strategies for conditioning recipients for stem cell transplantation.
Introduction
Hematopoietic stem cell transplantation is curative for red blood cell disorders, inherited disorders of metabolism, and numerous autoimmune disorders and induces donor-specific tolerance to organ transplants. An understanding of the mechanism of bone marrow cell (BMC) rejection has allowed reduced intensity conditioning approaches to be developed, resulting in significantly less morbidity and mortality (25,50). T-cells have been implicated as the primary effector cells in alloreactivity (21). Recent studies in a presensitized model of BMC rejection demonstrated that humoral immune responses predominate over T-cell responses, although both contribute (49). The role of innate immunity in bone marrow transplantation (BMT) has not been fully explored.
The innate immune system acts as a sentinel for the immune system and functions to rapidly respond to infection. The molecular patterns on pathogenic cells are exquisitely distinguished from mammalian cells. Therefore, research in transplantation on rejection, tolerance, and immunosuppressive agents has been directed toward the adaptive immune response. The role of innate immunity in transplantation, with the exception of natural killer (NK) cells, has generally been ignored. However, not all clinical observations are explained by adaptive immune responses, suggesting factors other than recognition of major histocompatibility complex (MHC). For example, more immunogenic allografts such as skin, lung, and intestine are in close contact with the environment and commensal organisms (17,26). The overall survival for these grafts is significantly inferior to that of renal transplantation (26). The “danger signal” theory has been proposed to explain immune recognition and reaction to damage signals in the body regardless of their origin (26).
In transplantation, the danger signals, which may contribute to the initiation of an inflammatory response, are considered to be antigen-independent stimuli and include ischemia-reperfusion, surgical injury, systemic stress, and brain death (22,40).
There is growing evidence that innate immunity plays a role in many aspects of organ transplantation. In recent decades, the role of innate immune responses in the control of adaptive immunity has been revealed with the study of the toll-like receptors (TLRs), one important recognition receptor of the pathogen-associated molecular patterns (10,20). Emerging evidence suggests that in addition to responding to pathogen-associated molecular patterns of microorganisms, TLR can be activated by endogenous ligands expressed by mammalian cells (3). TLR activation has been shown to be a barrier to the induction of transplantation tolerance (2,3,21). A critical role for the toll-like receptor signaling adaptor protein myeloid differentiation factor 88 (MyD88) was recently reported in the rejection of organ allografts (11). Clinical studies suggest that activation of TLR4 signaling in lung transplant recipients contributes to the development of acute rejection (31).
In the present study, we show for the first time that the signaling through the TLR4/toll-interleukin-1 receptor domain-containing adapter inducing interferon-β (TRIF) pathway plays a critical role in allogeneic BMC rejection. The innate immune system acts as the first-line barrier that precedes the fully appreciated adaptive immune barrier in BMT. We demonstrate that innate immune cells of NK cells and macrophages both elicit potent cytotoxicity against allogeneic BMC. This effect is independent of Tand B-cell responses in early allogeneic BMC rejection. These findings may have direct application to novel strategies to condition recipients for BMT.
Materials and Methods
Animals
Four- to 6-week-old male C57BL/6 (B6; H-2b), BALB/cJ (BALB/c; H-2d), B10.BR/SgSnJ (B10.BR, H-2k), B6.129P2-Tcrbtm1MomTcrdtm1Mom/J [T-cell receptor b/d knockout (TCRb/d-/-), B6 background], B6.129S7-Rag1tm1Mom/J [recombination activating gene knockout (Rag-/-), B6 background], B6.B10ScN-Tlr4lps-del/JthJ (TLR4-/-, B6 background), C57BL/10, B6;129S1-Tlr3tm1Flv/J (TLR3-/-), and B6129SF2/J (controls for TLR3-/- mice with the same genetic background) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). C57BL/6J-Ticam1Lps2/Lps2 (TRIFLps2/Lps2, B6 background) pairs were purchased from The Jackson Laboratory and bred at the University of Louisville. TRIFLps2/Lps2 mice express a truncated form of TRIF that is inactive due to a mutated lipopolysaccharide response sequence (14). MyD88-/- mice (B6 background) were kind gifts from Shizuo Akira (via Ross Kedl; 3M Corporation, Minneapolis, MN, USA) and bred at the University of Louisville. Animals were housed at the Institute for Cellular Therapeutics under specific pathogen-free conditions and cared for according to NIH guidelines. Animals were 6–10 weeks old when they were used in experiments. All studies were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Louisville.
Preconditioning and Bone Marrow Transplantation
Recipient B6 mice were conditioned with agents targeting innate immune cells in combination with T-cell targeting-based strategies and 0–100 cGy total body irradiation (TBI, γ-cell 40; Nordion, Ottawa, ON, Canada). Agents targeting innate immune cells included anti-mouse Gr-1 [RB68C5, Rat IgG2b, κ, generated in our laboratory from the hybridoma that was kindly provided by Dr. Emil Unanue (Washington University School of Medicine, St. Louis, MO, USA); 500 μg on day –3 and day –2] to deplete neutrophils, anti-mouse NK1.1 [PK136, mouse IgG2a, generated in our laboratory from hybridoma that was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), 1 mg on day –3] to delete NK cells, and clodronate liposomes (CloLip, Vrije Universiteit, Amsterdam, Netherlands, 1.2 mg/10 g of animal weight at day –2) to deplete macrophages. With T-cell targeting-based strategies, B6 mice were pretreated intraperitoneally with anti-ab-TCR (H57-597: hamster antimouse IgG, generated in our laboratory; 30 μl per mouse at day –3), anti-cluster of differentiation 154 (CD154; MR-1: hamster anti-mouse IgG3; BioXcell, Lebanon, NH, USA; 0.5 mg per mouse at days 0 and +3), and rapamycin (3 mg/kg, days 0 through +4; LC Laboratory, Woburn, MA, USA). Conditioned mice were transplanted with 15 × 106 untreated donor BMCs via lateral tail vein injection 4–6 h after TBI. The intravenous injection of CloLip suspension has been reported to induce the complete depletion of splenic and hepatic macrophages within 24 h (42,43).
Chimerism Testing
Recipients were characterized for chimerism using flow cytometry to determine the relative percentages of donor-derived peripheral blood leucocytes (PBLs) at 4 weeks. Peripheral blood was obtained from the tail vein and stained with Abs specific for MHC class I antigens of donor [fluorescein isothiocyanate (FITC)-conjugated anti-H-2Kd, 36-7-5, mouse IgG2a] and recipient (PE-conjugated anti-H-2Kb, AF6-88.5, mouse IgG2a) origin. All antibodies were obtained from BD/PharMingen (San Jose, CA, USA). Nonspecific background staining was controlled for using isotype-specific Ab directed against irrelevant Ag conjugated with the same fluorochrome. The engraftment of donor cells was defined as ≥1% donor cells in the peripheral blood lymphoid gate.
Flow Cytometric Analysis of TCR Vβ Families
BALB/c mice express superantigen I-E, resulting in the clonal deletion of superantigen-specific Vb5.1/2+ and Vβ11+ T-cells. As recipient B6 mice do not express I-E, they do not delete these two Vβ subfamilies. Peripheral blood (8–100 μl) from mixed chimeras and naive control mice was stained with anti-Vβ5.1/2-FITC (MR9-4), Vβ6-FITC (RR4-7), Vβ8.1/2-FITC (MR-5-2), or Vβ11-FITC (RR3-15) versus anti-host H2Kb-phycoerythrin (PE), anti-CD8-peridinin chlorophyll protein complex (PerCP), and anti-CD4-allophycocyanine (APC) (all from BD PharMingen). Relative expression indicates the percentage of Vb-positive cells within the CD8+ or CD4+ T-cell subsets of the host (H2Kb) lymphoid gate in peripheral blood.
In Vivo Cytotoxicity Assay
Target and internal control splenocytes or BMCs (100 × 106/ml) were incubated with 4.0 or 0.2 μM carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes, Leiden, The Netherlands), respectively, at room temperature for 10 min. An equal volume of fetal bovine serum (Gibco BRL, Grand Island, NY, USA) was added to quench the reaction. After washing, cells were mixed in a 1:1 ratio and resuspended in phosphate-buffered saline (PBS, Sigma-Aldrich, St. Louis, MO, USA), and 20 × 106 cells from each were injected intravenously. Peripheral blood was collected from individual mice at selected time points after cell infusion: 0.5 h, 1 h, 3 h, day 1, day 2, day 3, and day 7. After lysis of red blood cells, peripheral blood lymphocytes were analyzed for CFSE expression by fluorescence-activated cell sorting (FACS). The percentage of killing was determined by calculating the ratio between target and internal control cells. Wild-type F4/80+ macrophages used in in vivo cytotoxicity assay were sorted from cells prepared from spleens and collected from peritoneal cavities, and NK1.1+ NK cells were sorted from spleens of B6 mice after staining with F4/80 PE (BM8, rat IgG2a, k, BioLegend, San Diego, CA, USA) and NK1.1 FITC (PK136, mouse IgG2a, κ, BD PharMingen), respectively.
Enzyme-Linked Immunosorbent Assay (ELISA)
Cytokines are evaluated using ELISA MAX™ Set Deluxe (for IL-1β and IL-6; Biolegend), VeriKine™ Mouse ELISA Kit (for IFN-α and IFN-b; PBL InterferonSource, Piscataway, NJ, USA), ELISA Ready-SET-Go Kit [for monocyte chemotactic protein-1 (MCP-1); eBioscience, San Diego, CA, USA], and Platinum ELISA 96 Test Kit [for IFN-γ-induced protein 10 (IP-10); eBioscience] as per the manufacturer's directions. Briefly, the specific antibody for each cytokine was precoated. One hundred microliters of the serum sample or cytokine standard was incubated for 2 h. After washing, the detection antibody specific for the particular cytokine was added and incubated for 1 h followed by 100 μl of diluted avidin–HRP solution for 30 min. The color was developed by adding 100 μl 3,3′,5,5′-tetramethylbenzidine (TMB) substrate solution and incubated in the dark for 15–30 min. After adding a stop solution of dilute hydrochloric acid, the optical density of color product was measured at 450 nm using ELISA microplate reader.
Statistical Analysis
Data are presented as the average±SD. Means of two groups were compared by the two-tailed t test (two-sample, assuming unequal variances). Means of more than two groups were compared by ANOVA. Statistical analysis was carried out using SigmaStat software (Jandel Scientific, San Raphael, CA, USA). The difference between groups was considered significant at p < 0.05.
Results
Innate Immunity Mediates Early Allorejection of Donor BMC
To evaluate the role of innate immunity in BMC rejection, T-cell-deficient B6 mice (TCRb/d-/-) as well as wildtype B6 mice were used as recipients in in vivo cytotoxicity assays. CFSE-labeled allogeneic BALB/c donor (high CFSE fluorescence intensity) and control recipient (low CFSE fluorescence intensity) splenocytes or BMCs (all 20 × 106) were injected (Fig. 1A, B). Wild-type B6 mice that received only syngeneic CFSE-labeled cells with high or low intensity also served as a control. We found that the kinetics of cytotoxicity were very similar using either splenocytes or BMCs as target cells. Therefore, the results from splenocytes or BMCs were summarized together. Donor cells were rapidly eliminated in control wild-type B6 mice, and rejection was completed by day 3. The kinetics of donor cell elimination in TCRb/d-/- mice resembled wild-type controls. The cytotoxic activity observed in this experiment was not due to the CFSE labeling as the syngeneic controls of B6 cells labeled with CFSE high/low exhibited no cytotoxicity. These results indicate that BMC rejection in wild-type mice was T-cell independent. To rule out any potential involvement of naturally occurring antibodies in the cytotoxicity observed, Rag-/- mice, which produce no mature T- and B-cells, were used. A similar kinetic of donor cell killing was observed (Fig. 1C, D). The rapid loss of allogeneic cells from immunocompetent mice is comparable with that in T- or T-/B-cell-deficient mice, indicating that allogeneic BMCs are subject to Band T-cell-independent rejection early after transplantation (≤3 days). Thus, the effectors that mediate BMC rejection would presumably be innate immune cells.

Innate immunity is responsible for early allorejection of donor BMCs. To determine the role of innate immune cell populations in the rejection of donor BMCs, T-cell-deficient [T-cell receptor knockout (TCRβ/δ-/-), H-2b] (A and B) and T- plus B-cell-deficient mice [recombination activation gene knockout (Rag-/-), H-2b] (C and D) were used as recipients in vivo cytotoxicity assays. Carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled donor BALB/c (H-2d) splenocytes (as targeting cells, high CFSE fluorescence intensity) and recipient control splenocytes (as internal controls, low CFSE fluorescence intensity) (both 20 × 106) injected intravenously into T-cell-deficient mice and B6 wild-type control mice. Wild-type B6 mice that received only syngeneic CFSE-labeled cells with either high intensity or low intensity also served as a control. Donor cell survival was compared over time. Peripheral blood was collected from individual mice at selected time points after cell infusion: 1 h, 3 h, day 1, and day 3. Representative histograms from each group are presented (B and D). Spleens (SP), lymph nodes (LN), femurs for bone marrow (BM), as well as peripheral blood (PB) were collected from each recipient on day 1 after BMC infusion to compare the cytotoxic activity in these different tissues (E). Peripheral blood lymphocytes were analyzed for CFSE expression by a FACSCalibur after lysis of red blood cells. The results of TCRβ/δ-/- mice are the summary of three experiments. The results of RAG-/- mice are the summary of two experiments.
Donor BMCs Are Not Sequestered Preferentially in Tissues
To rule out sequestration of donor BMCs in other tissues, the cytotoxic activity in peripheral blood was compared with that for spleen, lymph node, and femur. CFSE-labeled BALB/c donor (high intensity) and B6 recipient (low intensity) BMCs (both 20 × 106) were injected into naive B6 mice. Spleens, lymph nodes, femurs, and peripheral blood were collected from each recipient on day 1 after BMC infusion. Significant killing rates were observed in these different tissues tested (Fig. 1E), resembling the results in peripheral blood. These results suggest that the killing rates of allogeneic BMCs in peripheral blood reflect the fate of donor cells in other tissues. The allogeneic BMCs are not trapped preferentially in specific tissues.
Allogeneic BMC Rejection Is Dependent on the TRIF and Not on the MyD88 Pathway
An effector role for innate immune cells in BMC rejection was suggested in our findings in T-deficient mice that BMC allorejection was T-cell independent. Innate immune system initiates defense mechanism with sensing function of the family of toll receptors. The signaling function of TLR depends upon intracellular adaptors. The majority of TLRs signal via MyD88 and TRIF. The adaptor MyD88 transmits signals emanating from all TLRs, except TLR3, while TRIF specifically mediates TLR3 and TLR4 signaling via type I IFN (30,52). To determine which TLR signaling pathway is involved in BMC rejection, MyD88-deficient (MyD88-/-) and TRIF-deficient (TRIFLps2/Lps2) mice were nonmyeloablatively conditioned with established conditioning strategies targeting T- and B-cells: anti-CD154/rapamycin/100 cGy TBI and transplanted with 15 × 106 BALB/c BMCs (Fig. 2A). Anti-CD154 mAb and rapamycin were synergistically used to control host T-cell response to allogeneic BMCs by blocking CD40:CD154 costimulatory pathway and late-stage T-cell activation by inhibiting IL-2 responsiveness, respectively. A threshold of alloengraftment was observed in our previous study (51) with anti-CD154/rapamycin/100 cGy in wild-type B6 mice that served as controls in our current experiment. As expected, engraftment was achieved in 20.0% of recipients at 1 month after BMT. Surprisingly, engraftment was achieved in only 30% of MyD88-/- recipients at 1 month, resembling that for wild-type B6 mice. In contrast, 100% engraftment was achieved in TRIFLps2/Lps2 mice (Fig. 2B). The level of donor chimerism in TRIFLps2/Lps2 mice was 5.1±0.1% at 1 month, which was significantly higher than in MyD88-/- (1.6±0.8%; p < 0.05) or in wild-type B6 mice (1.3±0.8%; p < 0.05) and thereafter throughout all time points tested up to 6 months (Fig. 2C) (p < 0.05). The levels of donor chimerism increased with time, reaching 17.1±8.6% in TRIFLps2/Lps2 recipients at 6 months. These results suggest that allogeneic BMC rejection is independent of the MyD88 pathway but requires TRIF signaling.
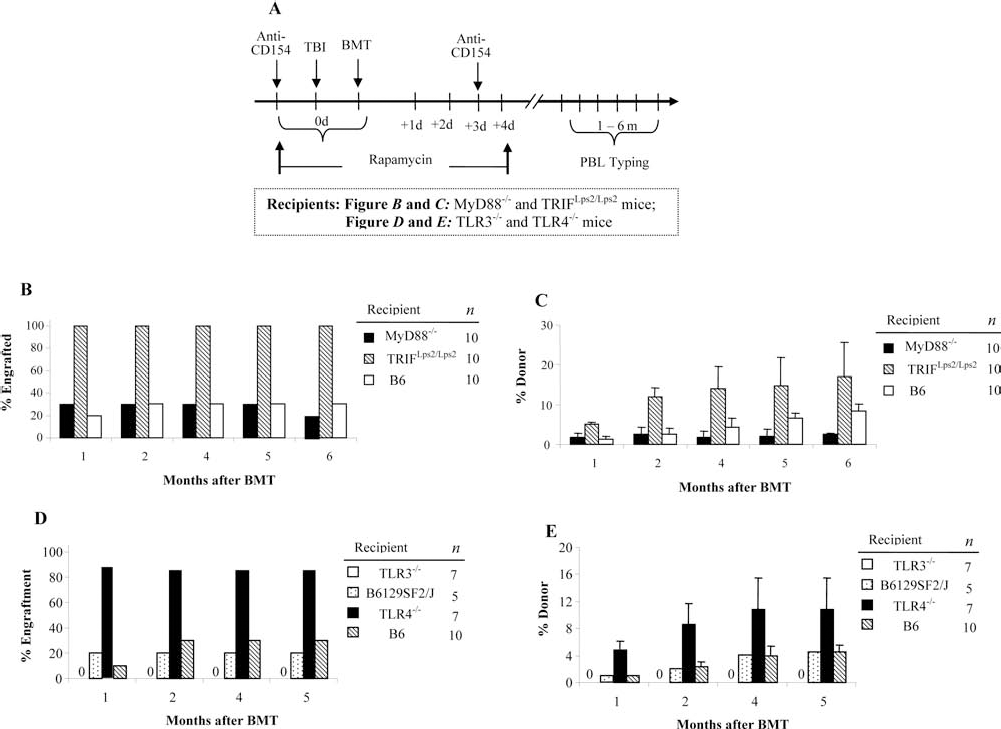
Allogeneic BMC rejection is dependent on the TLR4/TRIF signaling pathway. In order to define the innate signaling pathways active in allorejection, myeloid differentiation factor 88 knockout (MyD88-/-) and toll-interleukin-1 receptor domain-containing adapter inducing interferon-β lipoplysaccharide 2 mutation (TRIFLps2/Lps2) mice (B and C) were used as recipients of allogeneic bone marrow transplantation (BMT). Then toll-like receptor 3 knockout (TLR3-/-) and TLR4-/- (D and E) were recipients in allogeneic BMT. Wild-type mice with the same genetic background served as controls. Recipients were conditioned with anti-cluster of differentiation 154 (CD154), rapamycin, and 100 cGy total body irradiation (TBI) and transplanted with 15 × 106 untreated donor BMCs. (A) The schematic treatment of recipient is shown. Donor peripheral blood chimerism (PBL typing) was tested monthly. Figures show the frequency of engraftment from 1 to 6 months after BMT (B, D) and the level of chimerism in animals that engrafted from 1 to 5 or 6 months (C, E).
Allogeneic BMC Rejection Is Dependent on the TLR4 Pathway
TRIF cascade is a MyD88-independent signaling pathway specifically associated with TLR3 and TLR4. We therefore next determined whether the TLR3 or TLR4 pathway was involved in BMC rejection using TLR3-/- and TLR4-/- mice as recipients. TLR3-/- and TLR4-/- mice were conditioned with anti-CD154/rapamycin/100 cGy TBI for BMT (Fig. 2D, E). None of the TLR3-/-, 20% of the B6129SF2/J mice (TLR3-/- controls), and only 1 of the 10 wild-type B6 mice (controls for TLR4-/-) engrafted at 1 month. In striking contrast, 85.7% of the TLR4-/- mice engrafted. The donor chimerism was 4.2±1.8% at 1 month and increased with time, reaching 10.8±4.7% at 5 months. The levels of donor chimerism were comparable between TLR4-/- and TRIFLps2/Lps2 recipients over the 5-month follow-up (p > 0.1). Taken together, these results suggest that rejection of allogeneic BMCs is exclusively dependent on the TLR4/TRIF signaling pathway.
Depletion of Macrophages, NK Cells, or Both, But Not Granulocytes, From Recipients Delays Donor Cell Allorejection
The timing of acute rejection of BMCs in wild-type B6 was similar with T-cell-deficient recipients and occurred within 3 days prior to the time required for T-cell activation. Thus, the effectors that mediated BMC rejection at this period of time would be innate immune cells: macrophages, neutrophils, or NK cells. To differentiate the role for these innate immune cell populations in BMC allorejection, in vivo cytotoxicity assays were performed in mice from which macrophages, NK cells, or neutrophils had been depleted with CloLip, anti-NK1.1, or anti-Gr-1, respectively (Fig. 3A, B). The kinetics of elimination of donor cells in mice treated with anti-Gr-1 mAb was similar to saline-treated controls. Donor cells were totally eliminated by day 3. The rejection of donor cells was delayed in recipients treated with CloLip alone, anti-NK1.1 alone, or both and reached the most significance (p < 0.003) at day 1 after infusion compared with that in saline-treated controls (88.7±4.5%). At day 1, the killing percentages of CFSE-labeled BALB/c cells were 58.0±2.9%, 66.9±14.0%, or 42.6±7.5% in recipients treated with anti-NK1.1 alone, CloLip alone, or both, respectively. The impaired cytotoxicity remained significant (p < 0.05) at day 3. Subsequently, all donor cells were eliminated in all three groups by day 7, most likely representing the initiation of adaptive immunity. The decreased cytotoxicity in recipients treated with CloLip and anti-NK1.1 was additive, indicating that, in addition to NK cells, macrophages play an important and nonredundant role in early allogeneic BMC rejection. The depletion of NK cells and macrophages did not lead to a complete inhibition of donor cell killing, suggesting that other unknown mechanisms or cell populations existed.

Depletion of innate immune cells enhances allogeneic BMC survival. To determine the effect of innate immune cell populations on the rejection of donor BMCs, in vivo cytotoxicity assays were performed in mice treated with anti-NK1.1, anti-Gr-1, or clodronate liposomes (CloLip). CFSE-labeled donor BALB/c splenocytes (CFSE high) and recipient B6 splenocytes (internal controls, CFSE low) (both 20 × 106) infused as target cells. Recipients treated with saline served as controls. Peripheral blood was collected at selected time points after cell infusion: 1 h, 3 h, day 1, day 3, and day 7. After lysis of red blood cells, peripheral blood lymphocytes were analyzed for CFSE expression by a FACSCalibur. Representative histograms from a mouse that received CloLip and a control that received PBS treatment at days 1, 3, and 7 (A). The kinetics of killing is the summary of four separate experiments (B).
Depletion of Macrophages and/or NK Cells in Combination with T-Cell Targeting-Based Strategies Eliminates the Requirement for Irradiation for Engraftment
Targeting host-versus-donor reactive T-cells in recipients has significantly reduced the minimum TBI dose required for engraftment (47,48). Our data suggested that innate immune cells were effectors in early BMC rejection, and so we hypothesized that, by specifically targeting both macrophages and NK cells in addition to adaptive immune cells, engraftment could be achieved without irradiation as a novel approach to conditioning.
To evaluate the efficacy of targeting selected innate immune cells in conditioning for allogeneic engraftment, B6 mice received CloLip, anti-NK1.1 mAb, or anti-Gr-1 mAb. These agents were used in combination with anti-αβ-TCR/anti-CD154/rapamycin. Conditioned recipients were transplanted with 15 × 106 BALB/c BMC (Fig. 4A). Allogeneic engraftment was achieved in 45.5% of recipients conditioned with anti-αβ -TCR/anti-CD154/rapamycin plus 100 cGy TBI. The addition of CloLip alone, anti-NK1.1 alone, or both agents to the conditioning resulted in engraftment in 100% of recipients with donor chimerism of 23.5±8.0%, 19.1±11.4%, and 39.4±3.4%, at 1 month (Fig. 4B, C), respectively. Engraftment remained 100% at 6 months in all three groups (Fig. 4D). When anti-Gr-1 was utilized, only 33.3% of animals engrafted. When TBI was eliminated from preconditioning, none of the control mice engrafted when conditioned with anti-αβ-TCR/anti-CD154/rapamycin. However, engraftment occurred in 40% or 60% of recipients treated with an addition of anti-NK1.1 or CloLip, respectively. When both anti-NK1.1 and CloLip were added, engraftment occurred in 100% of recipients (Fig. 4B). Donor chimerism was 1.9±0.9% at 1 month, and the animals remained chimeric at ≤6 months (Fig. 4C, D). Moreover, specific Vβ5.1/2 and Vβ11 clonal deletion was detected in host original CD4+ T-cells in all chimeras (n = 23, donor percentage ranged from 2.5% to 46.7%), suggesting the induction of central tolerance to donor alloantigens.

Depletion of macrophages or/and NK cells promotes alloengraftment. To evaluate the efficacy of targeting different innate immune cell populations in promoting allogeneic engraftment, B6 mice received CloLip, anti-NK1.1 (NK) mAb, or anti-Gr-1 (Gr1) mAb. These agents were used in combination with conditioning strategies that target T- and B-cells: anti-αβ-TCR + anti-CD154 + rapamycin (Rapa) + irradiation (TBI). (A) The schematic procedures of recipient. Conditioned recipients were transplanted with 15 × 106 untreated BALB/c BMCs via lateral tail vein injection at least 4–6 h after TBI. Chimerism was followed by flow cytometric analysis monthly with peripheral blood staining of mAbs specific for donor [anti-H-2Kd fluorescein isothiocyanate (FITC)] and recipient [anti-H2Kb phycoerythrin (PE)] origin. (B) Percentage of animals with engraftment, (C) level of donor chimerism in engrafted recipients at 1 month, and (D) kinetics of donor chimerism. These results are the summary of five experiments.
We next assessed whether chimerism could be successfully achieved without the use of T-cell depletion. B6 mice were conditioned with CloLip and/or anti-NK1.1 mAb in combination with anti-CD154/rapamycin (Fig. 5A–C). Allogeneic engraftment was achieved in 30% of control recipients conditioned with anti-CD154/rapamycin plus 100 cGy TBI. With the addition of CloLip alone to anti-CD154/rapamycin/100 cGy conditioning, 72.7% (n = 11) of recipients engrafted with levels of donor chimerism 2.8±1.6% at 1 month after BMT. The addition of anti-NK1.1 alone or both of anti-NK1.1 and CloLip to anti-CD154/rapamycin/100 cGy conditioning resulted in 100% engraftment, with levels of donor chimerism 8.4±1.1% or 25.0±8.0%, respectively, at 1 month. With anti-NK1.1 and CloLip plus anti-CD154/rapamycin, without irradiation and host T-cell depletion, 100% engraftment was also achieved with 1.9±1.2% donor chimerism at 1 month and remained durable and stable during the 6-month follow-up (Fig. 5C).

Depletion of both macrophages and natural killer (NK) cells eliminates the requirement for T-cell depletion and irradiation for engraftment. In order to determine whether BMT could be successfully achieved without the use of T-cell depletion, B6 mice were conditioned with CloLip and/or anti-NK1.1 mAb in combination with anti-CD154 + rapamycin without anti-αβ-TCR and transplanted with 15 × 106 untreated donor BMCs. Chimerism testing was performed monthly after BMT: (A) engraftment, (B) level of donor chimerism at 1 month, and (C) kinetics of donor chimerism. The results are the summary of three experiments.
The Early Rejection of Allogeneic BMCs by Macrophages or NK Cells Is TRIF Signaling Dependent
To study the role of TRIF signaling in BMC rejection by macrophages and NK cells, TRIFLps2/Lps2 mice were used as recipients for in vivo cytotoxicity assays after adoptive transfer of wild-type macrophages and NK cells (Fig. 6). Wild-type F4/80+ macrophages were sorted from B6 spleens and peritoneal cavities, and NK1.1+ NK cells were sorted from B6 spleens. The doses of transferred NK1.1+ and F4/80+ cells were 370,000 and 140,000 per recipient, respectively. One day after transfer, 20 × 106 CFSE-labeled BALB/c target (high intensity) and internal control B6 (low intensity) BMC were injected. TRIFLps2/Lps2 mice that did not receive transferred cells and wild-type B6 mice treated with saline served as controls.
As expected, donor cells were rapidly eliminated in control wild-type B6 mice, and rejection was completed by day 3. The rejection of donor cells was significantly less in TRIFLps2/Lps2 mice without receiving transferred cells compared with that in wild-type B6 controls, from marginal significance (p = 0.04) at 3 h to the most significant (p = 0.0002) at day 3 after cell infusion. At day 3, the killing rates were 91.4±1.4% in TRIFLps2/Lps2 mice without transferred cells and 97.6±1.5% in wild-type B6 controls. The eliminating rates of donor cells were increased in TRIFLps2/Lps2 recipients that received either F4/80+ or NK1.1+ cells, and the kinetics of elimination of donor cells was shifted to resemble B6 controls with no significant difference between them at these three time points (p = 0.11–0.87). However, the eliminating rates of donor cells were significantly increased in TRIFLps2/Lps2 recipients that received either F4/80+ or NK1.1+ cells when compared with those in the TRIFLps2/Lps2 controls at all time points (p = 0.038–0.002), except the one at 3 h when compared between those in the TRIFLps2/Lps2 recipients that received NK1.1+ cells and those in the TRIFLps2/Lps2 controls (p = 0.058). At day 1, the killing percentages of CFSE-labeled BALB/c cells were 71.0±5.1%, 69.1±3.9%, or 61.1±5.8% in TRIFLps2/Lps2 recipients that received either F4/80+ cells, NK1.1+ cells, or none, respectively. Taken together, these data suggest that TRIF signaling is essential for macrophage- and NK cell-mediated allo-BMC rejection.

The role of TRIF signaling in BMC rejection by macrophages and NK cells. TRIFLps2/Lps2 mice were used as recipients in in vivo cytotoxicity assays after adoptive transfer of wild-type macrophages and NK cells. Wild-type macrophages and NK cells were stained with F4/80 PE and NK1.1 FITC, respectively. F4/80+ and NK1.1+ cells were sorted from spleens of B6 mice, and F4/80+ cells were also sorted from cells collected from peritoneal cavities. One day after transfer, target (BALB/c) and internal control (B6) BMCs were incubated with 4.0 or 0.2 μM CFSE, respectively. Cells (20 × 106) from each were injected intravenously. Peripheral blood was collected from individual mouse at different time points after cell infusion: 3 h, day 1, and day 3. After lysis of red blood cells, peripheral blood lymphocytes were analyzed for CFSE expression by fluorescence-activated cell sorting (FACS). The percentage of killing was determined by calculating the ratio between target and internal control cells. TRIFLps2/Lps2 mice that did not receive transferred cells and wild-type B6 mice treated with saline served as controls.
IL-6 and MCP-1 Production Is Significantly Increased Early and Transiently After Allogeneic BMC Infusion
TLR signaling mediates the activation of the NF-κB-dependent promoter to trigger the induction of inflammatory cytokines such as IL-1β and IL-6 (20,36). IL-6 also promotes Th2- and Th17-type responses to generate cytotoxic effectors and inflammation. Activation of type I IFN, IFN-α, and IFN-β is also involved in the immune response of the adaptor protein TRIF in innate immunity (13). Moreover, IP-10 and MCP-1 have been shown to be TRIF relevant chemokines (18,52). Therefore, the acute phase cytokines of IL-1β and IL-6, type I IFN (IFN-α and IFN-β), as well as chemokines of IP-10 and MCP-1 in recipients were assessed following allogeneic BMC infusion. IL-6 production was detectable very early at 0.5 and 1 h after BMT and then peaked at 4 and 8 h. The serum level of IL-6 decreased rapidly to undetectable levels at 12 h (Fig. 7A). Notably, MCP-1 levels significantly increased 1 h after allogeneic BMC infusion (p = 0.0034) when compared with the values pre-BMT (Fig. 7B). This peak was significantly higher than values at 4 h (p = 0.032). Other cytokines and chemokines, including IL-1β, IFN-α, IFN-β, and IP-10, were not detectable (data not shown). These data suggest that IL-6 and MCP-1 are critical cytokine and chemokine that may participate in mediating allorejection of BMCs.

Serum levels of cytokines and chemokines. B6 mice were transplanted with 15 × 106 BALB/c allogeneic BMCs. Serum was collected at 0.5, 1, 4, 8, and 12 h and days 1, 3, and 7 following transplantation. Control serums are from untransplanted B6 mice. The levels of IL-1β, IL-6, IFN-α, IFN-β, monocyte chemotactic protein-1 (MCP-1), and IFN-g-induced protein 10 (IP-10) were determined by ELISA. Production of each cytokine is represented in picograms per milliliter. There were four to five samples at each time point, and each sample was run in duplicate. Only IL-6 (A) and MCP-1 (B) were detectable and are shown.
Discussion
The barrier for rejection of allogeneic BMCs has been attributed primarily to adaptive immunity, especially T-cell immune responses. With T-cell targeting-based conditioning strategies, significant progress has been made in developing nonmyeloablative conditioning strategies to achieve mixed chimerism. It has been well known that T-cell depletion (47,48), combined with other conditioning strategies such as immunosuppressive drugs or costimulation-blocking agents (23,37,46), enhances allogeneic BM engraftment. The role of the innate immune system in BMC allorejection has not been adequately addressed. However, in addition to T-cell targeting-based conditioning, better alloengraftment still requires additional conditioning from nonspecific reagents, such as irradiation and immunosuppressive drugs. The need of other conditioning to improve alloengraftment may suggest the existence of another barrier in addition to T-cell barrier in BMT. As the humoral immunity is unlikely a barrier for BMT in unprimed naive recipients, innate immune system is most likely another barrier in BMT. In the present studies, we have evaluated the role of innate immunity in rejection of BMCs and show for the first time that the allorejection of BMCs is predominantly mediated by TLR4 signaling through the TRIF pathway.
T- and T-/B-cell-deficient mice were used as recipients to study the fate of allogeneic cells very early after transplantation. Donor cell elimination occurred within 3 days in TCRβ/δ-/- and Rag-/- mice, similar to that in wild-type B6 controls, indicating that the early rejection of allogeneic BMCs occurred before T-cell activation and was T- and B-cell independent. The T-cell-independent mechanism was also demonstrated in previous findings in that allogeneic BMC survived very similarly between recipients with T-cell depletion by monoclonal antibodies (mAbs) and naive recipients (12). The disappearance of allogeneic cells in peripheral blood was the result of cytotoxic activity and not due to sequestering in other tissues or the CFSE labeling as confirmed by the controls. These results implicate an effector role for innate immune cells in early BMC rejection.
The innate immune system represents the first line of defense against pathogens. In transplantation, the role of these defenses has not been fully appreciated until recently. Goldstein found that rejection of minor antigen (HY)-incompatible skin grafts is MyD88 dependent and is not mediated by TLR2 or TLR4 (11). They successfully extended their findings to an MHC fully mismatched model, but the costimulatory blockade was required (45). MyD88 is an adaptor molecule essential for signaling via most of the TLR to induce translocation of nuclear factor κ-light-chain enhancer of activated B-cells (NF-κB) and proinflammatory cytokine secretion (24,33). The possible mechanisms for these findings are that activation of innate immunity early during organ transplantation generates a proinflammatory environment that makes recipients more resistant to tolerance induction.
Whether and how the innate immune system recognizes or responds to allogeneic BMCs remain unknown. To our surprise, our findings in mice deficient in TRIF or MyD88 adaptor indicate that allogeneic BMC rejection occurred exclusively through TRIF signaling but not through MyD88 signaling. This is in contrast with prior reports demonstrating the critical role of the MyD88 pathway in the rejection of transplanted skin (45) and lung (31,35). This may reflect the specificity of BMT as cellular transplants compared to other transplants. Moreover, we found that mice deficient in TLR4, but not TLR3, had superior engraftment of allogeneic BMCs compared with wild-type controls. These results suggest that allogeneic BMC transplantation triggers TLR4 and, subsequently, the TRIF pathway, resulting in the activation of innate immune macrophages/NK cells. The innate immune response to allogeneic BMCs via TLR4, but not TLR3, may be a result of their biological locations. TLR4 is cell membrane bound, while TLR3 is associated with the intracellular endosomal membrane (32). Alternatively, it may be due to the fact that bone marrow transplants involve a single cell infusion while organ allografts are transplanted as a primarily vascularized unit.
In transplantation, TLR activation has been demonstrated to generate a proinflammatory environment that makes the recipient less susceptible to tolerance induction (9,11,28). TLR4 activation by lipopolysaccharide (LPS) at the time of costimulation blockade shortens the survival of skin, cardiac, and islet allografts (5,27,39). Although it has been difficult to absolutely exclude microbial contaminants to activate TLR, noninfectious endogenous inflammatory signals have been implicated to contribute to acute allograft rejection (2,10). The endogenous TLR ligands, including heat shock proteins, hyaluronan, and high mobility box group 1 (HMGB1), have been found to activate the innate immune system predominantly via TLR2 and TLR4 (29,38,41). The engagement of hyaluronan with TLR2 and TLR4 has been demonstrated to result in the activation of macrophages (16). However, the endogenous activators released after allogeneic BMT remain unknown and need to be further defined.
NK cells have been shown to play a major role in marrow rejection (7,12,15,19). They express inhibitory and activating receptors that are implicated in engraftment. The role of macrophages and neutrophils in rejection of BMCs has not been defined nor has the TLR signaling pathways. We observed that macrophages contributed significantly to the early elimination of transplanted cells. We further found that depletion of macrophages or NK cells combined with T- and B-cell conditioning-based strategies significantly enhanced alloengraftment. An additive effect was observed when both cellular populations were depleted, suggesting a nonredundant role. Targeting of innate immunity in addition to adaptive immunity completely eliminated the need for TBI to establish chimerism. Alternatively, depleting both NK cells and macrophages plus targeting T-cells may provide sufficient “space” in recipient marrow for the donor stem cells to populate. Our data strongly support the hypothesis that two barriers involving innate and adaptive immunity coexist in response to allogeneic BMCs. Thus, chimerism could be achieved with minimal regimen-related morbidity by targeting both the innate and adaptive arms of the immune response. This safe approach to condition recipients to establish mixed hematopoietic chimerism could have significant impact in the clinical application of BMT to treat hemoglobinopathies and autoimmune disorders and induce tolerance to solid organ and cellular transplants. Moreover, the restored cytotoxicity in TRIFLps2/Lps2 recipients that received either F4/80+ or NK1.1+ cells suggests that the early rejection of allogeneic BMCs by macrophages or NK cells is TRIF signaling dependent.
Innate immunity serves as an important link between antigen-dependent and antigen-independent responses. The cytokine environment plays a primary role to determine the link (44). When an organ or tissue is transplanted, an inflammatory response is induced through an intricate cascade of events initiated by cytokines. These signals are crucial for the generation of a potent adaptive immune response. We found that IL-6 was upregu-lated early within 12 h in response to allogeneic BMC infusion. IL-6 promotes T-helper 2 (Th2) responses and also plays a critical role in the promotion of Th17 differentiation in combination with transforming growth factor (TGF)-β (1,53). Modulation of macrophage function is linked to increased production of IL-17 by CD4 T-cells (34). A counterregulatory relationship between Th17 and regulatory T-cells (Treg) has been established. Thus, IL-6-mediated enhancement of Th17 would likely decrease the acceptance of transplants. Indeed, IL-17 secreted by Th17 cells has been demonstrated in acute transplant rejection (6,54). In addition, we found that the serum levels of MCP-1 increased in response to allogeneic BMT. MCP-1 gene is one of the IFN-inducible genes that function in a MyD88-independent manner (8,18). Type I IFN activation is involved in the innate immune response through TRIF (13). The increased MCP-1 may also chemoattract more macrophages and NK cells into the site of inflammation (4), amplifying the proinflammatory responses.
In summary, we show that BMC allorejection was critically dependent on TLR4/TRIF signaling. We further demonstrated that innate immune cell populations of macrophages and NK cells mediate early BMC rejection. By targeting both of these innate cell populations as conditioning, allogeneic BMC engraftment was achieved without irradiation. Based on these findings, we propose that the allogeneic BMT may cause the release of certain damage-associated molecular patterns into the extracellular matrix that subsequently trigger the TLR4 signaling pathway of both macrophages and NK cells. The activation of TLR4 leads to TRIF-dependent NF-κB activation and subsequently the release of MCP-1 and IL-6. The increased MCP-1 chemoattracts more NK cells and macrophages into the site of inflammation; thus, the activated innate immune cells by TLR4 and subsequent induction of proinflammatory cytokines eradicate allo-BMCs. The findings of the role of innate immunity in BMC rejection may allow the development of reduced intensity conditioning regimens to promote engraftment with reduced toxicity.
Footnotes
Acknowledgments
The authors thank Haval Shirwan, Thomas C. Mitchell, and Jill Suttles for the review of the manuscript and their helpfulcomments; Carolyn R. Casella and Paula M. Chilton for the experiment design on the TRIF pathway study; Carolyn DeLautre for the manuscript preparation; the staff of the animal facility for outstanding animal care; and the staff in Dr. Mitchell's lab for breeding the TRIFlps2/lps2 and MyD88-/- mice. This work was supported in part by NIH R01 DK069766, NIH 5RO1 HL063442, and JDRF 1-2006-1466; the Department of the Navy, Office of Naval Research; the Department of the Army, Office of Army Research [Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the Office of Army Research. This publication was made possible by Award No. W81XWH-07-1-0185 from the Office of Army Research]; the Commonwealth of Kentucky Research Challenge Trust Fund; the W. M. Keck Foundation; and the Jewish Hospital Foundation. S. Ildstad is the founding scientist and director of Regenerex, a biotech start-up company; it has not been capitalized. L. Bozulic is a research scientist and employee of Regenerex. All other authors declare no conflict of interest.