
Abstract
Allospecific T memory cell responses in transplant recipients arise from environmental exposure to previous transplantation or cross-reactive heterologous immunity. Unfortunately, these memory responses pose a significant barrier to the survival of transplanted tissue. We have previously reported that concurrent inhibition of CD154 and LFA-1 suppresses primary CD8-dependent rejection responses that are not controlled by conventional immunosuppressive strategies. We hypothesized that CD154- and LFA-1-mediated inhibition, by targeting activation as well as effector functions, may also be efficacious for the control of alloreactive CD8+ T-cell responses in sensitized hosts. We found that treatment with anti-LFA-1 mAb alone enhanced transplant survival and reduced CD8-mediated cytotoxicity in sensitized CD4 KO recipients. However, treatment with anti-CD154 mAb alone did not have an effect. Notably, when both CD4- and CD8-dependent rejection pathways are operative (wild-type sensitized recipients), LFA-1 significantly inhibited CD8-mediated in vivo allocytotoxicity but did not correspond with enhanced hepatocyte survival. We hypothesized that this was due to alloantibody-mediated rejection. When anti-LFA-1 mAb treatment was combined with macrophage depletion, which we have previously reported impairs alloantibody-mediated parenchymal cell damage, in vivo cytotoxic effector function was significantly decreased and was accompanied by significant enhancement of hepatocyte survival in sensitized wild-type recipients. Therefore, LFA-1 is a potent therapeutic target for reduction of CD8-mediated cytotoxicity in sensitized transplant recipients and can be combined with other treatments that target non-CD8-mediated recall alloimmunity.
Introduction
Memory T-cells pose a significant barrier to the induction and maintenance of long term survival of allografts in sensitized patients. Whether sensitization occurs through previous transplant or cross-reactive heterologous immunity (1,49), cluster of differentiation 8-positive (CD8+) T-cell memory responses have been linked to poor allograft survival in both clinical and experimental studies (6,59). Novel immunotherapeutic approaches that successfully regulate immune responses in sensitized recipients are necessary to achieve long term allograft survival in this growing transplant population.
Many strategies to achieve immune tolerance have employed a variety of approaches to interfere with CD8+ T-cells including T-cell depletion (22,23), costimulation blockade (38,66), inhibition of T-cell proliferation (32,40, 50), and induction of regulatory T-cells (54). Unfortunately, memory T-cells are more difficult to control than naive T-cells due to their reduced threshold for activation, enhanced magnitude of effector function, and enhanced survival in circulation (12,15,21,29,57,60). Several experimental studies have reported the efficacy of treatment regimens that effectively prolong primary allograft survival but fail to prevent rejection in sensitized recipients. For example, costimulatory blockade with anti-CD154 (CD40L) monoclonal antibody (mAb) induces long term survival of cardiac allografts in naive hosts, but not in recipients previously sensitized with donor-matched skin grafts (70). Similarly, cardiac allograft survival was not prolonged by costimulatory blockade immunotherapy consisting of donor-specific transfusion (DST) in combination with anti-CD154 mAb treatment when recipients received prior adoptive transfer of memory T-cells (13,61). Furthermore, immune tolerance induced by mixed chimerism is blocked by memory CD8+ T-cells in nonhuman primate kidney allograft recipients (34). CD8+ T-cells have also been a barrier to tolerance induced by mesenchymal stem cells transfer (14). Jones et al. has recently shown that memory CD8+ T-cells can be slowed, but not abrogated, by regulatory T-cells following the depletion of polymorphonuclear cells (30). It has been suggested that any unrestrained phase of alloreactive memory T-cell responses, including activation or effector function specific to memory cells, can contribute to graft loss (59). In addition, memory CD4+ T-cell responses can provide help to B-cells and lead to alloantibody production in the absence of CD40/CD154 interaction (52). Consequently, it is important to understand the mechanisms of T memory cell destruction of allografts for future design of immunotherapeutic strategies that effectively regulate recall alloimmunity.
We have reported that hepatocyte allografts induce CD8+ T-cell-mediated rejection responses including CD4-independent CD8+ T-cells. We and others have shown that CD4-independent CD8+ T-cells are resistant to therapies that readily control CD4-dependent rejection responses (4,7,10,31,67). Hepatocytes also induce CD8-independent, CD4-dependent rejection responses, which mediate graft damage through alloantibody-dependent macrophage-mediated cytotoxicity (26,28). Despite resistance to conventional therapies, previous work in our laboratory has shown short-term treatment (days 0–7 posttransplant) targeting lymphocyte function-associated antigen 1 (LFA-1) and CD154 effectively inhibits primary hepatocyte allograft rejection, CD8+ T-cell-mediated cytotoxicity, and antibody production in wild-type and CD4-deficient recipients. In fact, short-term combination therapy resulted in synergistic long term graft survival (>90 days) in the majority of recipients (39,67). Similar results have been reported with LFA-1 and CD154 blockade inhibiting primary rejection in other transplant models such as islet, bone marrow, and nerve allografts (36,37,41,58,65). The induced graft acceptance outlasts clearance of the treatment antibodies and resists challenge with adoptively transferred naive and primed CD4+ and CD8+ T-cells [unpublished observations and (45,46)]. The surprisingly strong efficacy of the combination immunotherapeutic strategy for control of rejection in recipients adoptively transferred with primed T-cells raised the issue of whether this treatment strategy could suppress rejection in sensitized recipients.
Following a second transplant, prompt secondary rejection responses in vivo occur through CD4-dependent and CD4-independent CD8+ T-cells as well as CD8-independent responses (i.e., alloantibody) (25,26,28). In the current studies, we utilized the hepatocyte allograft model to assess the efficacy of targeting CD154 and LFA-1 on CD8+ T-cells in both wild-type and CD4-deficient sensitized mice. These studies are the first to report that short term immunotherapy by interfering with LFA-1 successfully suppresses rejection by CD8+ T-cells in sensitized CD4-deficient recipients. This enhanced graft survival correlates with the nearly complete inhibition of alloreactive CD8+ T-cell cytotoxic effector activity. LFA-1 interference also inhibited the cytotoxic effector activity of CD8+ T-cells in sensitized wild-type recipients. However, hepatocyte allograft survival was not prolonged in sensitized wild-type recipients treated with anti-LFA-1 mAb alone likely due to the presence of alloreactive antibody. The existence of preformed and/or memory response alloantibody in sensitized wild-type recipients led us to target macrophage-mediated antibody-dependent cellular cytotoxicity (ADCC). Since we have recently published that alloantibody-mediated parenchymal cell damage is macrophage-dependent (28), we tested the effect of macrophage depletion in combination with anti-LFA-1 mAb immunotherapy on in vivo cytotoxic effector function and hepatocyte transplant survival in sensitized wild-type recipients. We found that treatment with anti-LFA-1 mAb in combination with blocking macrophage-mediated ADCC successfully prolonged graft survival in sensitized recipients and abrogated in vivo cytotoxic effector function. This novel approach of inhibiting CD8- and non-CD8-mediated in vivo cytotoxic effector activity appears to be a promising intervention to inhibit graft rejection in sensitized transplant recipients.
Materials and Methods
Experimental Animals
FVB/N (H-2q, Taconic, Hudson, NY, USA), C57BL/6 (H-2b, Jackson, Bar Harbor, ME, USA), and CD4 knockout (KO) (H-2b, Jackson) mouse strains were used in this study. Transgenic FVB/N mice expressing human α-1 antitrypsin (hA1AT-FVB/N, H-2q; created, bred, and maintained at the W. M. Keck Genetics Research Facility of the Neurobiotechnology Center, The Ohio State University) were the source of “donor” hepatocytes, as previously described (7,8). Mice that were 6–9 weeks of age were used in all experiments. All experiments were performed in compliance with the guidelines of the Institutional Laboratory Animal Care and Use Committee of The Ohio State University (Protocol 2008A0068).
Hepatocyte Isolation and Purification
Hepatocyte isolation and purification were performed as previously described (7,8). Briefly, the donor liver was perfused with 0.09% (EGTA)-containing calcium-free salt solution followed by 0.05% collagenase type 4 in 1% albumin (all from Sigma, St. Louis, MO, USA). Liver tissue was minced, filtered, and purified on a 50% Percoll gradient (Pharmacia Biotech, Uppsala, Sweden). Hepatocyte viability and purity were both consistently greater than 99%.
Hepatocyte Transplantation and Monitoring of Hepatocyte Graft Survival
Donor FVB/N (H-2q) hepatocytes (2×106) were transplanted into wild-type (C57BL/6, H-2b) and CD4 KO (H-2b) recipients by intrasplenic injection with circulation of donor hepatocytes to the host liver, as previously described (8). Graft survival was determined by detection of the secreted transgenic reporter product hA1AT in serial recipient serum samples by ELISA as previously described (7,8). However, the capture antibody, rabbit anti-human A1AT, is now obtained from Sigma. Sustained serum hA1AT levels indicated continued graft survival, while graft loss was determined by the time point at which host serum hA1AT was less than 0.5 mg/ml.
Sensitized Hosts
In sensitization studies, mice were transplanted with hepatocytes on day 0 and allowed to reject the transplant without treatment. A subsequent transplant of hepatocytes was performed 28 days after the primary hepatocyte transplant.
In Vivo Cytotoxicity Assay
Assay for cytolytic T-cell function in vivo was modified from published methods (48,64), as previously described (39). Syngeneic target splenocytes were isolated from wild-type C57BL/6 mice and were stained with 0.2 mM carboxyfluorescein diacetate succinimidyl ester (CFSElow, Molecular Probes, Eugene, OR, USA). Allogeneic target splenocytes were isolated from FVB/N mice and were stained with 2.0 mM CFSE (CFSEhigh). Allograft recipient mice and control naive mice received 20×106 CFSE-labeled syngeneic and 20×106 CFSE-labeled allogeneic target splenocytes by tail vein injection. Splenocyte target cell CFSE staining and ratios were verified by flow cytometric analysis prior to injection. Eighteen hours after CFSE-labeled target cell injection, splenocytes from hepatocyte recipients were retrieved and analyzed by flow cytometry (FACS Calibur, Becton Dickinson, Franklin Lakes, NJ, USA) with gating on CFSE-positive splenocytes. Percent allospecific cytotoxicity was calculated as previously described (25).
Assay of Allospecific Antibody
IgG antibody from recipient serum was tested for allospecificity first by incubation with allogeneic FVB/N target splenocytes. The percent binding of total IgG to splenocyte targets was determined by a second incubation with FITC-conjugated goat anti-mouse IgG Fc (Organon Teknika, Durham, NC, USA) and analysis by flow cytometry. Alloantibody level is represented as the percentage of target cells labeled by secondary fluorescent Ab as described previously (26).
In Vivo Host Treatments
CD4 KO and wild-type sensitized hosts (prior hepatocyte allograft rejection) were treated with monoclonal antibodies (mAbs; Bioexpress Cell Culture Services, West Lebanon, NH, USA) targeting CD154 (MR1, 1 mg IP, days 0, 2, 4, 7 relative to secondary transplantation), LFA-1 (M17/4.4.11.9, 300 μg IP, days 0–6), and/or CD4 (GK1.5; 250 μg, days -2, -1, 7, 14) prior to and after transplantation with a donor-matched secondary hepatocyte allograft that occurred 28 days post-primary transplant.
Treatment at the Time of Peak In Vivo Cytotoxicity
In separate studies, CD4 KO and wild-type primary hepatocyte allograft recipients were treated 24 h prior to the in vivo cytotoxicity assay. CD4 KO recipients were treated with anti-CD154 mAb (1 mg, IP, days 4 and 5) posttransplant, and/or anti-LFA-1 mAb (300 mg, IP, days 4 and 5) preceding the in vivo cytotoxicity assay on day 5 posttransplant. Wild-type recipients were treated with anti-CD154 mAb (1 mg, IP, days 6 and 7) posttransplant and/or anti-LFA-1 mAb (300 mg, IP, days 6 and 7) posttransplant previous to the in vivo cytotoxicity assay conducted day 7 posttransplant. The in vivo cytotoxicity assay was also performed following secondary transplantation in both CD4 KO and wild-type mice on day 7 (maximal cytotoxicity known to be sustained from day 3 to day 21 in retransplanted mice) (25).
To determine the contribution of in vivo allocytotoxicity dependent on alloantibody (and which we have previously reported to be macrophage dependent) (28), wild-type hepatocyte recipients were depleted of host macrophages (0.2 ml liposomal clodronate, IP; a kind gift of Roche Diagnostics GmbH, Mannheim, Germany) 24 h prior to the in vivo cytotoxicity assay, as previously reported. To determine the role of host macrophages in the effector phase of recall hepatocyte rejection, sensitized hepatocyte recipients were transiently depleted of host macrophages (0.2 ml liposomal clodronate, IP on days 5, 9, 13, 17, 21 posttransplant) while monitoring survival of a secondary hepatocellular allograft. The macrophage population is significantly reduced in the transplant site (liver) and is confirmed by analysis of F4/80+ (CI:A3-1 Caltag Laboratories, Burlingame, CA, USA), as previously documented (28). Liposomal clodronate and control liposomes containing only PBS were prepared as previously described (62).
Statistical Analysis
Graft survival between experimental groups was compared using Kaplan–Meier survival curves and log-rank tests. Other comparisons between experimental groups were performed using analysis of variance (ANOVA). SAS software (version 9.2, 2008) was used for both log-rank test and ANOVA (SAS Institute, Cary, NC, USA). All multiple comparisons were adjusted for appropriately using Bonferroni's method of adjustment and an adjusted p < 0.05 was considered significant. Summary statistics are listed as the mean plus or minus the standard error.
Results
CD8+ T-Cell-Mediated Graft Rejection in Sensitized CD4 KO, But Not Wild-Type Recipients, Is Delayed by Combination Anti-CD154 and Anti-LFA-1 mAb Immunotherapy
We have previously reported that combination immunotherapy with anti-CD154 mAb and anti-LFA-1 mAb successfully prevents primary rejection by immunoresistant CD4-independent CD8+ T-cells (CD4 KO mice) or by CD8-mediated rejection in wild-type mice (39,67). To investigate the efficacy of combination therapy targeting CD154 and LFA-1 for suppression of CD8-mediated rejection in sensitized recipients, FVB/N donor hepatocytes (H-2q) were first transplanted into CD4 KO and wild-type C57BL/6 hosts (H-2b). Primary hepatocyte allografts were monitored for graft survival. All grafts were rejected within 7–14 days posttransplant in these untreated hosts [median survival time (MST) = 14 days for CD4 KO and 10 days for wild-type recipients]. We have previously shown that rejection in CD4 KO recipients is CD8+ T-cell-dependent and concurrent with the development of in vivo CD8+ T-cell-mediated allospecific cytotoxic activity (25). Approximately 2 weeks following rejection of the primary graft, these rejector hosts were retransplanted with a second FVB/N allogeneic hepatocyte transplant (28 days after the primary transplant) and treated with an 8-day regimen of anti-CD154 and anti-LFA-1 mAbs (anti-CD154 mAb, 1 mg IP, days 0, 2, 4, and 7; anti-LFA-1 mAb, 300 mg IP, days 0–6, relative to the second transplant). A separate group of untreated sensitized recipients served as controls. Graft survival was monitored in both groups. Control sensitized recipients rejected hepatocyte allografts with enhanced kinetics in comparison to the primary graft, consistent with a recall response (MST = 10 days vs. 14 days for CD4 KO; MST = 7 days vs. 10 days for wild-type mice; p < 0.05 for both). The CD4 KO recipient group treated with anti-CD154 and anti-LFA-1 mAb immunotherapy demonstrated prolonged graft survival in all sensitized hosts (MST of 28 days; p = 0.050 compared to secondary rejection in untreated hosts) (Fig. 1A). On the other hand, wild-type recipients cotreated with anti-CD154 and anti-LFA-1 mAb immunotherapy did not manifest any enhanced graft survival (MST = 7 days) (Fig. 1B).
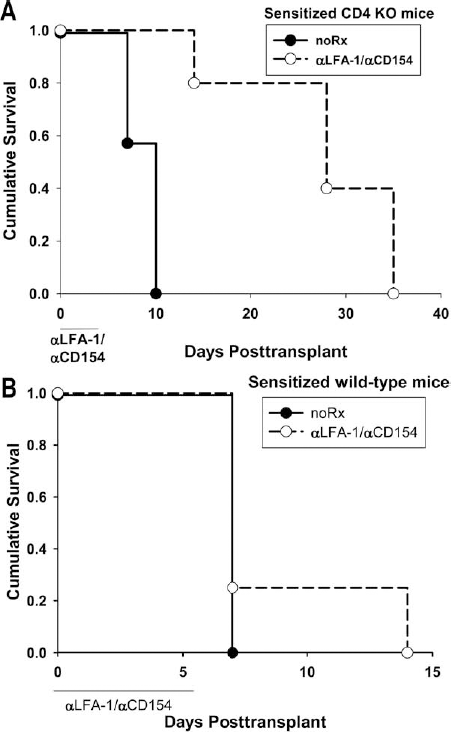
Combination treatment with anti-CD154 and anti-LFA-1 mAbs delays CD8+ T-cell-mediated hepatocyte allograft rejection in sensitized CD4 KO recipients but not in sensitized wild-type recipients. FVB/N (H-2q) allogeneic hepatocytes were transplanted into two groups of untreated naive wild-type and cluster of differentiation 4 knockout (CD4 KO; H-2b) hosts on day 0. Primary hepatocyte allografts were rejected in wild-type and CD4 KO hosts with median survival time (MST) of 10 and 14 days, respectively (p = ns between the groups). Following rejection, all host groups were transplanted with a second FVB/N hepatocellular allograft (day 28 post-primary transplant), and secondary graft survival was monitored. Cohorts of wild-type and CD4 KO rejectors were treated with anti-CD154 and anti-lymphocyte function-associated antigen 1 (LFA-1) mAbs (0–7 days post-secondary transplant). The untreated wild-type and CD4 KO hosts rejected the second graft with MST of 7 and 10 days, respectively (p < 0.05 relative to primary rejection kinetics). (A) CD4 KO hosts treated with aCD154/αLFA-1 mAb (n = 5) demonstrated prolonged allograft survival with MST = 28 days (p = 0.05 in comparison to allograft survival in untreated sensitized hosts; n = 7). (B) Cotreatment had no effect on rejection in sensitized wild-type mice (n = 4, p = ns; n = 8 for untreated controls).
Targeting LFA-1 (But Not CD154) Alone Delays Second Set Rejection in CD4 KO Recipients
In order to clarify whether or not enhanced graft survival observed in sensitized CD4 KO recipients treated with anti-CD154 and anti-LFA-1 mAbs required combination immunotherapy, we tested the efficacy of each of the agents alone. Previous work in our laboratory has shown that short-term monotherapy targeting CD154 or LFA-1 effectively controls primary CD8-dependent hepatocyte allograft rejection in CD4 KO hosts with MST of 32 and 77 days, respectively (20,67). Combination immunotherapy with both agents resulted in synergy and induced long term survival (MST > 90 days) of hepatocellular allografts in the majority of primary recipients (67). In order to test the efficacy of monotherapy with anti-CD154 or anti-LFA-1 mAb alone in sensitized recipients, CD4 KO recipients were first transplanted with FVB/N hepatocyte allografts without treatment. After rejection, sensitized recipients were then treated with either anti-CD154 mAb (days 0, 2, 4, 7 relative to secondary transplant) or anti-LFA-1 mAb (days 0–6 relative to secondary transplant) and received a secondary donor-matched hepatocyte allograft (day 28 post-primary hepatocyte transplant). Sensitized hepatocyte recipients treated with anti-CD154 mAb did not demonstrate enhanced survival of secondary allografts (MST = 7 days) in comparison to untreated sensitized CD4 KO transplant recipients. In contrast, treatment with anti-LFA-1 mAb alone significantly prolonged survival of secondary hepatocyte allografts to a similar degree as combination therapy (MST = 35 days for both anti-LFA-1 mAb; p = 0.039) (Fig. 2A). Thus, the observed efficacy of combination therapy for suppression of CD4-independent, CD8-dependent rejection in sensitized recipients can be attributed to the effect of anti-LFA-1 mAb treatment alone. Similar results of enhanced allograft survival following anti-LFA-1 treatment were observed in CD4-depleted sensitized wild-type mice (MST = 24 days, p = 0.031) (Fig. 2B).
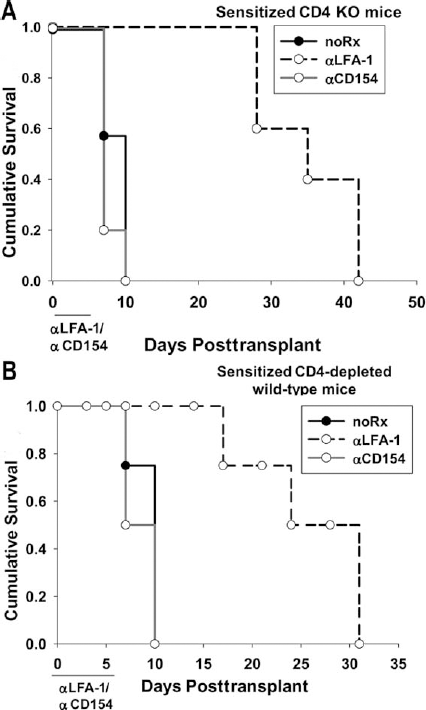
Interference with LFA-1, but not CD154, delays CD8+ T-cell-mediated allograft rejection in sensitized CD4 KO recipients. (A) FVB/N (H-2q) hepatocytes were transplanted into two groups of untreated naive CD4 KO (H-2b) hosts. Following hepatocyte rejection, the groups were transplanted with a second FVB/N hepatocyte allograft on day 28 and were treated with either anti-CD154 or anti-LFA-1 monoclonal antibody days 0–7 post-secondary transplant. Secondary graft survival was monitored. Anti-CD154 mAb treatment did not prevent second set rejection (MST = 7 days). Anti-LFA-1 mAb-treated hosts demonstrated prolonged survival of secondary allografts in sensitized hosts (MST = 35 days; p = 0.039 compared to untreated sensitized hosts; n = 5–7 for all groups). (B) Similar results were found with CD4-depleted (GK1.1, 100 mg on days -2, -1, 7, 14) wild-type recipients where anti-LFA-1 mAb significantly enhanced allograft survival (MST = 24 days; p = 0.031; n = 4 for all groups).
Targeting LFA-1 Inhibits CD8-Mediated Cytotoxic Effector Activity in Wild-Type and CD4 KO Primary Transplant Recipients
LFA-1 signaling has been shown to contribute to CD8+ T-cell immune reactivity at multiple steps, such as lymphocyte trafficking, activation, and/or effector function (17,47,63,71). However, these studies did not explicitly clarify the role of anti-LFA-1 mAb on the control of effector function in vivo, as the treatments were used in vitro or given prior to transplantation and thus could have indirectly affected downstream effector mechanisms. We hypothesized that the effective control of CD4-independent CD8+ cytotoxic T lymphocytes (CTLs) in sensitized recipients by treatment with anti-LFA-1 mAb is due to abrogation of CD8+ cytolytic effector function. We have previously shown that in vivo CD8+ T-cell-mediated cytotoxicity develops during hepatocyte allograft rejection in both wild-type and CD4 KO recipients. This response is detectable by day 3 in both recipient groups and continues to rise in magnitude in wild-type hosts, peaking by day 7 posttransplant (25). To investigate the role of LFA-1 on in vivo CD8+ T-cell-mediated allospecific cytolytic activity and effector function, FVB/N hepatocytes were transplanted into untreated wild-type primary recipients. Recipients were treated with anti-LFA-1 mAb on posttransplant days 6 and 7 (after maximal cytotoxicity has developed). In vivo cytolytic activity was assessed within 24 h of the antibody treatment. As shown in Figure 3, high levels of in vivo allocytotoxicity are generated in control wild-type hosts 1 week posttransplant (89.8 ± 4.6%). This cytotoxic effector function is LFA-1-dependent since treatment with anti-LFA-1 mAb significantly reduced allospecific in vivo cytotoxic activity in comparison to untreated controls (17.5 ± 6.1%; p < 0.0001). Similarly, in vivo allospecific cytotoxic effector function mediated by CD4-independent CD8+ T-cells [in CD4 KO mice; tested on day 5 peak cytotoxicity (peak cytotoxicity occurs days 3–5 in CD4 KO recipients)] is also inhibited by treatment with anti-LFA-1 mAb alone (8.7 ± 3.5%; days 4 and 5) as compared to untreated controls (26.9 ± 3.9%, p = 0.0114) (Fig. 3). In contrast, treatment with anti-CD154 mAb alone 24 h prior to the in vivo cytotoxicity assay did not reduce cytotoxicity in wild-type (74.7 ± 5.5%) or CD4 KO (22.7 ± 3.9%) recipient mice (p = ns for both). It is not surprising that delayed CD154 inhibition did not inhibit cytotoxicity as it is largely considered to block costimulation (67). Based on these observations in primary recipients, we investigated the effect of anti-LFA-1 mAb on in vivo cytotoxic effector function in sensitized recipients.

Targeting LFA-1 inhibits primary in vivo CD8+ T-cell-mediated cytotoxic effector function in both wild-type and CD4-deficient recipients. FVB/N hepatocyte allografts were transplanted into wild-type (H-2b) or CD4 KO (H-2b) recipients, which develop peak in vivo CD8-mediated allocytotoxicity on days 7 and 5 posttransplant, respectively. Wild-type recipient groups were treated with anti-CD154 mAb or anti-LFA-1 mAb on days 6 and 7 posttransplant. At 24 h after treatment (on day 7 posttransplant), recipients were tested for in vivo cytotoxic effector function. Some recipients were left untreated for comparison (NoRx). Treatment with anti-LFA-1 mAb significantly inhibited allospecific cytotoxicity (17.5 ± 6.1%, n = 4) as compared to untreated controls (89.8 ± 4.6%, n = 8; p < 0.0001). Effector phase treatment with anti-LFA-1 mAb on days 3–5 posttransplant in CD4 KO recipients significantly reduced CD8+ T-cell in vivo effector function assayed on day 5 posttransplant (8.7 ± 3.5%, n = 5) as compared to untreated controls (26.9 ± 3.9%, n = 4; p = 0.0114). In contrast, treatment with anti-CD154 mAb alone 24 h prior to the in vivo cytotoxicity assay did not reduce cytotoxicity in wild-type (74.7 ± 5.5%, n = 5) or CD4 KO (22.7 ± 3.9%, n = 4) recipient mice (p = ns for both). ∗Statistically significant results. A line is used to depict the groups compared.
Targeting LFA-1 Inhibits CD8-Mediated Cytotoxic Effector Activity in Wild-Type and CD4-Deficient Sensitized Transplant Recipients
Targeting LFA-1 in CD4-deficient sensitized recipients prolongs graft survival (Fig. 2). However, treatment with anti-LFA-1 mAb has no effect on the survival of hepatocytes in sensitized wild-type mice (Fig. 1). We hypothesized that sensitized CD8+ T-cells that mature in a wild-type recipient may develop effector mechanisms that are independent of LFA-1 signaling, while CD8+ T-cells that mature in a CD4-deficient recipient develop CD8-mediated cytotoxicity that is LFA-1-dependent. To investigate the role of LFA-1 on CD8+ T-cell-mediated allospecific cytolytic activity in sensitized recipients, FVB/N hepatocytes were transplanted into untreated wild-type or CD4 KO rejector mice. On days 6 and 7 after the second transplant, sensitized recipients were then treated with anti-LFA-1 mAb. It is important to note that maximal cytotoxicity is achieved by day 3 and is maintained over time (≥day 21) in both wild-type and CD4-deficient sensitized recipients, exhibiting a memory-like cytotoxic phenotype (25). In vivo cytolytic activity was assessed on day 7, within 24 h of anti-LFA-1 mAb treatment. As shown in Figure 4A, high levels of cytotoxicity are generated in untreated wild-type (97.7 ± 4.3%) and CD4 KO recipient hosts (31.4 ± 3.0%). Treatment with anti-LFA-1 mAb, however, significantly reduced cytolytic activity in vivo in sensitized wild-type recipients (32.9 ± 4.3%; p < 0.0001) and abrogated cytotoxicity in sensitized CD4 KO mice (6.1 ± 3.7%, p = 0.0004) (Fig. 4A). We postulate this is a direct inhibition of CD8+ CTL effector function as the cytotoxicity is already maximal at day 3 postretransplant (after sufficient CD8+ T-cell activation and proliferation), which occurs prior to the administration of anti-LFA-1 mAb treatment in this experiment. Unlike results for sensitized CD4 KO recipients, despite the efficacy of anti-LFA-1 mAb treatment for significant reduction of in vivo allospecific cytotoxic effector function in sensitized wild-type recipients, this did not translate into enhanced graft survival (Fig. 1B). We postulated that the failure to prolong graft survival in sensitized wild-type recipients could be due to alloantibody-mediated cytotoxicity or resistant CD8+ T-cells not regulated by anti-LFA-1 mAb.

Targeting LFA-1 inhibits secondary in vivo CD8+ T-cell-mediated cytotoxic effector function in both wild-type and CD4-deficient sensitized recipients. FVB/N hepatocyte allografts were transplanted into wild-type (H-2b) hosts or CD4 KO (H-2b) mice. After 28 days, hepatocyte recipients were retransplanted with FVB/N hepatocytes. (A) Sensitized wild-type recipient groups were depleted of macrophages (liposomal clodronate, lipo clod) on day 5 and/or treated with anti-LFA-1 mAb on days 6 and 7 postretransplant. At 24 h later, (on day 7 posttransplant) recipients were tested for in vivo cytotoxic effector function. Some recipients were left untreated for comparison (NoRx). Treatment with anti-LFA-1 mAb results in significant reduction of cytotoxic activity (32.9 ± 4.3%; n = 4) as compared to untreated controls (97.7 ± 4.3%, n = 5; p < 0.0001). Effector phase treatment with anti-LFA-1 mAb days 3–5 posttransplant in CD4 KO recipients significantly reduced CD8+ T-cell in vivo effector function assayed on day 5 posttransplant (6.1 ± 3.7%, n = 4) as compared to untreated controls (31.4 ± 3.0%, n = 6; p = 0.0004). Treatment with both anti-LFA-1 mAb and liposomal clodronate resulted in significant reduction of allocytotoxicity in sensitized wild-type hosts (16.4 ± 4.3%, n = 5; p = 0.0468) in comparison to sensitized wild-type recipients treated with anti-LFA-1 mAb alone (32.9 ± 4.3%). In contrast, allocytotoxicity in anti-LFA-1 mAb-treated sensitized CD4 KO hosts was already greatly reduced and combination therapy with both liposomal clodronate and anti-LFA-1 mAb did not further reduce allocytotoxicity (3.7 ± 3.7%, n = 4; p = ns) compared to those treated with anti-LFA-1 mAb alone (6.1 ± 3.7%). CD8 depletion reduced in vivo cytotoxicity to 12.1 ± 4.8% in wild-type recipients (n = 4; p < 0.0001) and to 2.5 ± 3.3% in CD4 KO recipients (n = 5; p < 0.0001). (B) A cohort of sensitized wild-type recipients was analyzed for alloantibody production on day 3 (prior to treatment or the in vivo cytotoxicity assay). Mice then were subsequently treated with anti-LFA-1 or anti-CD8 mAb and tested for in vivo cytotoxicity. The degree of in vivo cytotoxicity following treatment with anti-LFA-1 or anti-CD8 mAb is significantly higher among those sensitized recipients that have the highest amounts of alloantibody (average cytotoxicity = 35.6 ± 7.3%, average alloantibody 32.5 ± 7.4%, n = 4) as compared to lower alloantibody producers (average cytotoxicity = 14.1 ± 4.0%, average alloantibody = 5.6 ± 1.2%, n = 5; p = 0.026). ∗Statistically significant results. A line is used to depict the groups compared.
Combination therapy with anti-LFA-1 mAb and macrophage depletion results in synergistic suppression of in vivo cytotoxic effector function and prolonged graft survival in wild-type sensitized transplant recipients. To determine whether the observed residual cytotoxicity in anti-LFA-1 mAb-treated sensitized recipients was mediated by resistant CD8+ T-cells, CD8+ T-cells were depleted in sensitized wild-type mice and assayed for in vivo cytotoxic effector function. In vivo cytotoxicity was reduced from 97.7 ± 4.3% in untreated mice to 12.1 ± 4.8% in CD8-depleted recipients (clone 53.6.72; p < 0.0001) (Fig. 4A), which is lower than cytotoxicity observed in anti-LFA-1-treated recipients (32.9 ± 4.3%), suggesting the existence of LFA-1-independent CD8+ T-cells. However, CD8-depleted sensitized recipients still reject hepatocytes without enhanced survival (MST = day 7, p = ns). We have previously shown that, even in the absence of CD8+ T-cells, donor-specific antibodies mediate rejection of allogeneic parenchymal cells in vivo through a macrophage-ADCC mechanism (28). This possibility is supported by the observations that the inhibition of cytotoxic effector function in anti-LFA-1 mAb or CD8-depleted treated sensitized wild-type recipients was incomplete and also by the detection of alloantibody in the serum of these recipients. Of note, there is no detectable alloantibody in sensitized CD4 KO recipients as antibody production is CD4-dependent in our model (26). In addition, CD4+ T-cells do not contribute directly to in vivo cytotoxic effector function in wild-type recipients (25,28). Anti-LFA-1 mAb or anti-CD8 mAb treated sensitized wild-type recipient mice exhibited residual in vivo cytotoxicity that correlated with alloantibody levels, in a dose-dependent fashion, of these respective groups (Fig. 4B). To determine whether graft loss in anti-LFA-1 mAb treated sensitized wild-type recipients could be due to alloantibody-dependent, macrophage-mediated allograft damage, sensitized mice were retransplanted and treated with anti-LFA-1 mAb (300 μg, IP) and concurrently depleted of macrophages (liposomal clodronate; 0.2 ml, IP) or treated with control liposomal PBS as a vehicle control (0.2 ml; negative control) 48 h prior to the in vivo cytotoxicity assay. Macrophage depletion was confirmed as previously described (28). The addition of liposomal clodronate to anti-LFA-1 mAb treatment decreased the in vivo cytotoxicity from 32.9 ± 4.3% in anti-LFA-1 treated sensitized recipients to 16.4 ± 4.3% in combination treated sensitized wild-type recipients (p = 0.0468) (Fig. 4A). As expected, the addition of liposomal clodronate to anti-LFA-1 mAb treatment did not significantly affect in vivo allocytotoxicity (3.7 ± 3.7%) in sensitized CD4 KO recipients, which was already low (6.1 ± 3.7%). Liposomal clodronate treatment alone did not reduce cytotoxicity in sensitized wild-type (91.6 ± 5.5%) or CD4 KO recipients (27.5 ± 4.6%).
Given the significant inhibition of in vivo cytotoxic effector function, we hypothesized that liposomal clodronate and anti-LFA-1 mAb cotreatment would significantly enhance the survival of transplanted hepatocytes in sensitized wild-type recipients. We found that sensitized wild-type recipient mice treated with both anti-LFA-1 mAb (days 0–6) and liposomal clodronate (days 5, 9, 13, 17, 21) demonstrated enhanced hepatocyte survival as compared to controls treated with anti-LFA-1 mAb and liposomal PBS (MST = 30 days vs. 7 days, p = 0.012) (Fig. 5A). Interestingly, the previously reported enhanced hepatocyte survival in CD8 KO recipients that are treated with liposomal clodronate (MST = 25 days) closely mimics that of anti-LFA-1 mAb treatment combined with liposomal clodronate treatment in sensitized wild-type CD8-sufficient recipients (28). As expected, due to low antibody levels in CD4 KO recipients, this combination therapy did not further enhance hepatocyte survival in CD4 KO sensitized recipients as compared to anti-LFA-1 mAb treatment alone (MST = 35 days vs. 35 days) (Fig. 5B). Liposomal clodronate treatment alone did not enhance hepatocyte survival in sensitized wild-type (MST = day 7 vs. day 7) or CD4 KO recipients (MST = day 10 vs. day 10).

Cotreatment with anti-LFA-1 mAb and macrophage depletion results in enhanced hepatocyte survival in wild-type sensitized recipients. Wild-type and CD4 KO mice were treated with liposomal clodronate (days 5, 9, 13, 17, 21) and/or anti-LFA-1 mAb (days 0–6) cotreatment. A separate group of untreated sensitized recipients served as controls. Hepatocyte grafts were monitored for survival. (A) Cotreatment with anti-LFA-1 mAb and liposomal clodronate (lipo clod; n = 5) to deplete macrophages enhanced hepatocyte median survival time (MST) as compared to anti-LFA-1 mAb treatment alone (n = 6) in sensitized C56BL/6 mice (MST = 30 days vs. MST = 7, p = 0.012). (B) Cotreatment with anti-LFA-1 mAb and liposomal clodronate (n = 5) did not further enhance survival as compared to anti-LFA-1 mAb treatment alone (n = 5) in sensitized CD4 KO mice (MST = 35 days vs. MST = 35 days). Liposomal PBS was utilized as a vehicle control in both wild-type (n = 4) and CD4 KO recipients (n = 5) and show no efficacy in combination with anti-LFA-1 mAb versus anti-LFA-1 mAb alone in either recipient.
Discussion
T-cell memory responses exacerbate organ transplant rejection and interfere with the efficacy of conventional immunosuppressive treatment strategies (2,16,24,44,61,70). T memory cells have features such as a low threshold for reactivation, higher magnitude effector response, programmed inflammatory phenotype, and enhanced survival relative to naive T-cells (60). For these reasons, it is believed that costimulatory blockade strategies, which have successfully induced long term graft survival and donor-specific tolerance in unsensitized hosts, are generally not successful in preventing rejection in hosts with primed or memory T-cells (61,70). This pattern has been reported using various allograft tissues and treatment protocols, mostly targeting CD4-dependent, CD8+ T-cell-mediated allograft rejection. We have observed that recipients which are sensitized by rejection of prior hepatocyte transplants demonstrate accelerated rejection in response to a second hepatocyte transplant which is accompanied by enhanced in vivo allospecific cytotoxic effector function (25). Therefore, we used this model to determine if sensitized CD8+ T-cells, which mature in wild-type or CD4-deficient conditions, manifest CD154 and/or LFA-1-dependent cytotoxic effector function in vivo and whether successful suppression of in vivo allocytotoxicity would promote prolonged allograft survival in sensitized recipients.
Xie et al. have reported efficacy of anti-LFA-1 mAb in combination with other agents for prolongation of cardiac allograft survival in the presence of CD44+ alloreactive memory cells (68). Additionally, Xu et al. has recently reported that arsenic trioxide, in combination with anti-LFA-1 and anti-CD154 mAbs, prolongs cardiac allograft survival in alloantigen-primed mice (69). Badell et al. has also recently shown that short-term induction therapy with LFA-1 blockade in combination with basiliximab and sirolimus or belatacept blocks memory T-cell proliferation and prolongs islet graft survival in rhesus macaques (3). Kitchens et al. reported that memory OT-1 cells [derived from transgenic mice that have a T-cell receptor specific for an major histocompatibility complex (MHC)-class-I-restricted peptide derived from ovalbumin (OVA)] mediated OVA+ skin graft rejection despite the presence of costimulatory blockade [cytotoxic T-lymphocyte antigen 4 immunoglobulin (CTLA-4Ig) and anti-CD154 mAb]. This rejection was abrogated when costimulatory blockade was coupled with LFA-1 inhibition (33). However, the current study is the first to demonstrate significant immune consequences of treatment with anti-LFA-1 mAb alone on in vivo cytotoxic effector function and graft survival in sensitized recipients. Treatment of sensitized recipients with anti-LFA-1 mAb both suppressed in vivo allospecific cytotoxic effector function and prolonged survival of secondary allografts in CD4-deficient recipients. Thus, hepatocyte rejection by CD4-independent CD8+ T-cells in sensitized recipients can be suppressed by targeting LFA-1 and correlates with suppression of in vivo allocytotoxicity.
However, the same treatment regimen did not prolong hepatocellular allograft survival in sensitized wild-type recipients. We postulated that this could be due to increased numbers of (LFA-1 bearing) CD8 effector cells in second set responses or increased expression of LFA-1 on CD8 memory cells (55). However, when higher doses of anti-LFA-1 mAb (500-750 mg/day) were administered to CD4-deficient and wild-type sensitized recipients, allocytotoxicity was not further reduced nor was hepatocyte survival enhanced (data not shown). Furthermore, depletion of CD8+ T-cells in sensitized wild-type recipients was not sufficient to prolong allograft survival. Since sensitized wild-type recipients had detectable alloantibody, we pursued a combination treatment strategy to target both CD8+ cytolytic T-cells and alloantibody dependent rejection. Significant delay of rejection in sensitized wild-type recipients was achieved when macrophage depletion (to interfere with macrophage-mediated alloantibody dependent cellular cytotoxicity) was combined with LFA-1 inhibition. An argument could be made that macrophage depletion (by liposomal clodronate treatment) inhibits CD8+ T-cell priming by the elimination of antigen presenting cells. However, this possibility is unlikely since liposomal clodronate treatment did not occur until day 5 posttransplant (beyond peak CD8-mediated cytotoxicity which occurs on day 3 posttransplant). Furthermore, in control studies, we demonstrate that liposomal clodronate treatment alone does not inhibit CD8-mediated in vivo cytotoxicity or enhance hepacellular allograft survival.
Diab et al. found that anti-LFA-1 mAb, in combination with CTLA4-Ig and CD154 blockade, was not sufficient to enhance islet allograft survival in sensitized wild-type recipients (18). In this study, wild-type mice were sensitized by islet transplantation under the kidney capsule. We have previously reported that cellular transplantation to the kidney capsule leads to high alloantibody levels in primary recipients (27). Therefore, as observed in the current study, it is likely that alloantibody-mediated rejection inhibited the efficacy of LFA-1- and costimulation blockade in these sensitized transplant recipients.
LFA-1 has multiple potential mechanisms of immune suppression and several different binding partners. Evidence to date in our studies suggests that in vivo anti-LFA-1 mAb treatment targets CD8 cytotoxic effector function likely mediated by interference of LFA-1/intercellular adhesion molecule 1 (ICAM-1) interactions. Anti-LFA-1 mAb is nondepleting (11,39,45,51) and is known to inhibit activation, trafficking, signaling, and cytotoxicity of primary activated T-cells (43,56,69). In addition, we previously found that in CD4 KO primary recipients (39), anti-LFA-1 mAb treatment results in upregulation of CD8+CD25high regulatory T-cells that likely contributes to the enhanced survival of hepatocytes. However, given the kinetics of rejection in sensitized recipients (day 77 in primary CD4 KO recipients and day 35 in sensitized CD4 KO recipients and the eventual rejection of all allografts in the sensitized CD4 KO recipients) (67), we do not favor regulation as the mechanism for prolongation of transplant survival. It is likely that CD8+CD25high T-cells are insufficient to inhibit a memory-like response long term, as previously described in other models (54). Instead, our data support suppression of effector mechanisms as the basis for prolongation of allograft survival by combined modalities of immunotherapy. In a recent review by Ford and Larsen, inhibition of lymphocyte trafficking was not a likely mechanism for the prolongation of graft survival by LFA-1 blockade in primary allograft recipients (19). Furthermore, in contrast to findings by Setoguchi et al., which show that anti-LFA-1 mAb blocks the infiltration of memory CD8+ T-cells into cardiac allografts (53), the trafficking of CD8+ T-cells to the liver sinusoids (site of hepatocellular transplantation) is not dependent on LFA-1 in our model (39). LFA-1 is known to deliver costimulatory signals that are important for induction of T-cell activation and cytotoxic T-cell function (17,47,63,71). In previous studies, since anti-LFA-1 mAb is administered early posttransplant, it is not possible to distinguish between its effects on activation and effector function. However, in the current study, since anti-LFA-1 mAb is administered late after transplant (days 6 and 7, after development of peak allocytotoxicity), the results are most consistent with the interpretation that targeting LFA-1 effectively inhibits in vivo CD8+ CTL effector function. The main binding partner, ICAM-1, is expressed by donor hepatocytes, as well as splenocyte targets used in the in vivo cytotoxicity assay (9). We have observed that ICAM-1 blockade is required to detect in vitro target cell lysis by primary alloreactive CD8+ T-cells (9). In addition, detection of in vivo allocytotoxicity is dependent on target cell expression of ICAM-1 since in vivo cytotoxic effector function is significantly inhibited against ICAM-1 KO targets (11.0 ± 4.6%; p = 0.001) as compared to wild-type targets (51.5 ± 22.3%) in wild-type FVB/N recipients (unpublished observations). Based on the critical role of ICAM-1 on in vivo clearance of allogeneic target cells, LFA-1-mediated inhibition of CD8-dependent cytotoxic effector function in sensitized (as well as primary) recipients is likely to be due to interference of LFA-1/ICAM-1 interactions important for target recognition, adhesion, and destruction. This conclusion is consistent with reports by others, which have shown that LFA-1 is an important adhesion molecule for the in vitro target binding by CD8+ human leukocyte antigen (HLA)-A2-specific CTL clones (5).
The current experimental studies are the first to demonstrate that treatment with anti-LFA-1 mAb is efficacious for inhibition of in vivo allospecific CD8+ cytolytic effector function in sensitized recipients. Our studies also show that the efficacy of anti-LFA-1 mAb for prolongation of allograft survival can be significantly enhanced when combined with specific immunotherapies targeting LFA-1-independent immune responses, including alloantibody-mediated rejection. Thus, our results suggest that treatment with anti-LFA-1 mAb to inhibit CD8+ T-cell effector function could be effective as an acute intervention to interrupt ongoing CD8-mediated rejection. Furthermore, the failure to achieve immune tolerance using a variety of immunosuppressive strategies is often attributed to the activity of alloreactive CD8+ T-cells. Treatment with an agent such as anti-LFA-1 mAb that blocks effector function of CD8+ T-cells without depleting host CD8+ T-cells (17,39) could provide a window for administration of immune tolerance therapy (35,42) without the need for lifelong immunosuppression.
Footnotes
Acknowledgments
This work was supported in part by grants from the American Society of Transplantation Basic Science Physician Scientist Award (to P.H.H.), Roche Organ Transplantation Research Foundation (to G.L.B.), the American Society of Transplant Surgeons (to G.L.B.), and National Institutes of Health grants DK072262 and AI083456 (to G.L.B.) and F32 DK082148 (NIDDK; to J.M.Z.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes and Digestive and Kidney Diseases or the National Institutes of Health. The authors declare no conflicts of interest.