
Abstract
Unrestricted somatic stem cells (USSCs) derived from human umbilical cord blood represent an attractive cell source to reconstitute the damaged heart. We have analyzed the cardiomyogenic potential and investigated the fate of USSCs after transplantation into rat heart in vivo. USSCs demonstrated cardiomyogenic differentiation properties characterized by the spontaneously beating activity and the robust expression of cardiac α-actinin and troponin T (cTnT) at protein and mRNA level after cocultivation with neonatal rat cardiomyocytes. To study the fate in vivo, eGFP+ USSCs were injected transcoronarily into immunosuppressed rats via a catheter-based technique. Nearly 80% USSCs were retained within the myocardium without altering cardiac hemodynamics. After 7 days, 20% of the transplanted cells survived in the host myocardium and showed elongated morphology with weak expression of cardiac-specific markers, while some eGFP+ USSCs were found to integrate into the vascular wall. After 21 days, only a small fraction of USSCs were found in the myocardium (0.13%); however, the remaining cells clearly exhibited a sarcomeric structure similar to mature cardiomyocytes. Identical results were also obtained in nude rats. In addition, we found some cells stained positively for activated caspase 3 paralleled by the massive infiltration of CD11b+ cells into the myocardium. In summary, USSCs can differentiate into beating cardiomyocytes by cocultivation in vitro. After coronary transplantation in vivo, however, long-term survival of differentiated USSCs was rather low despite a high initial fraction of trapped cells.
Introduction
A typical human heart infarction involves the loss of approximately 1 billion cardiomyocytes; therefore, many investigators have sought to identify endogenous or exogenous stem cells with the capacity to differentiate into committed cardiomyocytes and to repopulate lost myocardium (24, 43). As a result of these efforts, dozens of stem cell types have been reported to have cardiac potential, including pluripotent embryonic stem cells and various adult stem cells from bone marrow, peripheral tissues, and the heart itself (26, 33). However, despite encouraging results from more than 140 worldwide clinical trials (24, 43), considerable disagreement exists regarding the long-term efficiency and even the reality of cardiac differentiation by many of these stem cell types, making these issues a continuing source of controversy in the field of stem cell therapy (26, 33). For donor cell types already in clinical studies (24, 43), the predominant in vivo effect of bone marrow or endothelial progenitor cells may be on neoangiogenesis and not cardiac specification. Moreover, skeletal myoblasts, despite integration and survival, are confounded by arrhythmia, perhaps reflecting lack of transdifferentiation (33). These obstacles underscore the need to seek cardiac progenitor cells beyond the few known sources, but so far the optimal choice of donor cell is still under intensive research (5, 33).
There is a growing body of evidence that unrestricted somatic stem cells (USSCs) derived from human umbilical cord blood exhibit great functional plasticity and can reprogram in a new environmental tissue niche to give rise to cell lineages specific for the new organ site (8, 21). In contrast to the adult stem/progenitor cells, these cells from the umbilical cord compartment are less mature and have a higher proliferative potential associated with longer telomeres and thus extended lifespan (38). Besides these biological advantages, USSCs are readily available and are routinely harvested without risk to the donor, and infectious agents such as cytomegalovirus (CMV) are rare exceptions, a definite advantage for the development of cell therapeutics in regenerative medicine (22). In a tissue-specific niche, USSCs were capable of giving rise to various types of cells (9, 21, 31). In earlier reports, surgically implanted USSCs into the infarcted heart in experimental porcine model were shown to improve regional and global left ventricular function (7, 17), but few human cells were detected in the myocardium after 4–8 weeks, and it is not known whether the beneficial effects of USSCs resulted from de novo formed functional cardiomyocytes together with vascular restoration and/or paracrine effect. Recently, USSC transplantation in a rat model was demonstrated to result in structural and functional improvement after myocardial infarction, which was dose-dependent, possibly due to concurrent cardiomyogenesis and vasculogenesis (16). However, the compelling functional effects were recently criticized by two negative reports showing both transcoronarily and intramyocardially delivered USSCs to eventually exhibit no improvement of the infarcted heart (30, 34). Besides small variations of the experimental protocol in terms of isolation, expansion, and transplantation of USSCs, the reason for these negative results remains unclear but may relate to cell retention after initial transplantation, cell fate adoption, and long-term engraftment of USSCs in the heart. Hence, mechanistic insights are required that address the mode of cell application, extent of cell delivery, and cell fate under conditions, which might later be applied clinically.
In the present study, we analyzed the cardiomyogenic potential of USSCs and systematically evaluated the fate of USSCs after coronary delivery in order to find out how many cells can be delivered by this route and to determine quantitatively how many cells differentiate into vascular cells and cardiomyocytes. Our data show that USSCs can differentiate into beating cardiomyocytes by cocultivation in vitro and in vivo after being transplanted into the rat heart. However, long-term survival of differentiated USSCs in the beating heart is rather low despite a high initial fraction of trapped cells, most likely due to apoptosis-mediated cell loss.
Materials and Methods
Isolation of Primary Cells and Cell Culture
USSCs from cord blood were isolated and cultured in accordance with informed donor consent and the ethical approval for the use of human-derived materials as described previously (21). In brief, for isolation of mononuclear cells (MNCs) from cord blood, a Ficoll gradient (Biochrom, Germany; density 1.077 g/cm3) followed by lysis of remaining red blood cells with ammonium chloride was applied. These MNCs were plated in T25 culture flasks containing Dulbecco's modified Eagle's medium (DMEM), 30% fetal calf serum (FCS; HyClone), dexamethasone (10–7 M), penicillin (100 U/ml), streptomycin (0.1 mg/ml), and ultraglutamine (2 mM) and incubated at 37°C in 5% CO2. For genetic labeling of USSCs, a lentiviral vector containing full-length enhanced green fluorescent protein (eGFP) under the control of the U3 promoter of spleen focus forming virus (SFFV) was transfected for stable expression of eGFP. Isolation, expansion, and growth curve, as well as the basic biological properties of USSCs, were described in detail previously (21, 22) and remained unaltered by the labeling procedure. They could be expanded up to passage 12 without spontaneous adipogenic and osteogenic differentiation. For the analysis of cardiomyogenic differentiation of USSCs in vitro and in vivo as in the present study, we employed rather naive USSCs at passages 6–7.
In Vitro Cocultivation
To explore the differential potential of cryopreserved USSCs toward cardiomyocytes, a cocultivation system with neonatal cardiomyocytes was used to induce cardiomyogenic differentiation of USSCs. Neonatal cardiomyocytes were isolated from newborn Wistar rats (P0–P3) as described previously (4). In brief, the ventricular muscle of the neonatal rats were minced and incubated in digestion medium (0.25% trypsin EDTA, Sigma) repeatedly at 37°C for 10 min. After two rounds of digestion, the supernatant was filtered through a sterile 100-μm nylon mesh and was seeded on 60-mm plastic tissue culture dishes containing DMEM supplemented with 20% fetal bovine serum (FBS; Biochrom, Germany), penicillin (100 U/ml), and streptomycin (100 μg/ml). After 2 h of preincubation at 37°C, the nonadherent cells were counted and cocultivated (ratio 1:1) with eGFP-labeled, cryopreserved USSCs and were transferred to 100-mm Petri dish (RT-PCR and FACS) or onto chamber slides (immunohistochemistry).
Microscopic visualization was performed intermediately after 3 days, and the beating activity of the eGFP+ cell was recorded using a digital video system (Zeiss, Axiovert 35). Seven days after cocultivation, cells were either detached for RNA extraction or fixed by applying 4% paraformaldehyde for 20 min for immunohistology and FACS analysis.
PCR Analysis
Total RNA was extracted from the coculture experiments with RNeasy mini kit (Qiagen, Germany). RNA for reverse transcription-polymerase chain reaction (RT-PCR) was converted to cDNA with a first-strand cDNA synthesis kit (Invitrogen, USA) according to the manufacturer's recommendations. Human-specific sequences were amplified by using designed primers for the following genes: GATA4; cardiac structural proteins: cardiac troponin T (cTnT), cardiac-actin; ion channel: hyperpolarization-activated cyclic nucleotide-gated potassium channel 2 (HCN2). A general internal control (18s rRNA) and species control for human cDNA (glyceraldehyde 3-phosphate dehydrogenase, GAPDH) were used. PCR primers were designed such that they would amplify the human but not the rat genes (31) (18S: for 5′-GTG GAG CGA TTT GTC TGG TT-3′, rev 5′-CGC TGA GCC AGT CAG TGT AG-3′; GATA4: for 5′-ATC TCG ATA TGT TTG ACG ACT T-3′, rev 5′-CCT CTT TCC GCA TTG CAA GA-3′; cardiac actin: for 5′-CTT CCG CTG TCC TGA GAC AC-3′, rev 5′-CCA GAC TGG AAG GTA GAT GG-3′; cTnT: for 5′-GCA AAG GAG GCT GAA GAT G-3′, rev 5′-TTC CTC CTG TTC TCC TCC T-3′; HCN2: for 5′-CGC CTG ATC CGC TAC ATC CAT-3′, rev 5′-AGT GCG AAG GAG TAC AGT TCT CT-3′; GAPDH: for 5′-TTG GTA TCG TGG AAG GAC TC-3′, rev 5′-CAC CAC TGA CAC GTT GGC A-3′).
FACS Analysis
For characterization of the immunophenotype of USSCs, native USSCs were incubated for 2 h with the fluorescence-conjugated antibodies (see Table 1). After washing, the cells were analyzed using a FACSCalibur instrument (Becton Dickinson, Germany). To determine the cardiomyogenic phenotype of USSCs, indirect immunostaining was performed. In brief, cells were stained for 2 h at 4°C with primary antibodies (mouse anti-α-actinin antibody mAB, 1:400, Sigma) and fluorescein isothiocyanate (FITC)-labeled secondary antibodies for 15 min at room temperature (goat anti-mouse-Ig, 1:400, Dako, Germany). Histograms of logarithmic fluorescence intensity versus cell number were recorded for 20,000 cells per sample. Antibody against human mitochondrial protein (pAB 1:1,000, Chemicon, USA) was used as a human marker and α-actinin as a cardiac marker.
Antibodies Used in Flow Cytometry

Animal Experiments
All animal experiments were approved by the local committee and conducted according to German animal care legislation. For in vivo transcoronary delivery of USSCs, Wistar rats (280–320 g) were intubated and anesthetized by mechanical ventilation with isoflurane (Abbott, Germany, 1.5% v/v) in 100% oxygen. An incision was made in the middle of the neck to expose the right carotid artery and the internal jugular vein. A homemade 3F balloon catheter was placed into the right atrium via the internal jugular vein to obstruct the venous return. A 2F through lumen balloon catheter built according to our specifications (Edwards Life Science, USA) was placed into the aortic root via the right carotid artery and was connected to a pressure transducer (Statham P23XL) to measure aortic pressure (AP). All data were recorded by a four-channel PowerLab system (ADInstruments).
After baseline measurements of physiological variables, the venous and the arterial balloons were simultaneously inflated to create right atrial and aortic occlusions. A cocktail solution containing a combination of the β-receptor antagonist esmolol (1 mmol/l, Brevibloc®, Gensia, UK) and acetylcholine (ACh, 1 μmol/l, Sigma) was then injected into the coronaries through the arterial catheter to induce a transient cardiac arrest. Thereafter, 1 × 106 eGFP-labeled USSCs in 500 μl were gently infused into the coronary vascular system. To facilitate cellular interaction, cardiac arrest was allowed to sustain for a period of 2 min. Spontaneous circulation was established by performing cardiopulmonary resuscitation and confirmed by the gradual recovery of the heart beat and the AP. One hour after reestablishment of stable arterial blood pressure, the wound was treated with lidocaine and sutured. The animals were weaned from the respirator, extubated, and placed in an oxygen-enriched environment. Of the 22 rats undergoing the protocol of cardiac arrest, all animals recovered and woke up after being weaned from ventilation. Only one rat died due to postoperative blood loss, resulting in the total mortality of less than 5% during follow-up. All animals received 15 mg/kg cyclosporin (Sandimmun®, Novartis, Germany) daily.
At indicated time points after cell transplantation, the animals were anesthetized and echocardiography was performed (Hewlett Packard Sonos 5500). Subsequently, the animals were euthanized, and hearts were fixed with 4% paraformaldehyde for 4–6 h and then rinsed in 0.1 M PBS for at least 24 h followed by storage for 12 h in PBS solution with 18% sucrose and frozen at −80°C.
Immunohistochemistry
Cryostat section of 10 μm was cut from the cryoprotected cardiac tissue and fixed with 4% paraformaldehyde for 30 min at room temperature. All chamber slides from cocultivation and the tissue slices were blocked with 5% bovine serum albumin (BSA) for 1 h at room temperature. The following primary antibodies were dilated in 0.8% BSA in TBS and incubated overnight at 4°C: rabbit anti-human mitochondria (hMito; pAB, 1:1,000, Chemicon, USA), mouse anti-α-actinin antibody (mAB, 1:400, Sigma, Germany), mouse anti-human nuclei (HN; mAB, 1:500, Chemicon, USA), mouse anti-troponin T (pAB, 1:400, Thermo, USA,), rabbit anti-human troponin T (mAB, 1:400, Abcam, USA), mouse anti-caspase 3 (mAB, 1:400, Thermo, USA), rabbit anti-smooth muscle actin (sm-actin; pAB, 1:400, Dako, Germany), mouse anti-CD11b (mAB, 1:400, Sigma, Germany).
The secondary antibodies biotinylated goat anti-rabbit-Ig and goat anti-mouse-Ig (1:400, Dako, Germany) were used in 0.8% BSA for staining of sections and chamber slides. A confocal laser scanning microscope (Zeiss, Germany) with wavelengths 488 and 543 nm, beam splitter HFT 488/543, and filters BP505-530 for eGFP and LP560 for Alexa 586 was used for imaging.
Calculation of eGFP+ USSCs in the Heart and Statistics
The method for calculating eGFP+ cells in the entire heart was outlined by us previously (4). In brief, the number (n) and the area (A) of eGFP expressing USSCs was measured under the microscope, and the volume was calculated by multiplying the thickness of the section (T). The total number of eGFP expressing USSCs was then calculated using the following formula:

Data are expressed as mean ± SEM. Statistical analysis was performed by using two-way ANOVA to compare the differences in cardiac function after cell delivery. A value of p < 0.05 was considered to be statistically significant.
Results
Immunophenotype of USSCs
FACS analysis revealed that USSCs stained positive for CD10, CD13, CD29, CD71, CD73, CD90, CD105, CD146, CD166, HLA-ABC, NG2, PDGFra and PDGFrb and negative for CD14, CD18, CD31, CD33, CD34, CD36, CD45, CD80, CD83, CD106, CD117, CD133, CD184, CD271, CCR7, cytokeratin, and HLA-DR. However, it has to be emphasized that there is no specific surface marker available that is able to distinguish USSCs from cord blood mesenchymal stromal cells (MSCs) and bone marrow mesenchymal stromal cells (BMMSCs). USSCs, however, have been recently shown by our group to be distinct from MSCs in umbilical cord blood and bone marrow by the “biological fingerprint” of the homeobox (HOX) gene family (27) and delta-like 1 (DLK-1) expression (18).
In Vitro Cardiomyogenic Differentiation of USSCs
To explore the cardiomyogenic potential of USSCs that can be expanded to a clinically applicable scale under good manufacturing practice (GMP) conditions (1), we cocultivated the eGFP-labeled USSCs with neonatal rat cardiomyocytes. Three days after cocultivation, USSCs were found to be morphologically similar to neonatal cardiomyocytes, and after 7 days cocultivation, a significant number of eGFP+ USSCs were stained positive for cardiac markers (α-actinin, Fig. 1A and cTnT, Fig. 1B). A clear striation pattern of cTnT in eGFP+ USSCs could be observed at higher magnification (Fig. 1B). Additionally, we used antibody that specifically recognizes human cTnT antigen and found positive immunostaining in cocultivation, suggesting the human origin of cTnT expression in USSCs (data not shown). These results clearly demonstrate that USSCs possess the competence to differentiate into cardiac myogenic lineage in vitro. To assess the efficiency of cardiomyogenic differentiation of USSCs, we performed FACS experiments to quantify the percentage of α-actinin-positive cells 7 days after cocultivation. As shown in Figure 1C, 43 ± 13% (n = 3) eGFP+ USSCs acquired a cardiac phenotype indicated by the positive staining for α-actinin. A consistent finding was that eGFP+ USSC-derived cardiomyocytes contracted spontaneously (data not shown). Some eGFP+ USSC-derived cardiomyocytes integrated into the rat cardiomyocyte cluster and contracted in a synchronized manner (data not shown).

Cardiomyogenic differentiation of USSCs in vitro. After 7 days cocultivation with rat neonatal cardiomyocytes, enhanced green fluorescent protein (eGFP)+ unrestricted somatic stem cells (USSCs) acquired cardiac phenotype indicated by the positive staining for cardiac α-actinin (A) and cardiac troponin T (cTnT) (B). A striation pattern of cTnT staining of eGFP+ USSCs was shown in higher magnification view (B, inserts). Fluorescence-activated cell sorting (FACS) analysis revealed that the differential efficiency was about 43%, stained positive for cardiac α-actinin (C). The gene expression profile showed the induction of transcriptional factor for carcinogenesis and cardiac markers at mRNA level (D). Scale bars: 50 μm (in A) and 10 μm (in B and insets). GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HCN2, hyperpolarization-activated cyclic nucleotide-gated potassium channel 2.
Gene expression profile analyzed by RT-PCR revealed the expression of the cardiac-specific transcription factor GATA4 and cardiac markers such as cTnT and cardiac actin 7 days after cocultivation with neonatal cardiomyocytes in three independent preparations (Fig. 1D).
Transcoronary Delivery in Rat
To study the potential of USSCs to differentiate under in vivo conditions, we employed a minimal invasive catheter-based approach recently described by our group (6) to deliver USSCs via the coronary system in a clinically relevant manner. Figure 2A shows the representative recording of the aortic pressure during the intervention. Induction of a transient cardiac arrest permitted the wash out of blood from the coronary system and, at the same time, enhanced the contact time of USSCs with the coronary endothelium. After cell delivery, the spontaneous heart beat was resumed by cardiopulmonary resuscitation. All animals (n = 20) recovered after being weaned from ventilation. We found a slight loss of body weight (18%) within 7 days after operation, which was similar to sham-operated animals (Fig. 2B). This was also true when we monitored heart function by echocardiography. As summarized in Figure 2C, ejection fraction and fractional shortening remained unaltered 7 and 21 days after the delivery of the USSCs (p > 0.05). In addition, we found no incremental rise in biomarkers of cardiac injury (serum cTnT) after cell delivery (data not shown).

In vivo transplantation of USSCs. Cells (1 × 106) were delivered into the coronary system during a cardiac arrest indicated by the fall of the aortic pressure as described previously (4) (A). Scale bar: 1 min. The animal received daily immunosuppression that caused slight body weight loss (18%) in both sham and USSC groups. At the indicated time points (arrows), function and structure of the heart were analyzed (B). Echocardiographic assessment of the ejection fraction and fractional shortening indicated that transcoronary delivery of USSCs was not associated with the alteration of the cardiac contractile function (C).
Distribution, Integration, and Survival of USSCs
In a first experimental series, we examined the distribution of eGFP+ USSCs within the heart 2 h after transplantation. The representative histological sections in Figure 3A show that the distribution of eGFP+ USSCs was homogenous throughout the ventricular free wall and the septum. The total number of all eGFP-expressing cells retained within the heart was calculated to be 79.5 ± 4.5% (n = 4) of the initially transplanted USSCs. The grafted cells were occasionally (three to five events in each section) found to be smooth muscle actin-positive and some still remained inside the vascular lumen 2 h after transplantation but adhered to the vessel wall (Fig. 3B).
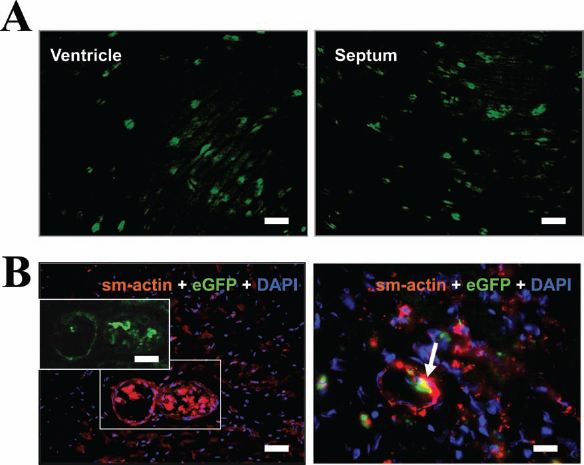
Distribution and cardiac retention of USSCs. In the representative section, eGFP+ USSCs were shown to be relatively homogenously distributed into both the ventricular free wall and the septum shortly after transplantation (A). Visualization of the vascular system by smooth muscle actin (sm-actin) staining gives evidence of intravascular localization (B, right) and adhesion to the vascular wall (B, left) of eGFP+ USSCs. Scale bar: 50 μm (in A and B, left and inset) and 20 μm (in B, right).
To determine whether the grafted cells integrated into the host myocardium and differentiated afterwards into cardiomyocytes similar to what we observed in coculture experiments, the fate of USSCs was determined 7 days after transplantation. Counting the number of green fluorescent cells revealed a significant diminution of the number of eGFP+ USSCs to 19.80 ± 5.5% (n = 5) of the initially transplanted cells. The surviving eGFP-expressing USSCs were found to intersperse within the host rat myocardium, and the grafted cells were increased in size showing a more elongated morphology (Fig. 4A). Histochemistry revealed the expression of the cardiac-specific marker α-actinin in the eGFP+ cells. However, those cells failed to show a clear sarcomeric structure, an indicator of mature cardiomyocytes (Fig. 4B). In addition, a small fraction of eGFP+ USSCs (less than 1%) were found to be integrated into the vascular wall (Fig. 4C).

Survival and integration of USSCs 7 days after transplantation. The surviving eGFP-expressing USSCs were found to intersperse within the host rat myocardium and increased in size, showing a more elongated morphology (A). Some cells exhibited a weak expression of the cardiac-specific marker α-actinin but failed to show a clear sarcomeric structure (B). A small fraction of eGFP+ USSCs were stained positive for smooth muscle actin and found to be integrated into the vascular wall (C). Scale bar: 20 μm.
In a further experimental series, we studied the fate of the grafted USSCs in the host myocardium 21 days after cell transplantation. The number of eGFP+ USSCs in the heart sections was found to be as low as 0.13 ± 1.1% (n = 4) of the initially transplanted USSCs. The identified eGFP+ cells, however, were well integrated into the host myocardium and acquired cardiomyocytic morphology (Fig. 5A). The survived eGFP+ USSCs stained positive for cardiac α-actinin and exhibited matured sarcomeric structure (Fig. 5A). To confirm that the differentiated USSCs in the heart retained their human identity, human-specific markers were used. As shown in Figure 5B, the cells stained positive for human mitochondrial proteins and human nuclei (Fig. 5B), suggesting differentiation of USSCs into cardiomyocytes.

Long-term engraftment of USSCs. Three weeks after transplantation, a small fraction of injected USSCs was found in the host myocardium. The remaining cells integrated well, exhibited a morphological similarity to mature cardiomyocytes, and expressed the cardiac marker α-actinin (A). Those cells stained positive for human mitochondrial protein (hMito) and human nuclei (HN) (B). Scale bar: 50 μm.
Immunobarrier and Apoptosis of USSCs
To exclude the possibility that the disappearance of the transplanted USSCs over time might have been due to inadequate immunosuppression by cyclosporine, an additional series of experiments (n = 5) was carried out in nude rats lacking T-cell immunity. Using the same experimental procedure as before, we found that eGFP+ USSCs in the heart declined in the same manner as was observed in Wistar rats (Table 2). In order to further study the mechanism underlying the disappearance of transplanted USSCs, we examined the role of caspase 3 activation in the heart 7 days after USSC transplantation. As shown in Figure 6A, a large proportion of eGFP+ USSCs were positive for active caspase 3, indicative of undergoing apoptosis. In addition, we found substantial infiltration of CD11b+ cells into the myocardium 7 days after USSC transplantation (Fig. 6B), but not in the sham-operated heart (data not shown).
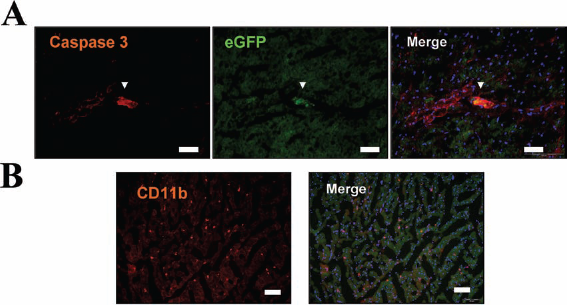
Apoptosis of USSCs and cardiac inflammation. Representative section showed that 7 days after transplantation, most of the surviving USSCs stained positive for active form of caspase 3, suggesting those cells went into apoptosis irreversibly (A). This led to a massive infiltration of CD11b+ cells (macrophages) in the myocardium (B). Scale bar: 50 μm.
Long-Term Engraftment of USSCs in Wistar and Nude Rats

USSCs showed a large fraction of cardiac retention after transcoronary delivery but diminished to <1% 21 days after transplantation. This phenomenon was observed both in immunosuppressed Wistar rats and nude rats.
Discussion
Given the divergent reports in the literature on the therapeutic potential of USSCs (7, 16, 17, 30, 34), the present study was designed to characterize in detail the cardiomyogenic potential and the fate of eGFP-labeled USSCs after transplantation into the recipient rat heart. To this end, we transplanted eGFP-labeled USSCs via the coronary arteries into rat heart using a catheter-based technique that was initially developed for efficient cardiac gene transfer (6) and later modified for cardiac-specific cell delivery (3). A major finding of our present study was that approximately 80% of the initially infused USSCs were retained within the heart immediately after transplantation without altering cardiac hemodynamics. Yet the retained USSCs underwent apoptosis so that the long-term engraftment of USSCs was rather low (0.13%). However, the surviving USSCs adopted a cardiomyocytic phenotype, and some even incorporated into the vascular wall.
Transcoronary Delivery and Cardiac Retention
The intracoronary route provides theoretical merit over direct intramuscular injection because there is less myocardial damage during the engraftment procedure. However, retention of infused stem cells in the vascular territory is generally considered to be rather low (1.3–17.8%) both in experimental models (28, 41) and in clinical settings (12, 14). Similarly, the initial entrapment of intracoronarily applied 111indium-oxine-labeled human umbilical vein endothelial cells (HUVECs) was found to be about 18% of infused cells, as determined from single photon emission computed tomography (SPECT) measurements and microscopic analysis (4). We were therefore surprised to find in the present study that about 80% of all delivered USSCs were trapped by the heart (Fig. 3 and Table 1). This effect might have several reasons. One important factor appears to be the size of the USSCs being ≤20 μm in diameter (30), which compares to only 7-μm diameter of capillaries (40). Similarly, myoblasts and MSCs, which have a similar size to USSCs, have also been reported to be largely entrapped (<90%) in capillaries after intracoronary infusion (37) or at the precapillary level of the cremaster vasculature (40). Along the same line, increasing size of cell aggregates by cross-linking of endothelial progenitor cells with phytohemagglutinin increased the fraction of transplanted cells (3). In contrast, bone marrow stem cells with a diameter of about 7 μm—which have been used in the majority of clinical trials—are rapidly washed out during the first passage, leading to a relatively low retention in the heart (14, 23). Aside from cell size, it is also well conceivable that the interactions of the delivered cells with the coronary endothelium might play a role in the adherence and transmigration through the endothelial barrier. In addition, trypsinization of USSCs prior to transplantation might have activated protease-activated receptors by cleaving their N-terminus, which could have acted as its own ligand by activation of various intracellular signaling pathways in USSCs (4). In summary, it appears that physical size and cellular adhesion of USSCs may play a crucial role for the highly efficient cardiac retention in the present experiments.
Giving the high fraction of USSCs initially deposited in the heart, it is conceivable that this might have critically obstructed coronary blood flow (30). Comparison with microsphere (15 μm) deposition into the myocardium—used to measure regional blood flow—reveals that a total of approximately 5 million microspheres/kg body weight did not impair cardiac function (36). In the present study, we used 1 × 106 USSCs per animal, and it can be calculated that maximally 5% of all capillaries of the rat coronary system were blocked immediately after USSC delivery. This low fraction can explain why transplantation of USSCs into the heart was not associated with any changes of cardiac hemodynamic parameters (Fig. 2C). Consistent with this, we did not find evidence for apoptotic or necrotic cardiomyocytes, and no incremental rise in serum cTnT after cell delivery (data not shown) was detected.
Cell Survival and Integration
As to the fate of transplanted USSCs, we found that the fraction of cells retained within the heart declined from initially 80% to only 0.2% after 3 weeks. This value is similar to what has been previously reported in a swine model after intracoronary delivery of USSCs (30). Even in immunosuppressed animals, CD3+ (T-cell antigen) cells and macrophages were observed in the area where USSCs were engrafted after intramyocardial delivery (17). Interestingly, a therapeutic benefit of stem cell therapy can be achieved even without immunosuppression (10, 15), which might be due to the fact that stem cells can indirectly inhibit T-cells by contact-dependent induction of regulatory APCs (antigen-presenting cells) with T-cell-suppressive properties and thus induce immunological tolerance (2).
As to our experiments, immunological rejection of USSCs because of insufficient immunosuppression by cyclosporine can be excluded, since we obtained similar results in nude rats subjected to the same protocol (Table 2), in line with previous data showing that allergenic and synergic graft yielded similar cell disappearance after cardiac transplantation (42). However, in both experimental series, we noted that a large fraction of the transplanted USSCs stained positive for activated caspase 3 together with the local accumulation of macro phages (CD11b) suggesting an inflammatory response. The reason why transplanted USSCs underwent apoptosis remains unclear (29) but may be related to the continuous physical strain exerted by the beating heart, local hypoxia, or a less appreciable microenvironment for cell survival. It is interesting to note that cotransplantation of USSCs, together with other cell populations, is alternative strategy to enhance the survival and integration of USSCs in the host myocardium.
Differential Potential Toward Cardiomyocytes
Our study shows that USSCs in cell culture differentiated into spontaneously beating cardiomyocytes, which is consistent with data of the literature (31). Nearly 43% of all USSCs acquired a cardiac phenotype after 7 days of cocultivation. The expression of cardiac markers was unlikely due to an uptake of cardiomyocyte materials by USSCs as the cardiac protein was shown by specific antibodies to be of human origin. Under in vivo conditions after transplantation, a small fraction of USSCs adopted a myocytic phenotype by the criteria of expression of cardiac-specific marker and morphological similarity to mature cardiomyocytes. As engrafted cells may have obtained a cardiac phenotype by means of cell fusion (32), we analyzed the gross morphology and the specific identity of eGFP+ cells in heart sections (Fig. 5A). The USSC-derived cardiomyocytes were comparatively small in dimension but retained the nuclear marker of human origin. This makes it unlikely that USSCs fused with preexisting cardiomyocytes to form hybrid cells. Together with the previous report (16), our data therefore strongly suggest that under in vivo conditions USSCs are also capable of differentiating into cardiomyocytes, which integrate well into the host myocardium. Despite a low number, the engrafted cells adopted a cardiac destiny and formed de novo myocardial cells.
Stem cells from human umbilical cord blood have previously been demonstrated to have high cardiomyogenic potential, but until recently there were no genetic or functional marker available to distinguish the different cell populations in cord blood (22). USSCs are a unique cell population that can be distinguished from mesenchymal cells in cord blood by the criteria of the expression of DLK-1 and the HOX gene family (18, 27). In contrast, previous studies used CD34+ (13) or CD133+ (25) and even unclarified mononuclear cells (11) from cord blood, which are mainly hematopoietic stem cells and immunologically distinct from the cell population in the present study (19, 21, 22). In 2007, Nishiyama et al. described an MSC population in cord blood expressing the transcription factors for cardiogenesis (GATA4 and Nkx2.5) and cardiac markers (cardiac actin and TnT) at default state, and thus, the cells were rather termed “cardiac progenitors” (31). They have a limited lifespan and need to be genetically modified for in vitro expansion (38). In contrast, the USSCs used in the present study grew adherently and could be expanded under GMP conditions (1). Here, we show that those cells were negative for cardiac transcription factor and markers at default condition (Fig. 1D), and the cardiomyogenic potential was able to be experimentally induced in vitro cocultivation and in vivo transplantation into the heart.
Implications of Clinical Benefits
There are several functional implications of our study. First, the number of surviving and differentiating USSCs after transplantation was rather small, so that the reported beneficial effect of USSCs to support the failing heart (7, 17) is unlikely to be due to a functional replacement of lost cardiomyocytes. Rather it appears that paracrine factors may be important to protect the ischemic cardiomyocytes from apoptosis and/or to facilitate the endogenous repair by the activation of resident progenitor cells (16). Second, we observed a massive loss of transplanted USSCs due to apoptosis together with substantial infiltration of inflammatory cells. Inflammatory mediators such as cytokines had also been reported previously to indirectly promote cardiac repair and injection of microspheres and the associated inflammation was shown to augment left ventricular function (35). A unique pattern of USSCs to secrete cytokines has been reported previously (20), but the mechanistic interlink to cardiac repair remains unclear so far. Third, myocardial neovascularization may be beneficial for the functional restoration of the diseased heart. The capacity of stem cells to form large coronary arteries was reported previously (39), and we observed integration of USSC transplantation into the vessel wall (Fig. 4B). Whether this rare event is of functional significance is presently not known. Differentiation of USSCs into vascular cell lineages has been reported (16), implying the supposition that USSC-induced vascular regeneration may be one of the beneficial effects for cardiac repair. In summary, the future therapeutic potential of USSCs may involve multiple levels including paracrine factors and inflammatory mediators that support cardiomyogenesis and neovascularization in the myocardium. Further understanding of such mechanisms at the cellular level may help to maximize the therapeutic benefits in a clinical setting.
Footnotes
Acknowledgments
This study was supported by the Forschergruppe (FOR717) from the Deutsche Forschungsgemeinschaft (http://www.dfg.de/). We thank Ms. Claudia Viethen for her kind help in the RT-PCR experiments. The authors declare no conflict of interest.