
Abstract
The feasibility of ex vivo blood production is limited by both biological and engineering challenges. From an engineering perspective, these challenges include the significant volumes required to generate even a single unit of a blood product, as well as the correspondingly high protein consumption required for such large volume cultures. Membrane bioreactors, such as hollow fiber bioreactors (HFBRs), enable cell densities approximately 100-fold greater than traditional culture systems and therefore may enable a significant reduction in culture working volumes. As cultured cells, and larger molecules, are retained within a fraction of the system volume, via a semipermeable membrane it may be possible to reduce protein consumption by limiting supplementation to only this fraction. Typically, HFBRs are complex perfusion systems having total volumes incompatible with bench scale screening and optimization of stem cell-based cultures. In this article we describe the use of a simplified HFBR system to assess the feasibility of this technology to produce blood products from umbilical cord blood-derived CD34+ hematopoietic stem progenitor cells (HSPCs). Unlike conventional HFBR systems used for protein manufacture, where cells are cultured in the extracapillary space, we have cultured cells in the intracapillary space, which is likely more compatible with the large-scale production of blood cell suspension cultures. Using this platform we direct HSPCs down the myeloid lineage, while targeting a 100-fold increase in cell density and the use of protein-free bulk medium. Our results demonstrate the potential of this system to deliver high cell densities, even in the absence of protein supplementation of the bulk medium.
Introduction
Red blood cell and platelet transfusions are routine life-saving procedures in medicine. While the relative ease of harvest and storage of these cell subsets enable this process, an adequate supply of suitable donor material remains problematic. The production of ex vivogenerated transfusible blood products, or Blood Pharming, remains one of the most promising, as well as perhaps one of the most distant, deliverables of current cell expansion technologies. The current technical limitations are both biological and engineering in nature. We recently reviewed the individual challenges associated with production of red blood cells (RBCs), platelets, and neutrophils (25). The most promising culture methods for producing each cell product were reviewed and critically analyzed for the feasibility of scaling these “best practice” methods such that clinically relevant cell numbers could be generated. A key conclusion was that generating the necessary cell numbers, utilizing current best practice cell densities and expansion protocols, would require culture volumes that preclude the feasibility of either RBC or platelet manufacture.
Our general inability to replicate the cell density and efficiency achieved within the human body is perhaps most strikingly appreciated by considering current methods for RBC production. Utilizing the protocols developed by Giarratana et al. (10), producing a single unit of RBCs (1012 cells) would require 660 L of medium (assuming a cell density of 3 × 106 cells/ml) or approximately 9,500 traditional laboratory T175 flasks (25). In contrast to RBC manufacture, the ex vivo production of neutrophils for temporary immune support may be more feasible given the reduced necessary cell number (1010 cells/dose) (27), as well as the economic motivation to offset the significant cost of hospitalizing neutropenic patients (25). In either case, one of the critical engineering requirements is the development of high-density cell culture strategies that reduce the working volume of blood product cultures, and thus the relative cost of producing such cell subsets for clinical use.
In a previous article we outlined the development and use of a membrane microbioreactor designed to enable the culture of cord blood-derived hematopoietic stem progenitor cells (HSPCs) at densities 100-fold greater than those traditionally realized in a static culture flask (7). This microbioreactor system enabled high cell density cultures to be achieved by enhancing mass transport through sandwiching the cells between a gas permeable membrane and a semipermeable membrane that permitted the exchange of medium components, less than 10 kDa in size, from a bulk medium reservoir. The primary objective of this previous work was to evaluate the feasibility and impact of high cell density culture on HSPC expansion. Our results indicated that, over the culture durations assessed, HSPC expansion was not compromised and could in fact be enhanced, in some cases, when the local cell density was increased 100-fold in the bioreactor. While we feel that this device represents a suitable tool with which to further optimize culture conditions for high cell density production of specific blood cell subsets, we acknowledge that its micronature does not fully address the engineering scaling challenges that currently hinder blood product production. It is likely that scaling will favor geometries such as those provided by hollow fiber bioreactors (HFBRs), rather than simple sandwich designs such as that described in our previous work.
While there are many HFBRs on the market, their volumes and system complexities tend to be too great for the “screening level” assessment and optimization of high-density HSPC cultures, where input cells are limited and growth factor supplements are costly. When hollow fiber dialyzers are utilized in the culture of cells for protein production, cells are cultured on the extracapillary side or on the intracapillary side if the cell suspension is being recirculated as in an alternating tangential flow (ATF) device (9). In either case pumps are required to drive perfusion.
In the first demonstrated HFBR, Knazek et al. cultured cells in the extracapillary space while perfusing medium through the intracapillary space (18). The geometry of hollow fiber systems is such that the intracapillary volume is always less than the extracapillary volume. Generally cells are cultured in the extracapillary space, and this obligates perfusion through the intracapillary space, as relative to the extracapillary space there is minimal volume in the intracapillary space to function as a medium reservoir. While intracapillary culture is uncommon, there are potential benefits in that the plug flow nature of fluid through the capillaries enables homogenous seeding as well as easy harvest. Previous studies have identified challenges in harvesting cells from extracapillary cultures (24). In addition, the maintenance of HSPCs within fibers overcomes problems associated with cell settling in the extracapillary volume—a significant concern with nonadherent cell types. In the HFBR described herein, the HSPCs are cultured in the intracapillary space with the fibers simply submerged in a static medium reservoir that supplies low molecular weight nutrients such as oxygen, glucose, and amino acids. HSPCs, and high molecular weight medium supplements, are loaded into the intracapillary volume, while the bulk extracapillary reservoir (99% of the system volume) contains protein-free medium. The 10-kDa molecular weight cut-off (MWCO) of the Cuprophan™ membrane (Membrana, Germany) retains high molecular weight supplements, such as cytokines, within the fiber lumen, thus preventing their dilution into the bulk medium.
In this study we use this simplified HFBR system to evaluate the potential of the hollow fiber geometry for use in high-density expansion of HSPCs. In these experiments HSPCs are directed down the myeloid lineage, which is perhaps a logical starting place as neutrophils, as a blood product, are most likely to be clinically available in the near future (25,26).
Materials and Methods
Fabrication of the HFBR
It is our opinion that the complexities and volumes of traditional HFBR systems limit “bench-level” screening, and are therefore not appropriate for the first pass evaluation of the expansion of HSPCs in a hollow fiber-based system. In order to minimize complexity, the hollow fiber system shown in Figure 1 was fabricated. Cuprophan™ (Membrana, Germany) was potted in polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning), where 5-ml syringes (Terumo Corp, Philippines) were used both as a mold for potting and as the intracapillary header or manifold. Each fiber bundle contained 187 fibers, having a length of 17 cm and inner diameters of 200 μm, providing a total intracapillary volume of 1 ml. Fiber bundles were prepared by first dipping the ends (terminal 5 mm) of the fibers in PDMS and curing the PDMS at 80°C for 20 min. This step was found to be necessary to prevent further flow of PDMS into channels in subsequent stages of the fiber bundle preparation. A 5-ml syringe body was then used as a mold to cast the fibers in a PDMS plug. The terminal ends of the bundles were placed into the syringe body molds and then backfilled with approximately 5 ml of liquid PDMS. PDMS was cured at 80°C for 20 min. The cast bundles were then cut from the syringe molds and a razor blade used to cleanly cut the majority of the cast PDMS and fibers from the bundle leaving an approximate 1.5 cm PDMS cast fiber bundle. The terminal ends of the bundles were then placed into fresh 5-ml syringe bodies that had been cut at the 1.5-ml mark. This small length of syringe body then became the header, with the male luer lock being the entrance/exit into the fiber bundle. The PDMS fiber bundle plug was pushed deep into the syringe body in order to minimize the header volume. While the PDMS plug formed a tight well-fitted seal with the syringe body, this seal was further enhanced by placing a small volume (~0.5 ml) of liquid PDMS into the back end of the syringe header. This additional seal was then cured at 80°C for 20 min. The fiber bundles were each mounted into individual T75 flasks (Becton, Dickinson and Company, Franklin Lakes, NJ). At the tail end of each flask 11-mm-diameter holes were drilled through the polystyrene and the syringe header fixed in place using silicone sealant (Selleys, Auckland, NZ). The finished bioreactor systems were gas sterilized using ethylene oxide and then permitted to degas for at least 1 month.

The image shows a static hollow fiber bioreactor. A bundle of hollow fibers is contained in a standard T75 flask. Cells are loaded into the fiber lumens from the syringe headers, while the fibers themselves are bathed in medium contained within the flask.
Preparation of the HFBR
Before use in culture both the intracapillary and extracapillary volume of the bioreactor were rinsed thoroughly with phosphate-buffered saline (PBS). The fibers were left wet for at least 30 min to ensure hydration and restoration of mass transport properties. The extracapillary volume was then drained to allow clear visualization of the fibers. Fluid volumes were introduced into the intracapillary side using a syringe and needle via a three-way valve and syringe bung as shown in Figure 2. A sterile syringe/needle was used every time and the syringe bung was sterilized before each addition using 70% ethanol. Fluid exited the opposite fiber bundle header into a closed reservoir. Prior to the addition of cells to the HFBR we purged the fibers with Stemline II medium (Sigma-Aldrich, St. Louis, MO). This purge was done purposefully and continuously until any air bubbles remaining in the fiber bundle were eliminated.
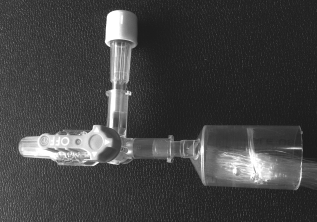
Fiber bundle header and inlet valve. Medium and cells are injected into the system through the syringe port, thus facilitating sterile fluid exchange. Following cell loading the three-way valve was used to isolate the fiber bundle and thus to prevent axial medium movement over the duration of the culture.
Cell and Medium Preparation
Umbilical cord blood from full-term deliveries was obtained with informed consent, from both the Royal Brisbane and Women's Hospital Human Research Ethics Committee and the University of Queensland Medical Research Ethics Committee. Mononuclear cells were isolated by centrifugation on Ficoll-Paque (Amersham Biosciences, Uppsala, Sweden) and enriched for CD34+ cells using Mini-MACS columns (Miltenyi Biotech, Bergisch Gladbach, Germany). We found the cell populations to be 95% CD34+ after two passes through the magnetic associated cell sorting (MACS) column.
At least 2 bioreactors and 16 control cultures were initiated in each study, thus requiring in excess of 106 input cells. To meet this requirement, and to mitigate the risks associated with biological variability, all cultures were initiated with cells from two or more unique cord blood isolations. These cells were then further expanded in flask cultures for 3 days to confirm cell viability and to ensure adequate cell number at day 0 of the bioreactor study. This was necessary to ensure statistical viability where the input cell number into each experiment was greater than 5 million cells, and we generally obtain fewer than 500,000 CD34+ cells from each cord harvest. In clinical applications where larger harvests are possible and statistical replicates are not necessary, this step could be eliminated, making the process a single expansion step in the bioreactor starting at day 0. In initial flask cultures the cells were suspended at 50,000 cells/ml in Stemline II medium supplemented with 100 ng/ml each of stem cell factor (SCF, Stemgen; Amgen, Sydney, Australia), thrombopoietin peptide (TPO; AusPep, Parkville, Australia) (4), granulocyte colony-stimulating factor (GCSF, Neupogen; Amgen), and 100 U/ml penicillin and 100 mg/ml streptomycin [1% penicillin streptomycin (PS); Gibco-Invitrogen, Carlsbad, CA].
Following 3 days of preculture, cells were harvested, concentrated by centrifugation (300 × g for 5 min) and then resuspended at 106 cells/ml in Stemline II medium supplemented with 10,000 ng/ml SCF, TPO, GCSF, 1% penicillin streptomycin, and 100 μl/1 ml (1:10) of insulin-transferrin-selenium-X (ITS; Gibco-Invitrogen) (7). Three milliliters of this cell suspension was used to fill the intracapillary volume of each reactor, such that we could be confident that the 1-ml fiber volume had been effectively exchanged with this inoculation cocktail. Great care was taken not to introduce bubbles into the fibers. The extracapillary volume of the flask was then filled with 100 ml of RPMI (Rosswell Park Memorial Institute) medium supplemented with 1% penicillin streptomycin.
Control cultures were made from the addition of 10 μl of the intracapillary inoculum into either 990 μl of protein-free RPMI or Stemline II. In doing so the ratios of protein in the RPMI control and in the HFBR were equal. The seeding density of these cultures was 10,000 cells/ml with growth factor concentrations of 100 ng/ml.
Cultures were then maintained for 7 days in 37°C, 5% CO2 incubators where the atmosphere contained 2% oxygen.
Cell Characterization
Cells harvested from bioreactors or from well plate cultures were enumerated, and then characterized by flow cytometry. Cell counts were performed using a Cell Lab Quanta SC (Beckman Coulter, Fullerton, CA). Flow cytometry (LSR Becton Dickinson, Franklin Lakes, NJ) was performed to identify the proportion of the cell population that had progressed down the myeloid lineage. The cells were labeled with monoclonal mouse anti-human CD15-fluorescein isothiocyanate (FITC; Miltenyi Biotech) and monoclonal mouse anti-human CD11b-phycoerythrin (PE; Miltenyi Biotech) as per manufacturer's instructions.
Statistical Analysis
SPSS 17.0 (SPSS Inc., Chicago, IL) was used for one-way analysis of variance (ANOVA) with Tukey post hoc tests to assess statistical significance, which was defined as p < 0.05. Number of replicates is stated in the figure captions.
Results and Discussion
Mass Transport
The use of the hollow fiber geometry in high cell density bioreactor systems is routine in protein production applications. However, as this particular system and application is unique we used a shell balance mass transport model to predict the local concentration of oxygen and glucose in a fiber packed with HSPCs. Assuming that the concentration of either metabolite is at a uniform concentration on the outside of the fiber, the analytical solution to the shell balance is:

Mass Transport Model Constants


Mass transport model through cell fibers packed with cells at a density of 109 cells/ml. Assuming an extracapillary oxygen concentration of 0.02 mM (approximately equivalent to 2% O2 atmosphere), the core fiber oxygen concentration would drop to 0 mM if a cell density 10-fold greater than intended was achieved. Glucose concentration would drop to approximately 11.2 mM or still in excess of twofold greater than what would be considered a low blood serum concentration. This result indicates that metabolite supply should not be limited in this system.
Note that the extracapillary medium in the HFBR is added without protein supplement, but that some components of the Stemline II and ITS supplement will pass through the fiber and be diluted into the bulk medium. For example, the selenium and insulin in the ITS supplement would be expected to be diluted into the bulk medium. In this case the ITS supplement would be diluted to 1:1,000 of the stock concentration (still within accepted working range), resulting in a final concentration equivalent to that in control cultures. In our previous work we demonstrated that this concentration of ITS was sufficient to expand HSPC cultures in a membrane system (7). Notably the membrane will exclude the passage of proteins having a molecular weight greater than 10 kDa such as albumin, transferrin, SCF, TPO, and GCSF. If a membrane having a molecular weight cutoff of less than 5.5 kDa was selected (such a cellulose-based fiber membrane is not, to our knowledge, commercially available) then all supplemented proteins, including insulin, would be retained in the fiber.
Cell Expansion and Analysis
All cultures were inoculated with cells suspended at 106 cells/ml in StemLine II medium containing ITS and growth factors. The cell inoculation volume was equivalent to 1% of the total culture volume. The inoculation was used to initiate cultures in either well plates containing StemLine II medium, or RPMI medium with no protein supplement, or into the HFBR where the extracapillary medium contained RPMI with no protein supplement. Thus, on a percentage basis the protein contents in the RPMI well plate control and in the HFBR were equivalent.
Images of the HSPC-derived cell culture within the fiber lumens are shown in Figure 4. These images immediately demonstrate the successful expansion of the cells in this device, while also revealing the heterogeneous distribution of the cells just prior to harvest. This distribution and the potential implications are discussed later.

(A) Fibers loaded with cells at day 0. Cells are reasonably uniformly dispersed. (B) Cells in fibers at day 7. Some sections of the fibers are packed with cells, while other sections appear to be void of cells over portions of the fiber length. The closed arrow points to a fiber filled with cells, while the open arrow points to adjacent fiber devoid of cells over the length shown in the image.
Actual cell expansion was determined by direct counting of the cell product generated from each of the cultures. Figure 5 shows the relative fold expansion of hematopoietic cells in each of the culture systems. Our results indicate that the preferred expansion environment was the well plate using StemLine II medium (approximately 30-fold expansion), with statistically equivalent results obtained in the bioreactor. In contrast, cultures maintained in well plates with RPMI medium performed poorly. The total system relative protein and cell content was identical in the RPMI well plate cultures and the bioreactor cultures, yet the bioreactor cultures performed significantly better. This initial result is significant, as it indicates that by sequestering the small quantity of protein into the fiber volume (1% of the system volume), it is possible to achieve significantly greater cell expansion than is possible when this same quantity of protein is diluted through the total culture volume as occurs in the RPMI well pate control. If we assume that the consumption of medium metabolites is similar in each system then the total medium consumption, no matter the culture methodology, will be similar. However, if it is possible, as shown here, to successfully generate a cell product where 99% of the medium utilized in the system is protein-free then this savings may justify the added complexity of membrane bioreactor systems, particularly as culture volumes approach scales such as 660 L per unit of cell product generated (10). While our experiments and discussion focus on the inclusion or exclusion of protein from the bulk medium, there may be the potential to replace albumin in the bulk medium with other less expensive large molecules such as Pluronic F68 (15). Such supplementation has been used successfully in the culture of immortal cell lines, but has not achieved similar success or popularity in the culture of HSPCs. However, the use of semipermeable membranes may enable advantageous exploitation of such protein-free alternative in HSPC culture.

The plot displays the fold expansion of cells in each culture system. The upper and lower panels provide example outcomes from two independent experiments performed on two separate occasions. Fold expansion in well plate: RPMI (no protein) cultures is statistically (ANOVA, p < 0.01) less than that observed in either StemLine II or Bioreactor cultures. Each of the four bioreactor, in the two experiments, was sampled four times (technical replicates), while each well plate condition has been replicated four times and sampled two times (two technical replicates). Bars indicate averages, while error bars represent 1 SD.
The maturation of cells was assessed via flow cytometry. Figure 6A–C shows the flow cytometry scatter plots of cells harvested from each culture system and stained for myeloid markers CD11b and CD15. We only screened these cultures for myeloid cells, as the selected growth factor cocktailed should effectively promote myeloid differentiation. In previous work we have shown that, over time, this combination of factors results in a virtually pure population of postmitotic neutrophils (26). The scatter plots (Fig. 6) indicate a similar level of maturation in both the StemLine well plate cultures and in the bioreactor cultures. In contrast, the scatter plot of the RPMI (protein-free) culture indicates that less maturation has occurred in these cultures. This is also shown quantitatively in Figure 6D, revealing that the StemLine cultures are in fact the most mature of the three culture conditions. Only the proportion of CD14+/CD15+ populations are similar between the bioreactor and StemLine II cultures, with maturation in the RPMI (no protein) cultures being significantly delayed. In HSPC cultures differentiation is generally coupled with expansion, so we would expect that in RPMI cultures, where reduced expansion was observed (Fig. 5), that maturation would also be delayed (Fig. 6).

Flow cytometry scatter plots of cells harvested from each culture system. (A) StemLine well plate cultures, (B) RPMI (no protein) well plate cultures, and (C) is bioreactor cultures. The scatter data suggest a similar level of maturation in the StemLine and bioreactor cultures, with less maturation in the RPMI (no protein) cultures. (D) Flow cytometry data demonstrating the relative proportion of maturing myeloid cells in each culture system. Plotted bioreactor data are generated from two separate bioreactors, each with two technical replicates (n = 4). Plotted well culture data are generated from n ≥ 3 independent cultures (ANOVA, *p ≤ 0.05, **p ≤ 0.01, each sample is of 10,000 events). Bars represent means, while error bars represent 1 SD.
In the experiments presented here the total cell expansion in the bioreactor system was approximately 30-fold (Fig. 5), resulting in a cell density in the fibers of approximately 3 × 107 cells/ml. In contrast, our static StemLine II cultures reached a cell density of approximately 3 × 105 cells/ml. While a cell density of 3 × 107 cells/ml is an impressive 10-fold greater than typical static culture confluence, the total culture cell number and fiber volume space should enable a further two more population doublings before reaching confluence in the bioreactor system. These results demonstrate that this culture methodology is viable. Future work will seek to extend the culture period and range of expansion by trialing lower cell seeding densities and perhaps evaluating densities greater than 108 cells/ml. We speculate that for such high densities to be achieved, and to avoid variation in results, the lumen contents will likely require mixing. In Figure 4 we noted that there was heterogeneity in the cell distribution at harvest. There are a number of factors that may have contributed to this. Our assessment of the bioreactor at time zero eliminates inconsistency in cell seeding as being the source. This implies that the heterogeneous distribution is the result of some phenomenon occurring during the expansion process. This may include the innate varied expansion potential of individual cells (28), or the indirect effects of local cell-cell signaling networks or cytokine supply. It is reasonable to expect that signaling networks would exist (17), and that due to the fiber geometry individual population networks could be distinct and isolated from other networks, including those spatially separated in the same fiber. To demonstrate that such a claim is rational, we consider the characteristic diffusion time of a 20-kDa signal molecule (this is comparable to recombinant human G-CSF which is 18.8 kDa and/or recombinant human SCF monomer which is 18.6 kDa; R&D Systems, Minneapolis, MN) over a distance of 1 cm. First, a molecule of this size would be excluded from passing through the fiber dialysis membrane (10 kDa MWCO) and therefore could not influence cells outside that same fiber. For a hypothetical signal molecule of 20 kDa the diffusion coefficient is estimated to be 7.44 × 10−7 cm2/s using a relation developed by Preston and Comper [Equation 1 (23)]:


For this system, the characteristic diffusion time over a 1-cm length is approximately 375 h. The accuracy of this estimate is a function of both the selected empirical relation for diffusion coefficient estimation and the generic nature of the time constant relation, which does not account specifically for hollow fiber geometry. While generic in nature, this estimate is similar to that generated using other relations (21). Critically these estimates put in perspective the qualitative isolation that would likely exist between two cell clusters separated by 1 cm in the same fiber. As the fibers in this bioreactor system are 17 cm in length, it is quite reasonable to assume that over the length of a single fiber a number of distinct and isolated microenvironments could develop. As a result, it is logical to expect that even if the fibers were initially loaded uniformly that distinct microenvironments would develop, further exaggerating the heterogeneity that exists in the CD34+-selected HSPC population (28). Prolonged diffusion times could generate scenarios where rapid cell expansion in one portion of a fiber might deplete local growth factor supply, despite the fact that adjacent fiber lengths might be void of cells and therefore remain rich in available growth factors.
Probable Future Developments
While we are able to present promising results regarding the high-density expansion of myeloid progenitors, the described observation provides opportunity for future improvement. It is likely that in order to fully exploit the potential of HSPCs in this culture system, and to consistently generate a uniform cell product, some sort of periodic mixing will have to be introduced. While the culture of cells inside the fiber lumen is virtually unreported (11), culture in the extracapillary space is very common and in such systems mixing of the extracapillary cell volume has been identified as a necessary step in some cases (22). Such a mixing protocol would likely involve displacing the cells from the fiber lumen, mixing the contents, and then returning the mixed contents back into the fibers. To our knowledge such a system where cells are cultured on the lumen side of the bioreactor, and mixed periodically, has not been reported. Such action would likely reduce potential heterogeneity in the soluble environment and better utilize factors that might otherwise never physically come into contact with cells in the fiber. Such a system could also be designed to facilitate sampling of the cell population and the temporal exchange of the cytokine cocktail. The inability to sample cell populations in a HFBR system, such as the one described here, could be consider a significant limitation because it compromises real-time culture optimization and quality control. Incorporating the proposed design modifications would likely improve the output and reliability of the HFBR in stem cell culture, which, when coupled with its ability to enable cell high-density expansion with less protein consumption, might well make it a preferred culture system for large-scale production of blood products.
Further Applications
For many decades considerable resources have been dedicated to achieving true self-renewal of HSPCs in vitro, with the understanding that provision of a greater HSPC dose would enhance cord blood transplant outcomes (13,19). While achieving extensive true HSPC self-renewal in vitro has proven exceedingly challenging, and transplant trials based on the provision of these expanded HSPCs provided minimal efficacy, the provision of progenitor cell populations, providing short-term engraftment and immune support, has proven to be beneficial (5,20). These results may indicate a shift in strategy, acknowledging that cotransplantation of an unmanipulated cord blood HSPC population with a transiently supportive blood product may provide the required efficacy to make this therapy suitable for adult patients. As such strategies evolve, it is likely that clinicians will request greater doses of support cells in order to maximize efficacy, thus creating a further need for bioreactor systems capable of producing large cell numbers efficiently. The system outlined in this article may be a suitable platform as the hollow fiber system, which provides a high surface area for mass transport, could also be used to for the attachment of support cells or bound ligands as used in the two recent clinical trials (5,20). As HFBR have been in use for decades, it is likely that hollow fiber-based expansion protocols could be rapidly scaled and even integrated into existing platforms.
Conclusion
Neutrophil transfusion for temporary immune support remains uncommon due to the challenge of obtaining sufficient functional donor cells, coupled with difficulty in storing this cell subset (14). Blood Pharming, or the in vitro production of blood products, could potentially overcome these limitations, providing an immediate source of pathogen-free donor material. In general, the critical engineering limitation currently preventing blood product production is the massive volumes required to generate even a single unit of cell product. In this article we contrast blood cell expansion, starting with CD34+ cord blood cells, in an optimized commercial medium, in our HFBR and in a traditional culture system where the total protein content is equivalent to that of the HFBR system. Our results demonstrate that while the optimized commercial medium in a conventional static culture performs best, the HFBR system's performance was virtually equivalent in terms of both total cell expansion and maturation. This result is promising as the HFBR system utilizes significantly less protein and achieves an approximate 100-fold greater cell density. By contrast, in traditional well plate cultures, where the total protein content was similar to that in the HFRB, cell expansion and maturation was significantly delayed. These results suggest that if the protein content in the culture system can be sequestered into the same volume faction as the cells, then it may not be necessary to supplement the remaining medium fraction to achieve acceptable expansion outcomes. We believe this work represents an important step towards Blood Pharming by achieving both an increase in cell densities and a reduction in protein consumption.
Footnotes
Acknowledgments
The authors would like to thank Inner Wheel Australia and the Queensland University of Technology's Vice Chancellor's Fellowship Scheme. The authors declare no conflict of interest.