
Abstract
Human hepatocyte transplantation is gaining acceptance for the treatment of liver diseases. However, the reagents used to isolate hepatocytes from liver tissue are not standardized and show lot-to-lot variability in enzyme activity and endotoxin contamination. For clinical application, highly purified reagents are preferable to crude digest preparations. A purified tissue dissociating enzyme (TDE) preparation (CIzyme™ purified enzymes) was developed based on the enzyme compositions found in a superior lot of collagenase previously used by our group for human hepatocyte isolation. The performance of this enzyme preparation was compared to collagenase type XI on 110 liver cases by assessing hepatocyte yield, viability, and seven other functional assays that included plating efficiency, basal and induced CYP450 activities, phase II conjugation activity, and ammonia metabolism. No statistically significant difference was observed between these TDEs when they were used to isolate hepatocytes from liver resections or organ donor tissue on 54 hepatocyte isolations with type XI enzyme and 56 isolations using CIzyme™. These results show that a highly purified and defined TDE preparation can be formulated that provides excellent performance with respect to viability, yield, and functional activity of the isolated cells. In addition to reproducible formulation, these purified enzyme products have only 2–3% of the endotoxin of crude enzyme preparations. These results show that purified enzymes such as CIzyme™ will be a safe and effective for the isolation of human hepatocytes for clinical transplants.
Introduction
The inability of orthotopic liver transplantation to meet the increasing demand for treating patients with acute or end stage liver disease indicates a need to develop alternative approaches to treat liver disease. Hepatocyte transplantation has been successfully used to treat chronic and acute liver failure and to correct inherited metabolic diseases (6,10,11,22,27,30,32,34,35). Translation of this cell therapy procedure from experimental to routine clinical practice requires development of standardized methods to generate a cGMP hepatocyte product (4,18). A critical component that should be addressed early in the standardization process is characterization and formulation of a purified tissue dissociation enzyme (TDE) reagent optimized for human hepatocyte isolation.
The present report describes the preparation of a purified TDE mixture that was formulated to replicate the most useful biochemical characteristics of a specific lot of an enriched TDE product with a proven history of successful human hepatocyte isolations (Sigma type XI, Lot 044K8638). Class 1 (C1) and class 2 (C2) collagenase and a neutral protease (thermolysin) have been shown to be the active components responsible for successful rat hepatocyte isolation (9). The new formulation described in this report was empirically determined by reverse engineering to produce an enzyme composition similar to those found in the type XI product. Several different formulations using purified components were tested for their ability to achieve similar or superior results to type XI collagenase in human hepatocyte isolation before defining a final formulation of collagenase C1 and C2 and a protease from Bacillus polymyxa. An extensive comparison of human hepatocyte yield and function from cells isolated with either the type XI or the final purified enzyme formulation showed that similar results were obtained with cells prepared with either TDE. The primary advantages of using a defined formulation in this application is consistency of manufacture, dramatic reduction in contamination with bioactive substances (e.g., endotoxin), and the ability to further optimize enzyme compositions to improve the quality and yield of hepatocytes. In this study we determined that there was no significant difference between the viability and function of hepatocytes isolated with either enzyme preparation when they were used to isolate hepatocytes from liver resections (LR) or organ donor (OD) tissue with 54 hepatocyte isolations (41 LR, 13 OD) and 56 isolations (42 LR and 14 OD) using type XI and CIzyme™ enzymes, respectively.
Materials and Methods
Tissue Dissociation Enzymes
VitaCyte's CIzyme™ Collagenase MA (MA), hereafter referred to as CIzyme™, was purified from C. histolyticum culture supernatants. The raw material used for purification was obtained after fermentation under anaerobic conditions in media that contained porcine gelatin and porcine pancreatic enzymes as the only animal sourced components. A three-column purification scheme led to the isolation of three different molecular forms of collagenase, all active in degrading native collagen: intact class 1 (C1116kDa), degraded class 1 (C1100kDa), and intact class 2 (C2; 114 kDa) (21). C1116kDa has two collagen binding domains (CBDs) while C1100kDa and C2 possess single CBDs (20). The three purified collagenases were blended by mass to achieve a 60% C1 (comprised of both C1116kDa and C1100kDa): 40% C2 ratio. The total collagen degrading activity (CDA) of the MA enzyme blend was targeted to match the total CDA measured in 125 mg of the reference lot of Sigma type XI. Vitacyte's thermolysin and BP protease from B. polymyxa were purified by column chromatography with the amount of neutral protease activity used in the hepatocyte isolation procedure being similar to that found in the Sigma type XI reference lot.
Five lots of Sigma type XI (Sigma, St. Louis, MO) and six lots of VitaCyte MA were analyzed by the biochemical procedures outlined below. The type XI product was reconstituted from the lyophilized powder by mass in water. The insoluble portion of type XI was clarified by centrifugation and the analysis performed on the clarified supernatant. The concentration of purified MA collagenases was determined spectrophotometrically assuming 1 mg of purified collagenase had an A280 = 1.41.
High-Performance Liquid Chromatography (HPLC)
Chromatography was performed on a Beckman System Gold HPLC using a Mono-Q anion exchange column (5 × 50 mm, GE Healthcare; Piscataway, NJ) with a Tris-Ca2+ mobile phase and a NaCl gradient as previously described (21). Peak assignments of C2, C1intact, and C1100kDa were made based on internal analysis of fractionated purified collagenase. Approximately 1 mg of the collagenase blends was passed over the Mono-Q column with ultraviolet detector monitoring at 280 nm with integration performed by 32Karat software. This analysis provides an excellent assessment of the quality of collagenase found in any TDE product since the retention times correlate with the molecular form and specific activity of the collagenase enzymes (21). The earliest eluting peak is C2 with a retention time of ~13–13.5 min. C1116kDa enzyme eluted next at ~19–19.5 min followed by the other two molecular forms of C1100kDa: C1b and C1c eluting at ~20–20.5 and ~21.5–22 min, respectively.
Assays for Enzyme Activity
The CDA fluorescence microplate assay procedure is described elsewhere (21). Specific activities were calculated by dividing the CDA enzyme units (CDA U = 1 fluorescent unit per min) by the milligram of protein. The specific CDA of the different molecular forms identified in the HPLC profile were found to be similar for the C2 and C1100kDa forms of collagenase. However, C1116kDa was found to have a specific CDA approximately 10-fold higher than the other molecular forms. The same assay format as the CDA assay was used to assess neutral protease activity (NP U) but 100 μg of fluorescein isothiocyanate (FITC)-bovine serum albumin was used in place of FITC fibril (5). The Wunsch activity of enzyme solutions was measured at 25°C as described previously (42).
Assays for Endotoxin
Endotoxin levels in each enzyme product were quantified using either the Limulus Amebocyte Lysate (LAL) kinetic assay (QCL-1000; Lonza, Walkersville, MD) or the Endosafe-PTS rapid assay (Charles River; Wilmington, MA) according to the manufacturers' instructions. Each product was tested for interference by measuring the recovery of a known amount of endotoxin spiked into each sample dilution.
Hepatocyte Isolation
All human tissues were collected with informed consent following ethical and institutional guidelines. The tissue dissociation and subsequent hepatocyte isolation procedures used were developed by Seglen and modified as described previously (8,24,29,36,37). A total of 110 liver tissues were used in these studies; 83 were derived from patients undergoing scheduled liver resection (LR). Residual tissue not needed for diagnostic purposes was transported to the laboratory from the operating rooms in cold Eagle's minimum essential medium (EMEM) (Lonza) within 30 min of removal. Most of the liver resections were the result of metastatic colon cancer. Organs not used for orthotopic liver transplantation (27) were obtained 3–24 h after cross-clamp and in situ perfusion. Two of the donor livers were from non-heart-beating donors and were reported to have 15 and 40 min of warm ischemia. Seven livers were from donors with extended anoxic periods: three from drug overdose victims, two from drowning, and two additional with cardiac arrests followed by resuscitation. Four cases were tissues remaining following organ transplantation. Remaining donor livers displayed moderate to severe steatosis (35–65% macrosteatosis) or were rejected for transplantation because of anatomical defects such as calcifications or plaques in the vessels. When whole organs were obtained, they were surgically reduced to smaller size for more effective perfusion. The left lateral segment was processed first after anatomical dissection from the right lobe, 2–3 cm from the falciform ligament. If necessary, the right lobe was divided in two parts and processed as separate tissues.
From this point forward, the procedure for hepatocyte isolation was the same for OD and LR tissues. Silicone catheters were inserted into the major portal and/or hepatic vessels and the tissue was perfused with Hank's balanced salt solution (HBSS; without calcium, magnesium, and phenol red; Lonza) to determine which vessel(s) provided the most uniform perfusion of the tissue. After the best vessels were identified, purse string sutures were used to encircle each vessel with its accompanying catheter and the sutures were drawn tight to prevent the leakage of buffer around the catheter during the perfusion. Excess suture material was tied to catheters to further secure them to the liver. Perfusion proceeded once all remaining major vessels on the cut surfaces were closed with sutures or surgical grade superglue. The liver tissue was then placed in a sterile plastic bag and connected to a peristaltic pump with flow rate dependent on the number of catheters and size of tissue (35–240 ml/min in this study). The bag containing the tissue was placed in a circulating water bath at 37°C and the tissue was perfused with HBSS supplemented with 0.5 mmol/L ethylene glycol tetraacetic acid (EGTA) without recirculation. Chelation of calcium by EGTA aids in the dissolution of intercellular junctions between hepatocytes and in the washing of hematopoietic cells. A second, nonrecirculating perfusion with HBSS without EGTA was performed to flush residual EGTA from the tissue since calcium is essential for TDE activity. Finally, tissues were perfused with a third buffer composed of EMEM supplemented with the TDEs and recirculated as long as needed to complete the digestion. Enzymes from both manufacturers were reconstituted per manufacturers' recommendations, filtered, and added to the EMEM just prior to perfusion. Collagenase type XI (Sigma-Aldrich) was used at a concentration of 250 mg/L and was supplemented with 50 mg/L DNase (Sigma-Aldrich). CIzyme™ collagenase MA (VitaCyte) concentration was 100 mg/L with 24 mg CIzyme™ BP Protease (VitaCyte) in 1 L of EMEM immediately before perfusion.
In general, hepatocyte isolations from OD required 1 L of each buffer solution for each lobe, while those from LR utilized half this volume. The duration that the tissue was exposed to the different buffers was dependent upon flow rate of the peristaltic pump, number of catheters, and quality of the tissue; but perfusion of the first two buffers usually resulted in sufficient rinsing and warming of the tissue within 20–30 min. Perfusion times for the EMEM-TDE solution varied among the tissue specimens and were determined subjectively by continual monitoring of the tissue integrity. Perfusion was stopped when the liver tissue beneath the capsule surface was visibly digested and separated from the capsule, which normally took about 25–35 min; thus, the total time of perfusion needed for cell isolation ranged from 40 to 65 min. The tissue was then transferred to a sterile plastic beaker that contained ice-cold EMEM and gently chopped with sterile scissors to release hepatocytes. The cell suspension was filtered through sterile gauze-covered funnels to remove cellular debris and clumps of undigested tissue. Hepatocytes were enriched by three consecutive centrifugation steps at 80 × g for 6 min each at 4°C. After three washes in EMEM, hepatocytes were suspended in cold Hepatocyte Maintenance Medium (HMM, Lonza). Cell viability was assessed by mixing an aliquot of the final cell suspension with an equal volume of 0.4% (w/v) trypan blue in phosphate-buffered saline and counting the number of viable (unstained) and dead (blue) hepatocytes with the aid of a hemacytometer. Viabilities are expressed as a percentage of the total cell number.
Plating Efficiency
Cells were resuspended in HMM supplemented with insulin (107 M), dexamethasone (10−7 M), and antibiotics-antimycotics (Antibiotic-Antimycotic Solution; BioGemini) containing 5% bovine calf serum (Sigma-Aldrich). One milliliter of the cell suspension (7.5 × 105 viable cells/ml) was seeded in each well of a six-well culture plate precoated with type I rat collagen (0.03 mg/ml concentration). Cells were cultured at 37°C in a humidified atmosphere of 5% CO2/95% air. Two hours after plating, media from half the wells was collected, centrifuged to a pellet (200 × g, 5 min), and stored frozen. Media from the remaining wells was discarded and all wells were replaced with HMM without serum. After 10–18 h, three wells were washed extensively with PBS to discard unattached hepatocytes and the remaining three wells were not. Cells were scraped and collected and plating efficiency (PE) was determined by measuring the protein content. The cell pellets derived from the three nonwashed wells were combined with their corresponding 2-h pellet. Protein content was determined with the Bio-Rad Protein Assay kit (Bio-Rad, Cat. No. 500-0006). Plating efficiency was defined as the ratio of total protein content in wells after washing (attached cellular protein) to that of nonwashed wells (total cellular protein) and is expressed as a percentage.
Drug Metabolism Studies
Each metabolite was measured in fresh 1-ml cell suspensions immediately after digestion and, when possible (n = 46), in adherent cultures after 3 days of exposure to specific prototypical inducers. To evaluate the long-term activity, cells were plated in collagen-treated 12-well plates with HMM and the medium was changed daily for 2 days to remove dead cells and to let the cytochrome p450 (CYP450) gene expression decrease to basal levels. Cells were then incubated for an additional 72 h in the presence of specific inducers [10 μM Rifampicin (rif) and/or 1 μM phenobarbital (PB) for cytochrome P450, family 3, subfamily A, polypeptide 4 (CYP3A4); 25 μM β-naphthoflavone (BNF) for CYP1A1/2; and 0.1% dimethylsulfoxide (DMSO) as vehicle control (all from Sigma-Aldrich) with replenishment every 24 h as previously described (17)]. Results are expressed as product mol/min and normalized to a million of viable cells for experiments conducted with fresh cells in suspension cultures or per milligram total cellular protein for cultured cells.
7-Ethoxyresorufin-O-Deethylase (EROD)
Cytochrome P450-1A1/2 phase I activity was assessed by the conversion of 7-ethoxyresorufin to resorufin as described previously (40). EROD activity was measured in 1 million viable cells in suspension immediately after isolation and in cultured hepatocytes after treatment for 3 days with a specific 1A1/2 inducer (BNF) or vehicle control (DMSO). Briefly, cells are incubated for 1 h at 37°C with 7-ethoxyresorufin solution (20 μM, Sigma-Aldrich) and salicylamide (1.5 mM, Sigma-Aldrich). Salicylamide was added to prevent conjugation of the fluorescent product. After 1-h incubation, the media was collected, clarified by centrifugation, and frozen. One hundred microliter of each sample, analyzed in triplicate, was added to a 96-well white plate and analyzed on a fluorescent spectrometer (Synergy HT, BioTek Instruments, Inc., Winooski, VT) equipped with Gen5 software at an excitation wavelength of 535 nm and an emission wavelength of 581 nm. Resorufin concentration was quantified by interpolating the A581 values for the unknowns from a standard curve of resorufin (Sigma-Aldrich) prepared in HMM [resorufin (μg/ml) = 617844 * A581 + 171.23; R2 = 0.9996]. Following sampling period, cells were harvested in phosphate buffer (0.1 M, pH 7.4) and stored at −20°C until protein content was determined. Results are expressed as picomoles of product formed/min normalized to 1 million viable cells (suspension cultures) or to total protein content (adherent cultures).
Testosterone Metabolism
Testosterone 6β-hydroxylase activity catalyzed by CYP3A4 was determined directly both in cell suspensions and in intact cultured hepatocytes, as described previously (17). Briefly, cells were exposed to testosterone (250 μM) for 60 min at 37°C. The media was collected, centrifuged to remove cells and fragments, and stored at −20°C until analyzed by HPLC. Metabolite was identified by comparing retention times with 6β-hydroxytestosterone standard prepared in HMM. Results are expressed as picomoles of product/min and normalized to 1 million viable cells (suspension cultures) or to the protein content (adherent cultures).
Resorufin Assay
Phase II activity was determined by the metabolism of the fluorescent compound resorufin after incubation for 30 min at 37°C and measurement of the decrease in fluorescent signal. A 50 ng/ml solution of resorufin was added to hepatocytes, and one well was immediately centrifuged and the supernatant retained as “time 0” and then stored frozen. Subsequent time point samples were centrifuged to remove cells and debris and then stored frozen until analyzed with the same conditions described for the EROD assay. Resorufin metabolism was quantified by comparing the fluorescent signal after 30 min to the time 0 value. All the results are expressed as percentage of resorufin metabolized or as picomoles of product formed/min and normalized to 1 million viable cells (suspension cultures) or to total protein content (adherent cultures).
Ammonia Metabolism
Cells isolated with enzymes from both vendors were evaluated for ammonia metabolism by direct in vitro colorimetric determination of ammonia (26). Hepatocytes were treated with 1 mM ammonium chloride (NH4Cl) for 2 h in culture medium without serum on uncoated plates. After deproteinization, each sample was prepared and read in triplicate (uQuant, BioTek Instruments, Inc.) at a wavelength of 560 nm and results was compared to a linear calibration curve. The NH3-N content is expressed as a percentage of ammonia metabolized after 2 h and as pmol/min normalized to 1 million viable cells (suspension cultures) or to total protein content (adherent cultures).
Statistical Analysis
Results are presented as Box and Whisker plots showing median, 25 and 75 percentiles or as histograms showing mean ± SEM. Data presented as mean ± SEM were compared by unpaired t-test. A value of p < 0.05 was chosen as the minimum level of significance. All data were analyzed by GraphPad Prism software (version 5.03, GraphPad Software Inc.). Although data from organ donors and liver resections are presented in the same figures or tables, statistical analysis was only conducted between the same tissue sources for each enzyme preparation. Data from organ donors were not compared to those obtained from liver resections.
Results
One lot of Sigma type XI collagenase (044K8638) was found to be exceptional for its ability to provide human hepatocytes with excellent viability and plating efficiency following isolation based on the experience with over 200 hepatocyte isolations, so we attempted to recreate the enzyme composition found in that particular preparation. The HPLC chromatogram and enzyme activity analysis of this particular material (Fig. 1, top) and two additional lots of the Sigma type XI product (Fig. 1, middle and bottom) revealed many contaminating components, as well as highly variable ratios of the collagenase molecular forms. The superior lot had unique characteristics: a relatively high specific CDA and neutral protease activity. After the supply of this lot was exhausted, Lot 118K8608 (Fig. 1, bottom) was purchased as a replacement, but it was found to provide lower hepatocyte viability and plating efficiency when compared to isolations performed with Lot 044K8638 (data not shown).

High performance liquid chromatogram and enzyme activity analysis of Sigma collagenase type XI. The major collagenase molecular forms are labeled. The variability in the ratios of the molecular forms of collagenase between lots leads to the variability in the specific collagen degrading activity (CDA) between lots. Unlabeled peaks represent uncharacterized pigments and other contaminating proteins. Variability in the amount of contaminating proteins also affects the specific neutral protease activity between lots. NP, neutral protease.
Type XI lot 044K8638 served as the reference material to define a purified enzyme blend for use in human hepatocyte isolation. In this and in all subsequent purified blend formulations described below, a 60:40 (C1116kDa + C1100kDa):C2 enzyme mixture was prepared and the dose used for hepatocyte isolation was adjusted to match the enzyme activity found in Lot 044K8638. The initial purified blend (blend A) was based upon matching the total Wunsch and neutral protease activity (as assessed by the azocasein assay) (28) contributed by 250 mg of the type XI reference material, a dose typically used for human hepatocyte isolation. This was equivalent to 200 Wunsch units and 332.5 azocasein units (equivalent to 7.2 mg thermolysin) for a 1-L volume of TDE solution. Thermolysin was the first choice as the source of neutral protease since this enzyme was used in an earlier enzyme mixture designed to isolate rat hepatocytes (9). Comparison of results from nine isolations using CIzyme™ or Sigma's enzymes showed significantly higher viabilities were obtained with the Lot 044K8638 but the cell yields were the same (blend A 72.7 ± 9.4% vs. Lot 044K8638 83.4 ± 5.5%, p < 0.05).
Reanalysis of the enzymatic activity for Lot 044K8638 using other assays of collagenase and neutral protease activities led to a second formulation of a purified blend (blend B) that matched the reference lot for CDA rather than Wunsch activity as a measure of collagenase activity and FITC-BSA rather than azocasein as a measure of neutral protease activity. To ensure the closest match of specific CDA to the reference material, blend B contained both molecular forms of C1 (C1116kDa and C1100kDa) and C2 with the total CDA U targeted to be the same as the reference lot (~5.0 million total CDA U/L solution). BP protease was used in place of thermolysin since it had a similar specificity as thermolysin but a turnover number 2.5× lower when the specific activities of the purified enzymes were assayed using FITC-BSA as substrate (5). The results from human hepatocyte isolations using blend B showed superior results when compared to blend A: higher cell viability and uniform dissociation of tissue. The time for completion of the tissue digestion was dependent on the concentration of neutral protease used in the digest with higher amounts of enzyme shortening the digestion time. After optimization of the BP protease activity, further adjustments were made to the collagenase activities, resulting in an optimized tissue dissociation enzyme formulation containing 2.5 million CDA U and 24 mg of BP protease/L of tissue dissociation enzyme solution.
The HPLC chromatograms for the three different lots of CIzyme™ are presented in Figure 2. These results demonstrate the purity of the individual components and consistency of product manufacture. The apparent difference in the chromatogram shown in Figure 2 (top) is a consequence of the inclusion of C1c in the formulation which, like C1b, is an active enzyme with a single CBD. Both of these molecular forms are effective in degrading native collagen and have similar specific CDA that are lower than that of intact C1. The presence of C1c in this formulation was intentional and does not represent a contaminating or an ineffective enzyme.
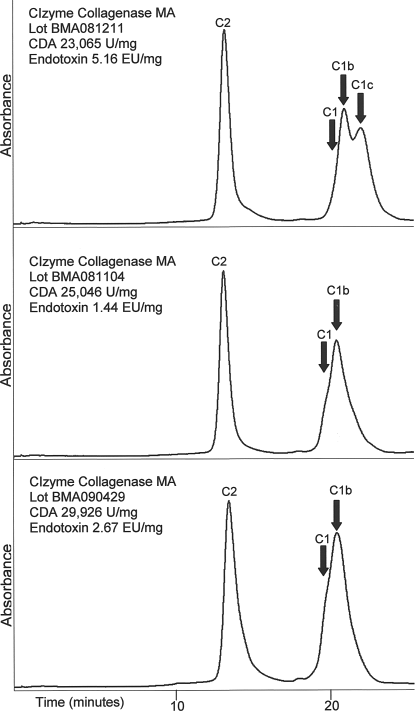
High performance liquid chromatogram and enzyme activity analysis of VitaCyte CIzyme™ collagenase MA. The major collagenase molecular forms are labeled. While there is variability in the C1 molecular forms between lots, these differences were intentional at the time of manufacture to achieve a more consistent specific CDA. The reduction in contaminating proteins results in lots of collagenase blends that are better characterized and are more consistent.
Table 1 summarizes the lot-to-lot consistency of Sigma type XI collagenase, CIzyme™, and Vitacyte BP protease as assessed by enzyme specific activities and endotoxin contamination. The specific CDA and neutral protease activities of these six lots of CIzyme™ and four lots of BP protease were less variable than similar enzyme activities measured in the various lots of the type XI product. There was also a substantial difference in the endotoxin contamination with the average EU/mg protein being 178, 6.32, and 32.85 for Sigma type XI, CIzyme™, and BP protease, respectively. These data illustrate the lot-to-lot consistency of a purified TDE product relative to an enriched collagenase product.
Characteristics of the Enzyme Preparations

CDA, collagen degrading activity; NP, neutral protease; BP, Bacillus polymyxa.
Hepatocyte Isolation
In this study, hepatocytes were successfully isolated from 110 of 115 total tissues acquired. Five cases were deleted from the study prior to data analysis for poor initial viability: four ODs for a prolonged cold ischemia, over 36 h, and one LR from a colangiocarcinoma with biliary obstruction and cholestasis. Data from the remaining 110 cases were used and the results presented here.
Tissue was randomly processed with either Sigma collagenase type XI (13 ODs and 41 LRs) or the CIzyme™ enzymes (14 ODs and 42 LRs). Table 2 summarizes patient demographics and isolation results including hepatocyte yield and cell viability.
Patient Demographics and Cell Isolation Results

LR, liver resection; OD, organ donors.
As shown in Table 2, the demographics of the two study groups were equivalent in terms of age, gender, and weight of tissue processed and the source of tissue (OD vs. LR). Figure 3 presents data on viability, cell yield, and plating efficiency on hepatocytes isolated with both enzyme sources. Figure 3a, c, and e presents data from both LR and OD for each enzyme source, while in Figure 3b, d, and f the data are broken out and presented separately for organ donor and liver resection tissue. No significant differences in viabilities were measured between hepatocytes isolated from LRs with Sigma type XI and CIzyme™: 80.2 ± 7.2% and 76.9 ± 8.9%, respectively. Viabilities of hepatocytes isolated from ODs were also similar (82.5 ± 9.0% and 80.5 ± 8.1%, respectively) for Sigma type XI and CIzyme™ (Fig. 3a, b). Yield of viable hepatocytes from LR did not differ between isolations performed with Sigma type XI and CIzyme™ (10.6 ± 7.8 and 8.2 ± 5.7 million cells/g tissue, respectively) (Fig. 3c, d). However, in OD the yield of viable hepatocytes appeared lower in isolations performed with CIzyme™ relative to those performed with Sigma type XI (4.5 ± 2.8 and 7.0 ± 5.5 million hepatocytes/g tissue, respectively); however, the difference was not statistically significant (Fig. 3b). These viability measurements and yields are in line with previous reports with similar tissue sources (1,23). The digestion times were similar between hepatocyte isolations from LR and OD performed with both enzymes (LR 23 ± 3 and 22 ± 2 min, OD 25 ± 4 and 27 ± 5 min, respectively, for CIzyme™ and Sigma type XI). The plating efficiencies were similar between hepatocytes isolated from LR and OD irrespective of the enzyme used in the tissue dissociation (73 ± 16% and 81 ± 15% for LR and 76 ± 16% and 75 ± 19% for OD with Sigma type XI and CIzyme™) (Fig. 3e, f). Attached cells showed typical hepatocyte morphology by light microscopy: polygonal shape, granular cytoplasm with vesicular inclusions, and one or more nuclei (Fig. 4).

Box-and-whisker plots and scatter plots for cell viability, yield, and plating efficiency. (a) Box-and-Whisker (a, c, e) or scatter plots (b, d, f) of human hepatocyte viability (a, b), yield (c, d), and plating efficiency (e, f) after isolation of cells from organ donors (OD) or liver resections (LR) with collagenase type XI (Coll.XI) and CIzyme™. Although data from organ donors and liver resections are presented in the same figures or tables, statistical analysis was only conducted between the same tissue sources for each enzyme preparation. Data from organ donors were not compared to those obtained from liver resections. Statistical analysis was only conducted on the means and SEMs, not on the median values.

Phase-contrast photographs after 2 days of culture of human hepatocytes isolated with each enzyme solution. Human hepatocytes isolated with collagenase type XI (left) or with CIzyme™ (right) plated as indicated in Materials and Methods on collagen-coated culture dishes. Media was changed at 4, 24, and 48 h. Photographs were taken with a phase contrast microscope after cells were cultured for 48 h. Scale bar: 40 μm.
Metabolic Activity in Freshly Isolated Cells
The metabolic activity measured in freshly isolated cells was analyzed in cells isolated with each enzyme preparation with substrates specific of CYP1A, CYP3A, conjugation reactions with phase II enzymes, and ammonia metabolism and the results are presented in Figure 5. The deethylation of ethoxyresorufin to resorufin (EROD) was used as a measure of CYP P450-1A1/2 activity. In freshly isolated hepatocytes, EROD activity was similar between cells isolated with each enzyme blend (1.9 ± 0.6 vs. 1.3 ± 0.6 pmol/min/million viable cells for the Sigma and CIzyme™, respectively) (Fig. 5a). The tissue source had no impact on 1A1/2 metabolism in freshly isolated hepatocytes: OD-sourced hepatocytes had comparable 1A1/2 activities whether the isolation was performed with CIzyme™ or Sigma type XI (1.6 ± 1.0 and 0.7 ± 0.3 pmol/min/million cells, respectively) (Fig. 5a). Similarly, hepatocytes isolated from LR tissue converted 2.4 ± 0.7 pmol/min/million cells when isolated with Sigma type XI collagenase versus 0.8 ± 0.2 pmol/min/million cells when CIzyme™ was used for the isolation (Fig. 5a).

Metabolic functions in freshly isolated hepatocytes isolated with collagenase type XI and CIzyme™. The indicated metabolic activity from hepatocytes isolated from LR or OD with each of the enzyme solutions. (a) Cytochrome P450, family 1, subfamily A, polypeptide 1/2 (CYP1A1/2); (b) CYP3A4, (c) phase II conjugation activity, and (d) ammonia metabolism. Activities are measured as pmol- or nmol/min and normalized to 1 million viable cells. Statistical analysis revealed no significant difference in the values obtained with the two enzymes in either the LR or OD groups. EROD, ethoxyresorufin-O-deethylase.
Testosterone metabolism by CYP3A4 measured immediately after isolation was similar between hepatocytes isolated with either Sigma type XI or CIzyme™ (0.2 ± 0.0 and 0.3 ± 0.2 nmol/min/million cells, respectively) (Fig. 5b). If the organ subgroups are analyzed separately, testosterone metabolism was not statistically different between the enzyme sources for LR (0.2 ± 0.1 nmol/min/million of hepatocytes). In the OD group the CIzyme™ isolated cells showed a higher but nonsignificant ability to hydroxylate testosterone when compared to the type XI group (0.5 ± 0.6 and 0.1 ± 0.2 nmol/min/million of viable hepatocytes, respectively) (Fig. 5b).
Conjugation of resorufin in freshly isolated cells was comparable between LR-derived hepatocytes isolated with enzymes from either vendor (3.45 ± 0.83 and 3.15 ± 0.97 pmol/min/million viable hepatocytes, respectively) (Fig. 5c). Similarly, hepatocytes isolated from OD with CIzyme™ had phase II activities comparable to those isolated from OD with Sigma type XI collagenase (5.43 ± 0.62 vs. 2.92 ± 1.02 pmol/min/million hepatocytes, respectively) (Fig. 5c).
Ammonia metabolism was analyzed in 29 preparations of cells (10 with type XI collagenase and 19 with CIzyme™). Average rates of ammonia conversion measured immediately after isolation were 43.9 ± 8.6 and 39.0 ± 12.8 pmol/min/million viable cells for hepatocytes isolated with Sigma type XI and CIzyme™, respectively, with no differences between hepatocytes sourced from OD or LR tissue for either enzyme combination (Fig. 5d).
Additional studies investigated the metabolic activity of the cells in longer term cultures (5 days) and also investigated whether the induction of specific CYP enzyme was affected by the source of enzyme used for cell isolation. Those results are presented in Figure 6. In normal hepatocytes, CYP1A1/2 activity can be induced by prior exposure to prototypical inducers such as BNF; therefore, we examined the induction of CYP1A in cultured cells. A 3-day exposure of cells to BNF resulted in a robust induction in CYP1A1/2-mediated EROD activity relative to noninduced controls in hepatocytes isolated from OD and LR (Fig. 6a). Basal and induced CYP1A1/2 activities did not differ between hepatocytes isolated with the enzymes from the two vendors (Sigma type XI 21.4 ± 4.9 pmol/min/mg vs. CIzyme™ 20.5 ± 2.6 pmol/min/mg) nor were differences between OD-and LR-derived hepatocytes detected for either enzyme preparation (Fig. 6a).

Long-term metabolic activity and CYP450 inductions in human hepatocytes isolated with collagenase type XI and CIzyme™. Values represent the activities, measured hepatocytes isolated from LR or OD on day 5 of culture and after 3 days exposure to the indicated inducers. Value are expressed as pmol- or nmol/min and normalized to total protein content. (a) CYP1A1/2 activity: numbers 1, 3, 5, and 7 are data from control cells while 2, 4, 6, and 8 are data from cells induced with β-naphthoflavone (BNF). (b) CYP3A4 activity: numbers 1, 4, 7, and 10 are data from control cells, numbers 2, 5, 8, and 11 are from cells pretreated with phenobarbital (PB), and 3, 6, 9, and 12 are data from cells pretreated with rifampicin (Rif). (c) Phase II activity and (d) ammonia metabolism activity. Statistical analysis revealed no significant difference in the values obtained with the two enzymes in either the LR or OD groups.
In cultured cells induced with Rif or PB for 3 days, both enzyme preparations showed an increase in metabolism of at least threefold compared to controls. The increase is most pronounced, and statistically significant, if compared for LR-derived hepatocytes isolated with either vendor when treated with PB for 3 days (0.67 ± 0.18 and 2.14 ± 0.36 nmol/min/mg, for type XI and CIzyme™, respectively; p = 0.026) (Fig. 6b). Although each individual human case responded with an increase in CYP3A4 activity when the cells were exposed to Rif, because of the wide range of response to Rif induction between cases, when all of the samples were analyzed together the difference was not statistically significant (0.78 ± 0.33 vs 2.30 ± 0.38 nmol/min/mg, respectively, for type XI- and CIzyme™-treated cells; p = 0.13) (Fig. 6b). In hepatocytes isolated from OD, the 3A4-mediated activity was similar when induced by either PB or Rif. The induced activity by PB in OD-derived hepatocytes appeared lower, but was not significantly lower in cells isolated with type XI enzyme compared to CIzyme™ (1.62 ± 0.79 and 3.14 ± 1.35 nmol/min/mg, respectively) (Fig. 6b). No difference was observed between the two enzyme preparations in cells from OD induced by Rif (2.85 ± 1.45 and 3.21 ± 1.29 nmol/min/mg, respectively, in type XI and CIzyme™ groups) (Fig. 6b).
Hepatocytes isolated from LR tissue and subsequently cultured for 5–6 days showed similar rates of resorufin conjugation (6.91 ± 2.25 and 5.67 ± 1.24 pmol/min/mg protein for Sigma type XI and CIzyme™, respectively) (Fig. 6c). Resorufin metabolism was also similar between OD-derived hepatocytes isolated with CIzyme™ and Sigma type XI collagenase (5.86 ± 1.16 and 6.80 ± 3.50 pmol/min/mg protein) (Fig. 6c). Resorufin conjugation activity of hepatocytes isolated from one OD with the Sigma enzyme was omitted from the mean presented in Figure 6c as this donor had a history of high alcohol consumption and exorbitantly high phase II activity (29 pmol/min/mg) relative to all others examined. Inclusion of this subject in the statistical analysis did not affect the statistical outcome but would skew the mean for this group to 14.07 ± 7.54 pmol/min/mg protein.
In hepatocytes isolated from LR and cultured as adherent monolayers, in vitro, ammonia metabolism was 102 ± 16.0 and 74 ± 21 pmol/min/mg in hepatocytes isolated with CIzyme™ and Sigma type XI, respectively (Fig. 6d). OD-derived hepatocytes displayed highly variable rates of ammonia metabolism with no significant differences between hepatocytes isolated with the two enzyme sources (112.8 ± 47.8 and 27.8 ± 15.9 pmol/min/mg, respectively, for CIzyme™ and Sigma type XI) (Fig. 6d).
Discussion
Little has changed in the quality of TDEs used in human hepatocyte isolation since Seglen reported his improvements to the Berry and Friend isolation procedure (3,29). Typically, the TDEs used for these isolation procedures are minimally purified biochemical products that are derived from C. histolyticum culture supernatants that are grown in culture media that usually contains animal protein (i.e., protease peptone) since early studies showed that rich media was required for expression of collagenase enzymes (2). This raises a concern of transmission of transmittable spongiform encephalopathy (TSE) since cells prepared with these enzymes are used in cell transplantation procedures. In 2007, the NIH suspended the use of Liberase HI in the Clinical Islet Transplantation Consortium clinical trial because bovine brain–heart infusion in the C. histolyticum culture media used to generate the crude collagenase posed a potential risk for TSE (16,25). However, subsequent analysis showed that the risk of transmission was minimal (≤1 in 10 million), assuming that the tissue was obtained from a clean US herd (25). The current suppliers of purified TDEs have further minimized this risk by using culture media containing bovine proteins that meet European directives for minimizing TSE (Serva); or porcine proteins (VitaCyte) since no evidence exists for naturally occurring TSE in pigs. Nor has TSE been transmitted when pigs were fed a dose of neural tissue infected with mad cow disease at a dose 50,000-fold higher than would normally be expected in a contaminated tissue (39). Finally, other suppliers use media free of any mammalian proteins in preparing crude collagenase (Roche).
However, these sources still contain considerable amounts of endotoxin. The commonly used commercial collagenase products can be subdivided into two grades: crude or enriched collagenase. These products differ by the degree of purification, which is reflected by the specific activity of the collagenase enzymes and by the amount of pigment in the final product. The high lot-to-lot variability in enzyme activity and endotoxin contamination in both classes of products directly reflects the enzyme activities found in the culture supernatants. This variability led many manufacturers to establish “lot qualification” programs that allowed customers to pretest small portions from a larger lot of product in their specific application prior to purchase.
Collagenase from C. histolyticum and a neutral metalloprotease from one of several different bacterial species have been shown to be the key enzymes responsible for releasing cells from mammalian tissue during an enzyme-mediated tissue dissociation procedure (12,13,15,33,38,41). Two reports in the literature confirm this observation for rodent hepatocyte isolation. Hatton and colleagues passed crude collagenase over several different resins and combined different chromatographic fractions to show that collagenase and neutral protease were required for effective rat hepatocyte isolation (12). Research studies performed during the development of the Liberase Purified Enzyme Blend products showed that enzyme mixtures that contained a 60:40 mixture of C1:C2 and thermolysin were as effective as a “good lot” of crude collagenase in isolating rat hepatocytes (9). There have been reports of using different Liberase purified TDEs for rodent (19,43), porcine (7), and human hepatocyte isolation (14,23,24,31), but no systematic study has been performed to optimize the enzyme composition for isolation of human hepatocytes.
This report used a novel, “reverse engineering” approach to define a purified TDE composition for human hepatocyte isolation. The enzyme activities used in this formulation were based on the enzyme activities found in an exceptional lot of Sigma type XI that was routinely used by our laboratory on over 200 human hepatocyte isolations.
During the course of this development, several formulations were assessed for their effectiveness to isolate human hepatocytes. The initial formulation contained a 60:40 C1:C2 collagenase and thermolysin (blend A). Comparisons of viability and viable cell yield per gram of tissue from cells isolated using blend A or type XI collagenase showed that use of either enzyme gave similar viable cell yields per gram of tissue but the cell viability was significantly lower when blend A was used in the isolation procedure. This led to reassessment of the type XI activities using a different set of enzymatic assays. Consequently, a second enzyme formulation (blend B) that contained CDA activity comparable to that of the reference type XI and used B. polymyxa neutral protease in place of thermolysin was evaluated. During this set of experiments, the amount of neutral protease used in the isolation procedure was shown to have a direct effect on the rate of tissue digestion. After an optimal dose of neutral protease was defined, the total CDA was reduced in half (similar amount used in blend A) with no adverse affect of the cell yield or viability. The defined enzyme formulations contained 2.5–5.0 million CDA U and 24 mg of BP protease/L of enzyme solution. Since these formulations did not affect hepatocyte yield or viability, these data were pooled for statistical comparison with the type XI enzyme.
In the current study, the performance of a purified enzyme blend in human hepatocyte isolation procedures was matched to that of an exceptional lot of enriched collagenase. Performance characteristics of the enzymes in the digestion itself (cell yield, viability, and digestion times) and functional characteristics (plating efficiency, basal and induced CYP450 activities, phase II conjugation activity, ammonia metabolism) of the hepatocytes immediately following isolation and after several days in culture did not differ. Moreover, CIzyme™ and type XI enzymes were both effective in isolating hepatocytes from tissue sourced from LR and OD.
The primary advantage of using defined enzyme compositions is the ability to control purity that in turn leads to manufacture of a consistent and stable product. Once the enzyme is purified, the consistency of the product can be confirmed by comparison of sequential lots of product for enzyme specific activity (enzyme U/mg protein) or from results generated by other analytical methods. Real-time or accelerated stability studies can be performed on individual purified enzyme products or on products that contain mixtures of purified enzymes to ensure that the product is stable on storage or to multiple freeze–thaw cycles.
The data presented in Figures 1 and 2 and in Table 1 illustrate the differences in the biochemical consistency of enriched collagenase versus a purified TDE product. Analysis of these products by anion exchange chromatography, for endotoxin contamination, or by specific enzyme activity illustrates the difference between lots for these two products. As expected, the variability of the collagenase and neutral protease activity found in different lots of type XI was higher than that found with the purified enzyme products. The endotoxin contamination per dose of enzyme used in an isolation was calculated to be nearly 40-fold higher in the type XI collagenase when compared to the purified enzyme products. Endotoxin was not detectable in the media of cells after the three wash steps, suggesting that cells prepared with either enzyme source would be acceptable for transplantation. However, there is still value in keeping the levels of endotoxin as low as possible in each step of the isolation process as endotoxin could readily activate the Kupffer cells found in liver sinusoids that in turn could lead to the release of cytokines that may influence hepatocyte engraftment or activate resident stellate cells.
The heterogeneity of human liver tissue cannot be controlled, but the conditions and reagents for hepatocyte isolation can be standardized, even continually improved by careful attention to each step of the isolation process. Results presented here demonstrate that a critical part of the isolation process, a digestive enzyme preparation, can be produced that is highly effective for the isolation of viable and functional human hepatocytes. Additionally, since the TDE is produced from highly purified components, one is able to attain consistent lot-to-lot characteristics with respect to enzyme activity while minimizing unwanted activities and contamination with byproducts such as endotoxin. Based on part to data presented here, the TDE described has been included on an IND approved by the United States Food and Drug Administration for the isolation and transplantation of human hepatocytes so the data will soon be available on the clinical effectiveness of hepatocytes produced with this product.
Footnotes
Acknowledgments
The project described was supported by grants from the National Institutes of Health (NIH), N01-DK-7-0004/HHSN26700700004C, and RC1DK086135 (SCS). VitaCyte supported by NIH grants R43DK065467 and R43DK070402, and NIH SBIR Match Awards and a 21st Century Research and Technology Award from the State of Indiana. We would like to thank the support of the COPEV Associazione per la Prevenzione e Cura dell'Epatite Virale “Beatrice Vitiello” ONLUS to R.G. Authors F.E.D. and R.C.M. have a financial interest in VitaCyte LLC; A. G.B. and M.L.G. are employees of VitaCyte. All other authors declare no conflict of interest.