
Abstract
Although the issue remains controversial, short-term culture is probably beneficial for islet graft quality. However, significant islet loss is invariably observed. This is related to reduced survival of large islets, which is compromised by hypoxia under standard culture conditions. We aimed to develop a method of culture, which would avoid exposure to relative hypoxia and hence maintain the quality of islets. Isolated rat islets cultured for 48 h in a liquid–liquid interface culture system (LICS) with a perfluorocarbon were compared to islets cultured under standard (C1) and suboptimal conditions (C2). Islets were tested for viability and response to a glucose challenge, and a marginal mass was transplanted into syngeneic diabetic recipients. The viability of islets after 24-h culture in LICS was higher than in C1 and C2 groups (89.0% vs. 77.5% and 64.6%, respectively) and decreased with time to reach 79.0%, 62.9%, and 53.4% after 72-h culture. The stimulation index in LICS-cultured islets was also significantly higher than in C1 and C2 groups (12.3 ± 0.4 vs. 5.8 ± 0.5 and 4.1 ± 0.2, respectively). Following transplantation of LICS-cultured islets 50% of recipients were rendered normoglycemic compared with 14.3% and 31.3% for C2 and fresh islets, respectively. Our liquid–liquid interface culture system using perfluorodecalin provides optimized culture conditions, which preserve both islet viability and their ability to engraft successfully after intraportal transplantation and could be used for islet transportation.
Introduction
The culture of isolated pancreatic islets prior to transplantation has advantages from the point of view of logistics and patient preparation for transplantation. It may also allow optimization of the metabolic state of both the recipient and the graft. However, controversy exists regarding the impact of islet culture on graft quality and its subsequent function following transplantation (10,14, 22,25,29). In particular, in large islets cultured in plastic vessels, oxygen diffusion to the islet cores is limited and may result in central hypoxia and necrosis (11,19). This is a particular problem if the islet seeding density is too high, if islet preparations are relatively impure, or if cellular oxygen utilization is increased (e.g., by exposure to high glucose concentrations). It may also occur if oxygen supply is restricted, as happens during culture in closed vessels or too high a volume of culture medium (during islet shipment for instance) (7,11,30).
In order to minimize exposure to hypoxia the density of islets cultured has to be limited and the total volume of culture medium kept low so that the depth of medium in the culture vessel is no greater than 2–3 mm (27). Some centers also use gas-permeable culture containers in order to improve oxygen delivery to the cultured islets (1,13). Despite these precautions, β-cells, which have a very high oxygen requirement, and other cells in the islet core, including endothelial cells, are at risk of hypoxic damage especially during islet shipment as both cell seeding density as well as oxygen provision are suboptimal.
Oxygen transporting agents such as perfluorocarbons are used in biotechnological culture systems (bacteria, plant cells) but have been little used for mammalian cell culture (17,24). Perfluorocarbons are able to dissolve large amounts of gases such as oxygen, nitrogen, or carbon dioxide but, because of their hydrophobic nature, they do not mix with water. They are chemically inert and are thus potentially ideal for many biomedical applications, including cell and tissue culture. In addition, a clinical grade material is available simplifying translation of research into clinical practice. The use of a liquid-liquid interface culture system (LICS), combining a PFC with a standard cell culture medium, could therefore have advantages for pretransplant islet culture both by providing a higher pO2 at the fluid/cell interface and by dissolving CO2 and hence increasing the buffering capacity of the culture medium.
We have previously described our liquid-liquid interface culture system (9) and in this preliminary study now report its effect on in vitro indices of islet viability and in vivo outcomes following intraportal transplantation of a marginal islet mass.
Materials and Methods
Animals
All experiments were carried out in accordance with the Animals (Scientific Procedures) Act, 1986 (UK). For in vitro experiments, islets from Sprague-Dawley rats (Comparative Biology Unit, Royal Free & University College Medical School) were used while for the in vivo studies inbred male Lewis rats (250–300 g) supplied by Harlan Olac (Bicester, UK) were used both as pancreas donors and islet graft recipients.
Induction of Diabetes
Diabetes was induced by an intraperitoneal injection of streptozotocin (55 mg/kg; Sigma Chemicals, Poole, Dorset, UK). Blood glucose was monitored using an AccuCheck® glucose meter and animals were included in the studies when their nonfasting blood glucose levels exceeded 20.0 mmol/L (360 mg/dl).
Islet Isolation
Pancreatic islets were isolated following enzymatic dispersion of the pancreatic parenchyma using our modification of the method described by Moskalewski and later adapted by Lacy and Kostianovsky (15,16,20). Briefly, pancreata were digested with collagenase solution (1.0 mg/ml; Collagenase P, Roche) and islets purified by centrifugation on a discontinuous gradient of sodium ditrizoate (1.085 g/ml; Histopaque, Sigma, UK) and HBSS buffer. Islets were then handpicked under an inverted contrast-phase microscope.
Islet Culture
Culture Medium and Culture Conditions.
Sodium bicarbonate-buffered RPMI-1640 (Gibco BRL), supplemented with L-glutamine (Sigma), penicillin & streptomycin (Sigma), and 10% fetal calf serum (Gibco BRL) was used as the culture medium. Islets were cultured under standard conditions (37°C, 5% CO2) for 48 h.
Liquid–Liquid Interface Culture System.
Islets were cultured in six-well, nontreated plates (Sarstedt, UK) at a density of 100 islets/ml of culture medium. Five milliliters of sterile, nonoxygenated perfluorodecalin (F2 Chemicals, UK) was placed in each well. Because of the high surface tension of the PFC, it was necessary to add 4 ml of culture medium to ensure an adequate depth of medium in each well. This produced a calculated depth of medium of 4.2 mm and a cultivation surface area of 9.51 cm2.
Control Islet Culture Conditions.
Control islets were cultured in six-well, nontreated culture plates (Sarstedt Ltd., UK) at a density of 100 islets/ml of culture medium. Because of the necessity to use a higher than normal volume of medium for LICS, a control group was included in which islets were cultured in an identical volume of medium (4 ml; 4.2 mm medium depth) as well as the standard volume (2 ml; 2.1 mm medium depth).
Measurement of Physical Parameters of Culture Systems
Partial oxygen pressure (pO2) and pH were measured in the culture medium either at the bottom of the culture vessel (standard culture system) or immediately above the interface between the medium and the perfluorocarbon (LICS). The partial pressure of oxygen was analyzed using a 10-μm O2 electrode connected to a transducer (Unisense, Denmark). The pH was measured using a standard laboratory pH-meter (PW9421, Philips).
Viability Assessment
Islet viability was assessed in three ways. Firstly, intact islets were stained with propidium iodide and acridine orange, and assessed under a fluorescence microscope. In order to reduce observer bias, we intentionally discriminated propidium iodide and double positive islets. Secondly, we performed quantitative propidium iodide assays using flow cytometry. Islets were handpicked from the culture and dispersed into a single-cell suspension using Accutase™ solution (Innovative Cell Technologies, CA). Following staining with propidium iodide, islet cells were analyzed on an Epics XL flow cytometer (Beckman Coulter). Finally, we performed a qualitative MTT assay. In living cells, a yellow MTT salt [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] is converted by succinyl dehydrogenase to an insoluble, purple formazan. After incubation with MTT (5 mg/ml), homogeneous staining indicated preservation of islet metabolic integrity/function.
Glucose Challenge Test (GCT)
Islets (30–50) were handpicked for each test, and each experiment was performed in triplicate. The test was performed with challenge media containing 2.5 and 25 mM glucose.
Extraction of Insulin From Isolated Islets
Islets were disrupted by sonication on ice using a Soniprep 150 sonicator (MSE Scientific Instruments, Crawley, UK) in the presence of glacial acetic acid and ethyl alcohol to disrupt the islets. The supernatant was diluted with distilled water and immediately assayed for insulin.
Assessment of Insulin Concentration
Insulin was assessed in the culture medium and acid-alcohol extracts of islets using ELISA kits (BioSource, Sweden). The color product of the reaction was analyzed on a microplate spectrophotometer (Biorad, UK) at 450 nm.
Electron Microscopy
Islets fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer were incubated in 1% osmium tetroxide, stained with 1% uranyl acetate, dehydrated, and embedded in acetonitrile/spur's resin. Ultrathin sections were analyzed on a TEM microscope.
Intraportal Islet Transplantation
Islets were transplanted intraportally using a nonselective technique as previously described (15). Following islet transplantation, tail capillary blood glucose was estimated with an AccuChek Advantage glucose meter. Posttransplant normoglycemia was defined as the achievement of nonfasting tail capillary blood glucose measurements on 2 consecutive days of less than 11.0 mmol/L, which is the upper limit of normal for nonfasting animals in this experimental model.
Statistical Analysis
Results were presented as mean ± SEM, unless stated otherwise. Data were analyzed using ANOVA with appropriate post hoc tests (Tukey) using a Statistica 6.0 package (StatSoft, Tulsa, OK). Transplantation outcome was analyzed using contingency tables. Changes in posttransplant glycemia were analyzed by comparison of areas under the curve (AUC) for each individual subject.
Experimental Design
Islets cultured under the following experimental conditions were compared: Control group 1 (C1), 2 ml of standard culture medium; Control group 2 (C2), 4 ml of standard culture medium; LICS group, 5 ml of perfluorodecalin overlain by 4 ml of standard culture medium. The C2 group was included as a control for LICS to account for suboptimal medium volume (4 ml).
In Vitro Measurement of Physical Parameters of Culture Conditions.
For changes in pH of culture medium (without added islets), six-well plates containing medium (HBSS buffered with sodium bicarbonate) or LICS (perfluorodecalin overlain with bicarbonate-buffered HBSS) were incubated for 120 min at ambient temperature (22–24°C) in atmospheric air (CO2 ~0.05%) in absence of islets. The perfluorodecalin used for LICS was pregassed with a mixture of 95% O2 and 5% CO2 for 30 min prior to use. The pH of the culture medium was measured 15 min before setting up the plates, immediately after set up, and then at 5, 15, 30, 60, and 120 min. The pO2 in the culture medium was measured after 24-h incubation of preoxygenated (99.9% O2) RPMI-1640 media (C1 and C2) and RPMI-1640/PFC (LICS with the PFC saturated with 99.9% O2) in standard conditions (37°C, 5% CO2). Measurements of oxygen partial pressure in the culture medium were performed as described above.
Assessment of Islet Function In Vitro.
After 48 h of culture, isolated islets were handpicked and viability and was assessed using flow cytometry, and glucose stimulation test was performed. An additional set of experiments assessed the viability (PI/AO staining) of cultured islets after 24, 48, and 72 h.
In Vivo Assessment of Islet Graft Quality.
For this part of the experiment, fresh (n = 16) as well as cultured islets from group C2 (n = 7) and LICS (n = 6) were used. A marginal dose of islets (2,000 islets/kg) was chosen based on our previous experience with the same experimental model, which showed that this number would be expected to correct diabetes in only ~30% (15). Animals were followed up for at least 28 days and then sacrificed.
Results
The pH Changes in the Medium
The pH of the HBSS buffered with NaHCO3 increased from 7.40 ± 0.01 to 7.68 ± 0.03 during the preparation of the culture plates. However, the addition of 4 ml of pregassed PFC to the culture plates (LICS) decreased the pH to 7.49 ± 0.04 after 5 min at 22–24°C. The pH decreased further to a nadir of 7.41 ± 0.06 after 30 min of incubation but then started to rise, reaching 7.82 ± 0.01 after 120 min. In contrast, the pH in the standard HBSS continued to rise over the incubation period to reach 8.05 ± 0.01 after 30 min and 8.35 ± 0.02 after 120 min.
Oxygen Partial Pressure
The pO2 measured at the bottom of culture plates in the control groups after 24-h incubation in standard conditions (37°C, 5% CO2; medium pregassed with 99.9% O2) was similar (150 ± 7.9 mmHg and 146 ± 3.4 mmHg for C2 and C1, respectively, p = NS). In contrast, the pO2 in the LICS plates, measured 24 h after oxygenation of PFC, was still some 20% higher (178 ± 2.3 mmHg, p < 0.001) compared to both control groups.
Islet Viability and Secretory Function
In all groups, islets were intact and round in shape at the end of the culture period and no significant islet loss was encountered. However, those cultured in 4 ml of culture medium had distinct necrotic cores unlike those cultured in either 2 ml of medium or LICS (Fig. 1).

Fluorescent viability assay with propidium iodide and acridine orange. Islets cultured for 48 h in a standard culture medium: C1, 1–4 (2 ml of culture medium); C2, 1–4 (4 ml of culture medium), liquid–liquid interface culture system (LICS, 1–4). Necrotic cores (PI-positive cells; stained in red), which are seen in islets from group C1 (2, 3, and 4) and prominent in C2 (2, 3, and 4), are smaller or completely absent in the majority of LICS cultured islets.
Viability of dispersed islet cells, measured by flow cytometry, was similar in all groups (85.0 ± 3.4%, 84.1 ± 4.6%, and 81.4 ± 4.1% for LICS, C1, and C2, respectively, p = NS) (Fig. 2A). However, viability tests performed on intact islets showed significant differences between the groups. In all groups the viability correlated significantly with culture time and culture conditions (R2 = 0.98, p < 0.01). After 24 h, islet viability in group C1 amounted to 77.5 ± 0.9?% compared to 64.6 ± 0.4% in C2 (Fig. 2B). The viability of islets cultured in LICS for the same time was significantly greater (89.0 ± 2.0?%, p < 0.01 vs. both control groups). A deterioration of viability was observed over the culture period and the percentage of viable islets decreased to 67.9 ± 0.9% and 54.2 ± 0.6% after 48 h and to 62.9 ± 0.8% and 53.4 ± 0.9% after 72 h for groups C1 and C2 (p < 0.01) (Fig. 2B). In contrast, the viability of islets cultured in LICS decreased only to 83.0 ± 1.1% and 79.0 ± 0.5% after 48 and 72 h, respectively (p < 0.05 vs. 24-h culture period). This was significantly greater than in either control group (p < 0.05) (Fig. 2B). Furthermore, the appearance of the islets as assessed by both MTT and by TEM was improved in LICS cultured cells (Fig. 3).
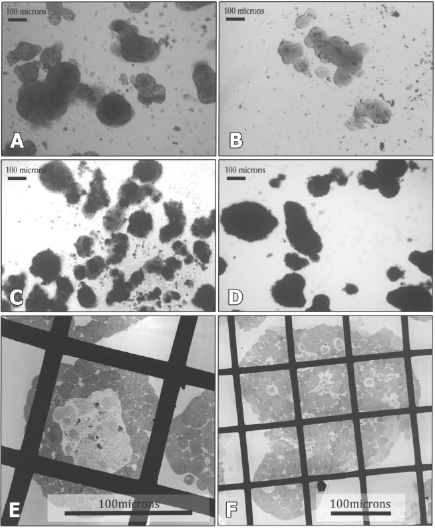
Light (A–D) and electron microscopy (E–F) images of cultured islets. In standard culture medium (A) necrotic, dark cores are noticeable after 48 h whereas in islets cultured on LICS (B) they are much less evident. Staining of islets cultured in standard culture medium with MTT reveals a “patchy” pattern of staining (C) whereas islets cultured on LICS exhibit homogenous staining (D). Cellular debris was also present in islets cultured under suboptimal conditions (C). Necrotic cores are seen even in 100-μm islets, cultured in standard conditions for 48 h (E) and they are rarely observed even in bigger islets cultured in LICS (F).

(A) Differences in viability of islets as assessed using different methods of counting. Flow cytometry: staining with propidium iodide only following islet dispersion; restrictive visual assessment as described in Materials and Methods (staining with propidium iodide and acridine orange) and permissive visual assessment (staining with propidium iodide and acridine orange) inclusive of islets with intermediate fluorescent staining. Data represent isolated islets following 48-h culture in experimental conditions. C1: 2 ml of culture medium, C2: 4 ml of culture medium, LICS: 4 ml of culture medium overlying perfluorocarbon. (B) Viability of islets cultured for 24, 48, and 72 h as assessed by fluorescent viability assay (staining with propidium iodide and acridine orange). C1: standard culture conditions, 2 ml of culture medium; C2: standard culture conditions, 4 ml of culture medium; LICS: liquid–liquid interface culture system.
Although islet insulin content was similar in all groups after 48-h culture (64.2 ng/islet on LICS, 57.3 ng/islet in C1, 58.3 ng/islet in C2, p = NS), the glucose stimulation index (SI) in islets cultured on LICS was more than twice as great as in either control group (12.3 ± 0.4 in LICS compared to 5.8 ± 0.5 in C1, p < 0.01 and 4.1 ± 0.2 in C2, p < 0.01 (Fig. 4).

Average stimulation indices (SI) for islets cultured for 48 h in different culture conditions (C1: standard culture conditions, 2 ml of culture medium; C2: standard culture conditions, 4 ml of culture medium; LICS: liquid–liquid interface culture system).
Graft Function After Intraportal Transplantation
During a 28-day follow-up period, 31.3% (5/16) of recipients transplanted with a marginal graft of freshly isolated islets became normoglycemic and mean blood glucose decreased from 27.6 ± 1.4 mM to 16.9 ± 2.4 mM (Figs. 5 and 6). In the C2 group, only 14.3% (1/7) of the animals regained normal glucose levels and the average blood glucose did not fall significantly (32.2 ± 0.7 mM vs. 28.3 ± 3.8 mM, p = 0.3) (Figs. 5 and 6). The AUC for glycemia in this group was significantly greater than in the fresh control group (821 ± 177 vs. 625 ± 106, p < 0.01).

Nonfasting glucose levels in the recipients of a marginal islet graft before (D0) and 28 days (D28) after transplantation. CTRL: fresh islets; LICS: islets cultured on LICS for 48 h; C2: islets cultured in suboptimal conditions (4 ml of culture medium) for 48 h.

Proportion of normoglycemic recipients after transplantation of a marginal islet mass (2,000 islets/kg). Fresh: freshly isolated islets; C2: islets cultured for 48 h in standard conditions; LICS: islets cultured in liquid–liquid interface culture system for 48 h.
In contrast, in the group transplanted with islets cultured on LICS, 50.0% (3/6) became normoglycemic, twice as many as in the group transplanted with cultured islets and some 60% more even than animals transplanted with fresh islets (p = NS) (Fig. 6). Blood glucose levels were reduced from 32.5 ± 0.5 mM to 19.7 ± 5.1 mM (p < 0.05) (Fig. 5), and the AUC was significantly lower than in the C2 group (590 ± 172 vs. 821 ± 177, p < 0.05) and was comparable with the AUC for control, fresh islets.
Discussion
The culture of islets prior to clinical transplantation has numerous potential advantages. It makes it possible to characterize the purity, sterility, and other properties of the intended graft, it has logistical advantages with respect to scheduling the transplant and cross-matching the patient, and it enables immunosuppression to be commenced a day or two prior to the transplant itself (3). Beneficial effects of islet culture at the molecular level include a decrease in the expression of proinflammatory molecules and recovery of energy stores and a better overall metabolic status (10,14). Unfortunately, however, islet culture may also have deleterious effects on the survival and viability of the islets themselves and hence on transplantation outcome.
Many of these detrimental effects are linked to hypoxia due to the high oxygen requirements of pancreatic β-cells and the slow diffusion of oxygen through the medium (4,11,22,27). This is further compounded by the three-dimensional structure of islets, which inevitably means that the area of islet surface in contact with the plastic floor of the culture dish is not involved in the diffusion of O2 and nutrients into the core of the islet, hence compromising their viability even further (4). Thus, large islets cultured under standard conditions develop dark cores, which relate to central necrosis/apoptosis (5,11), especially in the presence of a high cell density or high glucose concentrations (11,30). In our experiments, when islets were cultured in optimal and suboptimal conditions (an estimated fluid depth of 2.1 and 4.2 mm, respectively) central hypoxia resulted in dark centers within 48 h (Figs. 1 and 2). Electron microscopy confirmed the presence of necrotic centers within even small islets cultured in suboptimal conditions (Fig. 2). Staining of intact islets with MTT displayed a relatively patchy pattern (although this test is qualitative and subjective it complements other data obtained in this series of experiments) (Fig. 2).
In contrast, islets cultured on LICS can receive oxygen from all directions, because the perfluorocarbon cultivation surface can deliver as much oxygen as the overlying medium, which is in agreement with Buchwald's theoretical model of islet oxygenation (4). Indeed, central necrosis occurred in LICS-cultured islets to a much lesser extent despite a suboptimal volume of medium, presumably due to improved oxygen diffusion into the islet cores as a result of the increased ambient pO2 (Figs. 1 and 2). Electron microscopy confirmed that central necrosis was rarely observed even within the large islets (>150 μm) (Fig. 2). The viability of intact islets, as assessed by acridine orange and propidium iodide staining, was improved and preserved over the 72-h culture period when islets were cultured on LICS compared with controls. On the other hand, flow cytometry did not show any significant differences between the experimental groups. This discrepancy probably reflects a selective loss of necrotic cells during the preparation of the cell suspension for flow cytometry. Preservation of islet viability was also reflected by a homogenous and intense staining with MTT (Fig. 2).
Insulin secretion in response to glucose stimulation was significantly improved in LICS-cultured islets when compared with control islets. This positive effect seems to be related to preserved viability and improved secretory function of LICS-cultured islets rather than to increased insulin production, as insulin content was comparable in all groups.
In our experiments we have also shown that the composition of gases used to persufflate the perfluorocarbon ameliorates pH changes in the medium buffered with bicarbonate. Theoretically, this property of perfluorocarbons could increase the buffering capacity of the medium and hence aid gas exchange in cell culture. During islet transportation, CO2 accumulation may exceed the buffering capacity of a standard medium. This excess can be absorbed by a perfluorocarbon and hence eliminated from the culture medium. The persufflation of perfluorodecalin with a mixture of gases containing CO2 might not only reduce the amount of extra CO2 that can be absorbed from the islets in culture but theoretically might also limit the amount of dissolved oxygen in the PFC which has a higher affinity for CO2 than for O2.
The marginal graft size that we used rendered 50% of animals receiving LICS-cultured islets normoglycemic compared with only 31% of animals transplanted with freshly isolated islets. It may be that the postisolation culture of pancreatic islets may eliminate those islets that would otherwise die immediately following transplantation. This natural selection could theoretically improve transplantation outcome, though it should be noted that the same effect was not seen when islets were cultured for 48 h in conventional culture under suboptimal conditions. This is consistent with results published recently by Ihm et al. indicating that islets cultured in optimized conditions reverse diabetes in a greater proportion of animals than freshly isolated islets irrespective of islet loss during culture (14). In our experiments, control islets were cultured in suboptimal conditions (high medium depth), which resulted in poor transplantation outcome. Following optimization of culture conditions by using perfluorocarbon (despite keeping the volume of the medium identical in both experiments) a noticeable improvement in the outcome of intraportal transplantation was observed.
In addition to their oxygen carrying capacity, perfluorocarbons are able to dissolve other gasses such as CO2 and N2, and to dissolve lipid-soluble substances (6). The ability of perfluorocarbons to inhibit the formation of reactive oxygen species, to transfer lipid-soluble substances from the islet cells or pancreas (hydrophilic phase) to PFC (hydrophobic phase) acting as a sump for toxic compounds and hence remove them from the culture medium, has never been demonstrated in pancreas preservation or islet cell culture (12). Nevertheless, it is possible that these properties may be responsible for the positive effects observed in our experiments by diminishing the damage to cellular membranes and the extracellular matrix of cultured islet cells, resulting in improved viability and secretory function (18,21).
It is also possible that the benefit of exposure to PFCs may extend to the posttransplant period. It is known that a PFC may induce cell membrane changes as it incorporates into the membrane structure (8,21), and it is conceivable that these may facilitate transmembrane oxygen transport following transplantation into the hypoxic environment of the portal vein and hence modulate the islet response to the hostile intraportal environment which results from molecules such as free radicals and proinflammatory cytokines in the liver (21).
Surprisingly, given their physical characteristics, there are only a few reports of the application of perfluorocarbons to islet culture and transportation (2,9,28,31). Zekorn et al. first described improved islet secretory function, as gauged by a significant increase in the glucose stimulation index, using an emulsion of perfluorocarbon (FC-43) as a culture medium additive (31). Recently, Maillard et al. used a similar approach to Zekorn et al.'s and cultured islets in a perfluorocarbon emulsion. Application of this technique resulted in preservation of islet viability and function, and protection of extracellular matrix (18). In contrast, Bergert et al. described detrimental effects of perfluorocarbons on islet viability (2). However, they used perfluorohexane, a highly volatile perfluorocarbon not suitable for tissue culture systems. Because their biological and physical properties vary considerably between different PFCs, it is quite possible that the beneficial effects and lack of toxicity we observed are specific not only to the nature of the compound but also to the clinical grade of perfluorodecalin that we used. This PFC has previously been safely used for pancreas preservation prior to the isolation of islets for transplantation and has been claimed to have beneficial effects despite the fact that oxygen dissolved in perfluorodecalin has been found not to penetrate whole pancreas (23,26).
We have previously described our liquid–liquid interface culture system, and now show that it has beneficial effects not only on in vitro indices of islet function but also on the outcome of intraportal transplantation in vivo. Further studies are required to elucidate the exact mechanism of these beneficial effects and, in particular, to establish to what extent they relate to the ability of perfluorodecalin to provide oxygen or to its capacity to act as a sump for CO2 or lipophylic, organic substances derived from cultured cells (this applies equally to the mechanism of action of PFCs used for whole pancreas preservation).
Although our study represents only proof of principle at this stage, there are additional reasons to hope that the LICS technique can be developed to form part of clinical protocols. As the result of increasingly strict GMP criteria, as much islet manipulation as possible is now performed in a controlled and closed environment. The use of advanced gas-permeable membranes for culture vessels necessitates a controlled environment outside the vessel in order to create optimal conditions for culture or transportation. In contrast, the utilization of a perfluorocarbon-based LICS confines the environment, which needs to be controlled to the size of the culture or storage container. Furthermore, the use of two phases (PFC and water) allows for easy replenishment of the perfluorocarbon as required without the need for additional manipulation of the islets, thereby minimizing the risk of islet damage or contamination.
We have shown that islets cultured on LICS are able to engraft satisfactorily and correct diabetes in a greater proportion of animals than islets cultured in standard conditions or even freshly isolated islets. If similar benefits in the absence of PFC toxicity can be shown for human islets, especially with a high cell seeding density, there seems no reason why the technique should not be used clinically for the pretransplant storage and culture of islets and for their transportation to distant transplantation centers, thus ensuring the maximum benefit from an intraportal islet transplant.
Footnotes
Acknowledgments
This work was jointly supported by Diabetes UK Project Grant (BDA: RD03/0002694) and The Henry Smith Charity.