
Abstract
Hepatocyte transplantation (Tx) holds promise for curing genetic liver diseases. However, a limited number of donor hepatocytes can be transplanted into the host liver. Recipient preconditioning and donor cell engineering are under investigation to improve cell engraftment. In theory, genetically engineered cells secreting therapeutic proteins with superior function could compensate for poor engraftment efficiency. We have generated a bioengineered human coagulation factor IX (FIX) with augmented specific activity (named FIX-Triple). The aim of this study was to evaluate therapeutic efficacy of cell therapy using hemophilia B (HB) as a disease model by transplanting FIX-Triple-secreting hepatocytes. The donor hepatocytes were isolated from FIX-Triple knock-in (KI) or FIX-WT (wild-type) KI mice and transplanted intrasplenically into FIX knock-out (KO) mice. FIX-Triple KI recipients exhibited fourfold higher plasma FIX clotting activity than FIX-WT KI recipients. By repeated Txs, the clotting activity of FIX-Triple KI recipients even increased to more than 10% of normal mouse plasma. The engraftment and FIX production efficiencies of transplanted cells were equivalent between the FIX-WT KI and FIX-Triple KI donors. A hemostatic function assay showed that FIX-Triple KI recipients with repeated Txs had more enhanced clot kinetics and a greater maximum rate of thrombus generation than those with a single Tx. Moreover, FIX inhibitors in these recipients rarely developed. In conclusion, hepatocyte Tx with genetically engineered donor cells is an effective therapeutic strategy for HB.
Keywords
Introduction
Hepatocyte transplantation (Tx) is a promising approach for the treatment of genetic liver diseases (14, 39). However, a significant hurdle is the poor engraftment efficiency of 0.03–0.5% of transplanted cells in the host liver (19,42). Numerous strategies have been developed to increase cell engraftment and to stimulate hepatocyte proliferation. Recipient preconditioning methods (e.g., drug-induced hepatic injury, hepatectomy, and irradiation) were tested in animal models (18,52–54). We previously demonstrated that transplanted cells can be repopulated extensively in the host liver after recipient preconditioning treatment (52,53). Although recipient preconditioning has been successfully demonstrated in preclinical studies, these protocols are invasive and therefore inappropriate for clinical applications. To address the issue of recipient biosafety, donor cells genetically engineered for immortalization and enhanced growth advantages were established for cell therapy (6,24,50). Furthermore, there may be an alternative strategy for donor cell engineering. We hypothesize that genetically engineered donor cells secreting therapeutic proteins with superior function can compensate for poor engraftment efficiency.
Hemophilia B (HB) is a monogenic liver disease caused by a lack of functional coagulation factor IX (FIX) (23). The disease exhibits a range of clinical severity closely correlated with FIX clotting activity. Patients with >1% of normal FIX clotting activity after treatment, spontaneous soft tissue bleeds and excessive bleeds can be significantly improved, and those with >5% of normal FIX clotting activity, major bleeds and the development of chronic hemophilic arthropathy rarely occur. The current treatment for HB is frequent intravenous infusions with FIX protein, which is inconvenient and affects the quality of life. In vivo gene therapy is under investigation for long-term expression of FIX, but issues of tumorigenicity and immune responses still need to be overcome (13,32,37). Alternatively, cell therapy offers an opportunity to treat this bleeding disease. Tatsumi et al. pioneered the treatment of HB by hepatocyte Tx (49). In their mouse model, the donor hepatocytes contributed 1% and up to 1.85% of FIX clotting activity after single and repeated Txs, respectively. Their pilot study indicates that hepatocyte Tx is a potential HB therapy, but the poor engraftment efficiency needs to be improved.
FIX molecules with augmented specific activity are useful for treating HB (4,8,9,45). However, cell therapy in this field has never been investigated. We previously reported that a bioengineered human coagulation FIX (FIX-Triple) harboring V86A, E277A, and R338A re-placements exhibited 13-fold higher specific activity than FIX-WT (wild-type) (30). In this study, we produced bioengineered FIX-secreting hepatocytes driven by endogenous gene regulators and used these materials to test the therapeutic effects of cell therapy.
Materials and Methods
Animal Models
The recipient mouse model was FIX knock-out (KO) mice (B6.129P2-F9 tm1Dws , kindly obtained from Dr. Darrel Stafford) backcrossed to the C57BL/6 genetic background for more than 10 generations (31). The male FIX-KO mice were used at 6–8 weeks of age. The donor mouse model was human FIX knock-in (KI) mice carrying a single copy of the human FIX gene controlled by mouse endogenous gene regulators. Two strains of male FIX-KI mice were used at 12–16 weeks of age: FIX-WT (wild-type) KI and FIX-Triple KI mice backcrossed to the C57BL/6 genetic background for more than six generations [B6.129(Cg)-F9 tm1(hF9)Swl and B6.129(Cg)-F9 tm2(hF9-V86A/E277A/R338A)Swl ]. Inbred C57BL/6 male mice (National Laboratory Animal Center, Taiwan) at 6–8 weeks of age were used for the evaluation of engraftment efficiency at the early stage. All mice were treated according to the guidelines of the National Taiwan University in Taiwan.
Isolation and In Vitro Culture of Hepatocytes
The FIX-KI mice were anesthetized using sodium thiopental (Pentothal®, Hospira, Lake Forest, IL, USA). Mouse hepatocytes were isolated by collagenase perfusion as described previously (52). The isolated hepatocytes were seeded in a 6-cm petri dish with 1.5 million viable cells. After overnight culture, the hepatocyte growth medium (William's medium E containing 10% fetal bovine serum) (Gibco, Grand Island, NY, USA) was changed to 1 ml serum-free medium supplemented with insulin/transferrin/sodium selenite (I1884, Sigma, St. Louis, MO, USA) and vitamin K1 (10 μg/ml, Standard Chem. & Pharm., Tainan, Taiwan). The 24-h cultured media were collected for analysis of FIX clotting activity and antigen levels (see below).
Transplantation Procedures
Donor cells for Tx were used only when >85% excluded trypan blue dye. Thirty minutes before surgery, the FIX-KO mice were intravenously infused with recombinant FIX (BeneFIX®, Wyeth Pharmaceuticals, Collegeville, PA, USA) at 100 IU/kg. Under anesthesia, the spleen of the recipient was exposed by a left subcostal incision and immediately injected with ~1.5 million viable hepatocytes in 100 μl serum-free Dulbecco's modified Eagle medium (DMEM) (Gibco) through the splenic pulp. Hemostasis was secured by splenic ligature.
Enzyme Linked Immunosorbent Assay (ELISA)
Mouse blood (200 μl) collected from the retro-orbital plexus using capillary glass tubes was mixed with one-tenth volume of 3.8% sodium citrate (JT Baker, Phillips-burg, NJ, USA) and centrifuged at 4500 × g for preparation of citrated plasma. The plasma samples were stored at −80°C prior to assay. The amount of human FIX synthesized in vivo was quantified by using goat anti-human FIX antibodies as coating and detecting antibodies [GAFIX-AP160 and horseradish peroxidase (HRP)-conjugated GAFIX-AP190, Enzyme Research Laboratories, South Bend, IN, USA]. Normal human plasma (Dade Behring, Marburg, Germany) was used to generate standard curves for the quantification of test samples. Human FIX in the standard was assumed to be 5000 ng/ml.
Activated Partial Thromboplastin Time (aPTT)
FIX clotting activity was determined by using the aPTT reagent (Actin® FSL, Dade Behring) as the activator in a semiautomated blood coagulation analyzer following the manufacturer's instructions (CA-50, Sysmex, Kobe, Japan). Normal mouse plasma was made from inbred C57BL/6 male mice (n = 18) to generate standard curves for the quantification of test samples. FIX clotting activity in the standard was assumed to be 100%. Test samples were diluted at 1:5 and 1:10 in sodium barbital buffer prior to analysis. A one-stage clotting assay was performed by incubating 50 μl of test sample with 50 μl of human FIX-deficient plasma (American Diagnostica, Stamford, CT, USA) for 1 min at 37°C and adding 50 μl of activator for 3 min at 37°C. Then 50 μl of 25 mM CaCl2 was added and the time to clot was measured. Each value was reported after subtraction of the mean baseline level in the FIX-KO mice.
Engraftment Efficiency by Cell Labeling and FIX DNA and mRNA Quantification
For cell labeling, the isolated hepatocytes were labeled with a fluorescent mitochondrial marker (MitoTracker® Red CMXRos, Invitrogen, Carlsbad, CA, USA) at a dilution of 1:1000 and incubated at 37°C in a humidified incubator with 5% CO2 for 30 min. The labeled hepatocytes were then transplanted with 1.5 million viable cells into inbred C57BL/6 mice. At 3 and 6 h after Tx, the mice were sacrificed for collection of livers and spleens. Discontinuous 15-μm-thick tissue cryosections were counterstained with DAPI (Biogenix, San Ramon, CA, USA), and fluorescent images were taken using a fluorescent microscope (Leica DMR, Wetzlar, Germany). The number of the labeled donor cells in the portal areas was determined. Sixty tissue cryosections from three mice per group were used at each time point. For DNA quantification, genomic DNA was extracted from mouse livers (Wizard® Genomic DNA Purification kit, Promega, Madison, WI, USA) and quantified by quantitative PCR (Q-PCR) (iQ™ SYBR® Green Supermix and iQ5 Real-Time PCR Detection System, Bio-Rad, Hercules CA, USA) using human FIX primers (5′-GGAAGCAGTATGTTGATGG-3′ paired with 5′-TGGTTCACAGGACTTCTGGT-3′). Mouse sequences on chromosome X were used as an internal control (5′-TCTCCACTTCCTCTCACCAG-3′ paired with 5′-ACA CAGAAACCACCCTTGC-3′). For mRNA quantification, mouse livers were frozen in liquid nitrogen and ground into powder. Total RNA was purified using a commercial kit (Super RNApure™ kit, Genesis Biotech, Taipei, Taiwan) according to the manufacturer's protocol. First-strand cDNA was synthesized by reverse transcriptase (SuperScript™ III reverse transcriptase, Invitrogen) using oligo-dT primers. The cDNA was immediately quantified by Q-PCR using human FIX primers. The mouse β-actin gene was used as an internal control (5′-CACAGTGTTGTCTGGTGGTA-3′ paired with 5′-GACTCATCGTACTCCTGCTT-3′).
Hemostatic Function Assay
Whole blood mixed with one-tenth volume of 3.2% sodium citrate was assayed by thromboelastography (TEG® analyzer, Haemoscope, Braintree, MA, USA) using the citrated-kaolin mode. Analyses for the variables were conducted following the manufacturer's instructions.
Detection of FIX Inhibitors
Total IgG antibodies against human FIX were measured by an ELISA-based assay. Microtiter plates were coated with recombinant human FIX-Triple protein (1 μg/ml). Purification and quantification of recombinant proteins were performed as described previously (10). Mouse plasma samples were applied to wells at 1:40 dilution. Mouse plasma from the untreated FIX-KO mice (n = 9) were used as a baseline control, and those spiked with monoclonal anti-human FIX antibody (F2645, Sigma) were used as a positive control. Goat anti-mouse IgG conjugated with HRP (Chemicon, Temecula, CA, USA) was used as the detection antibody (1: 2000). Anti-FIX antibodies were quantified using standard curves from the wells coated with goat anti-mouse IgG antibodies (four subtypes, Sigma) and treated with mouse IgG (Sigma).
Statistical Analysis
Statistical analysis was done with SPSS 10.0.7C for Windows (SPSS Inc., Chicago, IL, USA). Data are shown as mean values ± SEM and were compared by two-tailed unpaired Student's t-tests or analysis of variance (one-way ANOVA) with LSD post hoc analysis. Statistical significance was set at p < 0.05.
Results
Study Design
We generated FIX-KI mice in which mouse FIX coding sequences were replaced with human FIX cDNA. In this mouse model, the interference of mouse FIX was eliminated, and human FIX was expressed under the control of mouse endogenous gene regulators based on the strategy described by Jin et al. (25). Two strains of FIX-KI mice were used in this study: those with bioengineered FIX with augmented specific activity (FIX-Triple KI) and those with wild-type FIX (FIX-WT KI) as a control. In our previous study, FIX-Triple KI mice showed sevenfold higher specific plasma FIX activity than FIX-WT KI mice (30). We used these mice to test in vivo cell therapy.
In Vitro Culture of Hepatocytes Isolated From FIX-KI Mice
To establish hepatocytes from the FIX-KI mice as a source of FIX, hepatocytes isolated from FIX-WT KI and FIX-Triple KI mice were separately cultured with 1.5 million viable cells. FIX in the culture medium was measured by human FIX-specific ELISA and aPTT assays (Fig. 1). The hepatocytes from FIX-WT KI and FIX-Triple KI mice secreted comparable amounts of FIX (14.80 ± 0.58 ng/ml for FIX-WT and 16.20 ± 1.32 ng/ml for FIX-Triple, p = 0.36). The clotting time of FIX-Triple-secreting hepatocytes was significantly shorter than that of FIX-WT-secreting hepatocytes (42.42 ± 0.57 vs. 59.92 ± 0.58 s, p < 0.001). These results indicated that the isolated hepatocytes were responsible for the production of FIX and were functionally intact in terms of FIX secretion and enzymatic activity.

Clotting time of in vitro cultured donor hepatocytes. The clotting time of FIX-Triple-secreting hepatocytes was significantly shorter than that of FIX-WT-secreting hepatocytes. FIX-KO hepatocytes were used as a negative control and as a positive control when supplemented with human FIX at physiological levels. n = 5 replicate cultures/group.
Single Tx with Hepatocytes From FIX-KI Mice
To test the hypothesis that bioengineered FIX with augmented specific activity is a useful tool for cell therapy, we intrasplenically transplanted the isolated hepatocytes (1.5 million cells/mice) from FIX-WT KI or FIX-Triple KI mice into FIX-KO mice. The recipients of hepatocytes from FIX-Triple KI donors exhibited 3.46 ± 0.40% to 5.75 ± 0.51% of normal mouse FIX clotting activity, whereas the recipients of FIX-WT KI donors exhibited only 0.83 ± 0.23% to 2.69 ± 0.24% of normal mouse FIX clotting activity (Fig. 2A). It was observed that FIX clotting activity in both groups declined to a plateau after day 5. Note that the clotting activity of FIX-Triple KI recipients, compared at each plateau level, was consistently fourfold higher than that of FIX-WT KI recipients. We also performed ELISA for FIX antigen detection. As shown in Figure 2B, the FIX antigen levels in recipient plasma were <1% (50 ng/ml) of normal human plasma (range of mean values for plateau levels: 16–21 ng/ml for FIX-WT and 14–15 ng/ml for FIX-Triple), and no statistical difference was found at any time point (p = 0.83). Collectively, genetically engineered hepatocytes boosted FIX clotting activity to therapeutic levels with low antigen levels of bioengineered FIX (FIX-Triple).

Tx with hepatocytes from FIX-KI mice. (A) FIX-Triple KI recipients (n = 10) consistently showed fourfold higher FIX clotting activity than FIX-WT KI recipients (n = 7). The clotting activity of FIX-Triple was elevated from 6.38 ± 0.54% to 11.87 ± 0.85% of normal mouse plasma after repeated Txs (n = 12). (B) Throughout the study, there was no significant difference in plasma antigen levels between FIX-WT and FIX-Triple. (C) Comparison of plasma antigen levels of FIX on 5 or 10 days after single/repeated Txs. The plasma antigen levels of FIX-Triple were significantly enhanced after repeated Txs.
Repeated Tx with FIX-Triple KI Donor Hepatocytes
To evaluate the potential therapeutic effects of FIX-Triple, we performed repeated Txs. We showed that FIX clotting activity increased approximately twofold (up to 11.87 ± 0.85% of normal mouse plasma) relative to a single Tx (Fig. 2A). The plasma antigen levels of FIX were also elevated after repeated Txs (Fig. 2C). Our data suggest that FIX-Triple-secreting hepatocytes have the potential to expand their therapeutic capacity by repeated Txs.
Engraftment Efficiency of the FIX-KI Donor Hepatocytes
Engraftment efficiency was demonstrated by cell labeling at the early stage (day 1) and quantitative PCR at the later stage (day 20). For the early stage, fluorescently labeled donor cells were used. The shapes and positions of the donor cells in the mouse liver after Tx were recorded. The numbers of labeled hepatocytes in the portal areas from FIX-WT KI and FIX-Triple KI donors were not significantly different at 3 h after Tx (0.88 ± 0.07 cell/portal area for FIX-WT and 0.88 ± 0.11 cell/portal area). The data were also indistinguishable at 6 h after Tx. At the later stage, there was no statistical difference in the ratios between donor and recipient liver cells (0.51 ± 0.14% for FIX-WT and 0.62 ± 0.06% for FIX-Triple, p = 0.44). Also, the engraftment efficiency of FIX-Triple KI donor hepatocytes was enhanced to twofold after repeated Tx (1.16 ± 0.11%). We also measured the mRNA levels of FIX in the liver. There was no statistical difference between the amounts of mRNA coding for FIX-WT and FIX-Triple (p = 0.96). These results indicated that the engraftment efficiency and amount of FIX transcripts of FIX-WT KI and FIX-Triple KI donor hepatocytes transplanted into the liver were comparable with those of normally distributed donor cells.
Hemostatic Function Assay of FIX-Triple KI Recipients
To access all the interacting cellular and plasma components in hemostasis, FIX-Triple KI recipients with single or repeated Txs were subjected to an overall assessment of ex vivo whole blood clot formation by thromboelastography (TEG). As shown in Table 1, compared with the nontransplant FIX-KO mice, FIX-Triple KI recipients exhibited shortened time to initial fibrin formation (R) consistent with the plasma aPTT assays. Most importantly, the TEG results provided evidence that the recipients with repeated Txs showed faster clot kinetics (K and Angle) (Table 1, Fig. 3A) and a greater maximum rate of thrombus generation (MRTG) than those with a single Tx (6.73 ± 1.49 vs. 2.69 ± 0.69 mm, p = 0.02) (Fig. 3B). We also established a coagulation index (CI) in inbred C57BL/6 male mice based on the human equation with a combination of R, K, Angle, and maximum amplitude (MA) (12). Values above +4.5 were considered hypercoagulable, whereas values below +2.5 were hypocoagulable. The values in the recipients were hypocoagulable but significantly improved (Table 1). These data suggest that FIX-Triple KI recipients with repeated Txs exhibit further enhanced hemostasis.

Representative plots of TEG tracing and thrombus generation in FIX-Triple KI recipients with single or repeated Txs on post-Tx day 12. (A) The TEG tracings showed that clot kinetics (K and Angle) of FIX-Triple KI recipients with repeated Txs were stronger than those with a single Tx. The definitions and means of each variable are indicated in Table 1. (B) FIX-Triple KI recipients with repeated Txs had a greater MRTG (maximum rate of thrombus generation) than those with a single Tx. MRTG and TTG (total thrombus generation) are indicated by arrowheads. B6, inbred C57BL/6 mice; HB, FIX-KO mice.
Effects of Whole Blood Hemostasis on FIX-Triple KI Recipients by TEG

B6, inbred C57BL/6 mice; HB, FIX-KO mice; R (reaction time), the time to initial fibrin formation; K, the speed to reach a specific level of clot strength; Angle, the rapidity of fibrin build-up and cross-linking; MA (maximum amplitude), the reflection of clot strength contributed by platelets and fibrin; CI (coagulation index), overall assessment of coagulability.
p < 0.001 compared with HB.
p < 0.01 compared with HB.
p < 0.05 compared between single and repeated Txs.
p < 0.01 compared between single and repeated Txs.
Long-Term Observation of FIX-Triple KI Recipients with Repeated Txs
The plasma FIX clotting activity in FIX-Triple KI recipients was continuously monitored after repeated Txs. As shown in Figure 4A, FIX clotting activity was measured to be 8% of normal mouse plasma for 2 months without a significant decline. Meanwhile, we performed ELISA for the detection of FIX inhibitors. FIX inhibitors did not develop in the FIX-Triple KI recipients except in one case with weak positive signals (Fig. 4B). These results demonstrated that the clotting activity of FIX-Triple remained at therapeutic levels for 2 months, and that FIX inhibitor in the recipients was rarely detected.
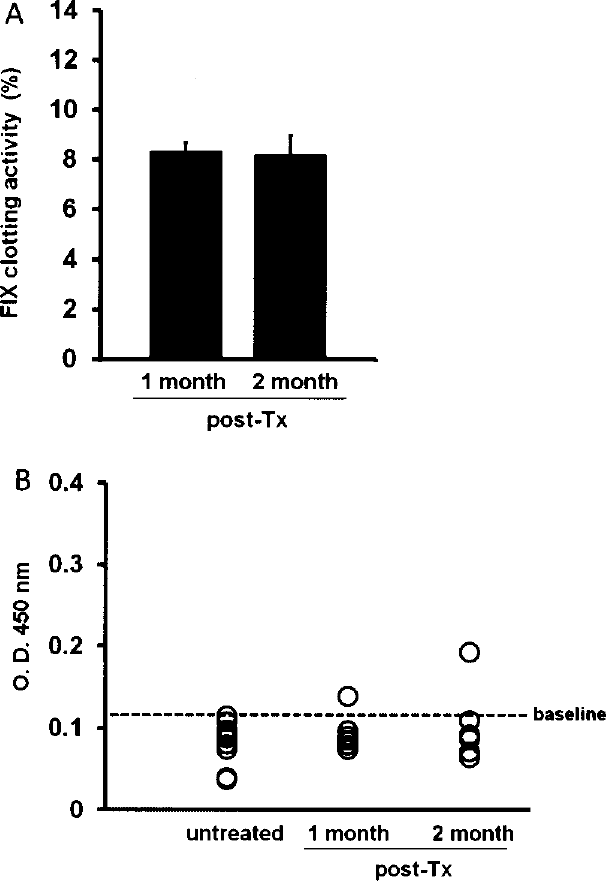
Long-term observation of FIX-Triple KI recipients with repeated Txs. (A) FIX clotting activity was measured to be 8% of normal mouse plasma at 1 (n = 8) and 2 months (n = 6) after repeated Txs. (B) FIX inhibitors were measured by ELISA and presented as optical density (O.D.) at the wavelength of 450 nm. The baseline (dashed line) is the maximum value measured in untreated FIX-KO mice (n = 11). Each open circle represents one mouse. Only one FIX-Triple KI recipient had a value above baseline at 1 and 2 months after repeated Txs (the same recipient).
Discussion
Hepatocyte Tx is a less invasive strategy compared to orthotopic liver Tx for the treatment of acute or chronic liver failure, and inherited metabolic or bleeding diseases (2,11). However, since the first hepatocyte Tx was performed on a Gunn rat with Crigler-Najjar syndrome, poor cell engraftment has always been a limiting step for further clinical applications (17,19,35,42). Therefore, methods of recipient preconditioning and donor cell engineering are under investigation to improve cell engraftment (6,18,24,50,52–54). Donor cell engineering is an attractive alternative for cell therapy because gene modification is done ex vivo, and the resultant changes in cell properties can be fully examined prior to applications. Immortalized cells were produced to improve cell engraftment, but their clinical applicability remains questionable due to unacceptable tumorigenic risks (44). We therefore set out to find another way to improve the efficacy of cell therapy.
This study was a proof-of-principle study in which engineered donor hepatocytes secreting therapeutic proteins with superior function were used to treat a genetic liver disease. We previously reported that purified FIX-Triple protein exhibited 13-fold higher specific activity than FIX-WT, and studies in FIX-KO mice showed that FIX-Triple was also effective in vivo (30). Although several bioengineered FIX molecules with augmented specific activity have been reported, to our knowledge there have been no publications up to now that investigated the influence of bioengineered FIX on hepatocyte Tx (4,8,9,45). Our study revealed that transplanting genetically engineered hepatocytes into FIX-KO mice effectively improved their hemophilia from severe (<1%) to moderate (1–5%) or mild (5–40%) by secreting FIX-Triple with normal cell engraftment. We also showed that the therapeutic effect of FIX-Triple can be further improved by repeated Txs.
FIX-Triple KI recipients showed fourfold higher FIX clotting activity than FIX-WT KI recipients, while purified FIX-Triple protein was reported to have 13-fold higher specific activity than FIX-WT, and FIX-Triple KI mice had sevenfold higher specific plasma FIX activity than FIX-WT KI mice (30).
In the current study, we presented our data with FIX clotting activity instead of specific activity calculated by dividing the clotting activity over its protein level. When we analyzed these data by specific activity for each plateau level, FIX-Triple KI recipients consistently showed five- to sevenfold higher specific plasma FIX activity than FIX-WT KI recipients. The reason that FIX-Triple protein purified from human embryonic kidney (HEK) 293 cells has greater specific activity is unclear. The simple difference is cell type: HEK 293-synthesized FIX-Triple versus mouse hepatocyte-synthesized FIX-Triple; that is, posttranslational modifications essential for protein maturation can be different in both the species and the cell type (27), and may affect the specific activity of the gain-of-function mutein. Also, it is theoretically possible that purified FIX-Triple protein is fractionated to homogeneity whereas FIX-Triple in in vivo expression system can be more heterogeneous in biological function. Even so, the evidence demonstrated that FIX-Triple is a good alternative substitute for FIX-WT in protein replacement therapy and gene-based therapy.
The maintenance of therapeutic levels of FIX clotting activity for 2 months was an encouraging result. Allogenic transplanted hepatocytes can only survive for 7–10 days without suppression of the host's immune responses (5,20). Because the FIX-KI and FIX-KO mice are incipient congenic and congenic strains of the C57BL/6 genetic background, respectively, it is more likely that the majority of donor cells were lost due to the innate immune system soon after Tx, and the remaining cells maintained their functions without clearance by the adaptive immune system. Furthermore, FIX inhibitors were detected with a low frequency in FIX-Triple KI recipients. Inhibitor development after FIX protein infusions occurs at a frequency of 1–3% in severe HB patients (3), and thus this issue should be taken into consideration for cell therapy. It was not surprising to detect no FIX inhibitors in almost all the mouse plasma samples. For one, there is increasing evidence that liver is an immunologically privileged organ (28, 29). Also, one residue with alanine replacement (E277A) of FIX-Triple is located within the highly immunogenic peptide region (L272-C289) of FIX-WT in inbred C57BL/6 mice (15). It is possible that altering the amino acids in this region may make the FIX molecule less immunogenic. The mouse strain we used is C57BL/6, which is known not to be very likely to make antibodies against human FIX protein or express FIX after gene transfer (36). As a consequence, the immunogenicity of FIX-Triple requires further investigation prior to clinical applications.
In this study, bioengineered FIX-secreting hepatocytes were isolated from the livers of FIX-KI mice in which the human FIX gene was controlled by mouse endogenous gene regulators. We presume that this transgene construct mimics the natural gene regulation environment at the FIX locus, unlike chimeric gene fusion of strong promoters or enhancers. Because high levels of FIX (>129%) in circulation is thought to increase thrombosis risk, the expression level of FIX should be noted, especially when using bioengineered FIX with augmented specific activity (46,51). Although FIX clotting activity was not improved to very high levels in this study, the strategy of FIX expression we used may minimize the possibility for occurrence of unexpected events (e.g., uncontrolled overexpression or ectopic expression of the gene product). Furthermore, modifying a gene of interest by gene targeting instead of viral-mediated random integration into the genome is more reliable, which avoids the potential problems caused by viral sequence-mediated tumorigenicity and poor transduction rate of virus vehicles (13,21,34). Consequently, the FIX-KI mice were thought to be an ideal model for investigating hepatocyte Tx.
In a clinical trial, an ex vivo gene therapy for homozygous familial hypercholesterolemia has been reported in which autologous hepatocytes were prepared from the resected liver and transduced with viral vectors encoding therapeutic gene products (low density lipoprotein receptor) (16,43). We hypothesize that it is also feasible and safe to treat HB with ex vivo genetically engineered autologous cells expressing bioengineered FIX with augmented specific activity. The use of autologous cells is thought to avoid potential adaptive immune responses. However, the source of autologous hepatocytes is limited, because the primary hepatocytes must be obtained through invasive methods and are not easy to cryopreserve (1,38). This problem may be relieved by introducing the concept of induced pluripotent stem (iPS) cells (48,55).
It is now known that human skin cells can be reprogrammed into an embryonic stem cell-like state (i.e., iPS cells), which serves as an inexhaustible cell source (41) and maintains the ability to differentiate into hepatocyte-like cells (47). Moreover, iPS cells can be generated in a virus-free system (26,40), and the defective gene of iPS cells can be corrected by gene targeting (22,56). By editing the defective FIX gene locus of the patient's iPS cells and then differentiating the iPS cells to hepatocyte-like cells, we think that iPS cell-based therapy in combination with bioengineered FIX-secreting hepatocyte-like cells may provide an effective individual therapy for HB. For instance, full-length cDNA coding for FIX-Triple could replace the defective gene of the HB patients caused by various mutations (point mutations, deletions, or additions downstream of the intrinsic regulatory elements) through zinc-finger nucleases (ZFN)-mediated gene targeting, the technique proven to greatly improve low efficiency of homologous recombination in human iPS cells (56). In this way, the resultant bioengineered iPS cells can be an inexhaustible cell source for differentiation into hepatocyte-like cells. The FIX-Triple secreting hepatocyte-like cells are unique for the patient and supposed to eliminate the risk of rejection and the need for immunosuppressive drugs. Besides HB, other inherited coagulation disorders like coagulation factor VII or VIII deficiency may be also suitable for this strategy because the variants with superior function have been verified (7,33).
Taken together, these results provide the first evidence that hepatocyte Tx for the treatment of HB can effectively improve therapeutic levels by using bioengineered FIX with augmented specific activity. Although there seems a long way to go before taking this idea into the clinic, this study preliminarily establishes a novel approach for the evaluation of possible personalized HB therapies using donor cells genetically engineered ex vivo.
Footnotes
Acknowledgments
We are grateful to the Transgenic Mouse Models Core of National Research Program for Genome Medicine (NRPGM) and the Gene Knockout Mouse Core Laboratory of NTU Research Center for Medical Excellence for generating the FIX-KI mice. We also thank Ms. Chia-Chi Chen for technical assistance. This work was supported by grants from National Taiwan University Hospital (NTUH96A15; NTUH97-00872) and from the National Science Council (NSC95-2320-B-002-054; NSC96-3112-B-002-023; NSC97-2752-B-002-001-PAE; NSC98-2627-B-002-015).